Obesity and diabetes are currently a major health problem worldwide with growing in prevalence. Obesity and diabetes induce secondary diseases with various pathophysiologic states including cardiovascular disease, neural disturbance, kidney disease, cancer and osteoporosis. Bone homeostasis is maintained through a delicate balance between osteoblastic bone formation and osteoclastic bone resorption. Aging and numerous pathological processes induce decrease in osteoblastic bone formation and increase in osteoclastic bone resorption, leading to osteoporosis with decrease in bone mass. Osteoblasts and adipocytes differentiate from a common precursor cell in the bone marrow mesenchymal stem cells. Type 1 and obese type 2 diabetes have been associated with increased fracture risk. Nutritional chemical factors are found to prevent diabetic osteoporosis. Zinc, Satsuma mandarin orange (Citrus unshiu MARC.) β-cryptoxanthin and plant component p-hydroxycinnamic acid was found to reveal stimulatory effects on osteoblastic bone formation and suppressive effects on osteoclastic bone resorption, thereby increasing bone mass. These factors were found to improve type 1 diabetic bone loss in vivo. Moreover, p-hydroxycinnamic acid suppressed adipogenesis in bone marrow cells in vitro. Such functional food factors may improve bone loss implicated with type 1diabetes and obese type 2 diabetes.
Obesity, diabetes, osteoporosis, adipogenesis, osteoblastogenesis, osteoclastogenesis, zinc, β-cryptoxanthin, p-hydroxycinnamic acid
Obesity and diabetes are currently a major health problem worldwide with growing in prevalence. The incidence of metabolic disease, including type 2 diabetes with obesity, is increased to epidemic levels [1,2]. Obesity and diabetes induce secondary diseases with various pathophysiologic states, which are important in clinical aspects including cardiovascular disease, neural disturbance, kidney disease, cancer and osteoporosis [3,4]. Diabetes is frequent in the elderly and therefore frequently coexists with osteoporosis. The incidence of metabolic disease including obesity and obese type 2 diabetes has increased to epidemic levels in recent years [2]. Obesity and osteoporosis are now thought to be closely related and to share several features. Low body weight has long been recognized as a risk factor for fracture in the elderly. Attention has only recently turned to fractures in overweight or obese individuals. In Western societies, mean body weight has increased dramatically in older people, and a similar trend exists in Asia.
One of these shared features is that osteoblasts and adipocytes differentiate from a common precursor cell in the bone marrow, the mesenchymal stem cells [5,6]. The pluripotency of mesenchymal stem cells is well known, and their ability to differentiate into osteoblasts, adipocytes, chondrocytes and myoblasts has been described extensively [7-14]. There is an inverse relationship between differentiation of mesenchymal stem cells to osteoblasts and adipocytes. Osteoporosis including obesity and diabetes are associated with bone marrow adiposity, which greatly produces tumor necrosis factor-α (TNF-α), an inflammatory cytokine [15-17]. TNF-α is found to suppress osteoblastogenesis and mineralization [18,19].
Thus, obesity, diabetes and osteoporosis are closely implicated. It is important to prevent and treat these diseases in clinical aspects. We found that some functional food chemical factors, novel osteogenic factors, reveal improvement effects on diabetic conditions associated with osteoporosis using animal models. This review has been written to outline our recent advances that have been made concerning preventive effects of novel osteogenic factors on bone loss that is induced in type 1 diabetes.
Obesity is based on stimulation of adipogenesis. Bone marrow mesenchymal stem cells are multipotent cells, which among other cell lineages, give rise to adipocytes and osteoblasts. Bone marrow mesenchymal stem cells are multipotent stromal cells, which among other cell lineages, that can differentiate into a variety of cell types including osteoblasts (bone cells), chondrocytes (cartilage cells), myoblasts (heart cells) and adipocytes (fat cells) [5,6]. This occurs through cross talk between complex signaling pathways including those derived from bone morphogenic proteins (BMPs), winglesstype MMTV integration site (Wnt) proteins, hedgehogs, delta/jagged proteins, fibroblastic growth factors, insulin, insulin-like growth factors, and transcriptional regulators of adipocyte and osteoblast differentiation including peroxisome proliferators-activated receptor-gamma (PPARγ) and runt-related transcription factor 2 (Runx2) [7-14]. Insulin, which is secreted by feeding, stimulates adipogenesis from bone marrow mesenchymal stem cells. In addition, bone marrow adiposity and mature adipocytes with obesity greatly produces tumor necrosis factor-α (TNF-α), an inflammatory cytokine [17]. This TNF-α may cause insulin resistance that leads to type 2 diabetes [21,22]. Differentiation of bone marrow mesenchymal stem cells may be involved in the development of osteoporosis.
Osteoporosis, which bone mass is dramatically reduced with menopause and aging [20], has also been shown to induce in obesity, diabetes (type I and II) and inflammatory disease [21,22]. Type 1 and type 2 diabetes have been associated with increased fracture risk. Osteoporosis and obesity are now thought to be closely related and to share several features [5,6]. One of shared features for obesity with osteoporosis is that osteoblasts and adipocytes differentiate from a common precursor cell in the bone marrow mesenchymal stem cells. Secondary causes of osteoporosis including obesity and diabetes are associated with bone marrow adiposity that greatly produces TNF-α [4,5].
Various hormones and cytokines, which include leptin, adiponectin, insulin, epinephrine, cortisol, glucagon, TNF-α and other factors, are well known as key molecules that relate to obesity and diabetes. Disturbance of these factors may play an important role in pathophysiologic conditions of obesity and diabetes. More recently, regucalcin, which is a suppressor protein of intracellular signaling systems, has been proposed to be a key molecule in obesity and diabetes associated with osteoporosis [23]. In addition, regucalcin has been demonstrated to stimulate adipogenesis in mouse bone marrow cell culture in vitro [24], suggesting an involvement as a stimulatory factor in adipogenesis. Thus findings may support the view that osteoporosis are implicated with obesity and diabetes.
Bone is a dynamic tissue that undergoes continual adaptation during vertebrate life to attain and preserves skeletal size, shape and structural integrity and to regulate mineral homeostasis. Bone homeostasis, which maintains bone mass, is skillfully regulated through a delicate balance between osteoblastic bone formation and osteoclastic bone resorption [25-27]. Osteoblasts are differentiated from bone marrow mesenchymal stem cells and stimulate bone formation and calcification. Osteoclasts are developed from hematopoietic progenitors and promote bone resorption. Physiologic process of bone turnover through these bone cells underpins development and maintenance of the skeletal system. Bone remodeling and modeling underpin development and maintenance of the skeletal system [25]. Bone modeling is responsible for growth and mechanically induced adaptation of bone, and it requires that the processes of bone formation and bone removal (resorption). Process of bone remodeling that makes bone unique among organs and tissues, and add so many levels of complexity with respect to interactions along remodeling sequence by systemic influences (hormones), stress action (physical activity/weight bearing), and growth factors and cytokines produced by the bone cells or factors that come from nearby cells in the marrow tissues.
Numerous pathological processes have the capacity to disrupt this equilibrium by leading to conditions where the rate of bone resorption outpaces and the rate of bone formation, leading to osteoporosis [20]. Osteoporosis is a disease characterized by loss in bone density and bone strength and deterioration of bone microarchitecture, resulting in increased risk for bone fractures. The most dramatic expression of osteoporosis is represented by fractures of the proximal femur for which the number increases as the population ages [20]. Osteoporosis is a common metabolic disease and generally affects people at an advanced age and suffering from other chronic diseases. It is more common in women and a significant loss of bone mass after menopause begins. Bone mass is dramatically reduced after menopause, which depresses secretion of ovarian hormone (estrogen) in women [20]. Deficiency of estrogen advances osteoclastic bone resorption. This is very important as a primary osteoporosis. Postmenopausal osteoporosis is the archetypal osteoporotic condition in women after menopause and leads to bone destruction through complex and diverse metabolic and biochemical changes.
Osteoporosis is a major cause of increased morbidity and mortality affecting the aging population. It has been estimated that osteoporosis affects at least 200 million women worldwide, one third of women aged between 60 and 70 years and two thirds over 80 years [28,29]. In 1995, incidence of osteoporotic fractures in the U.S. was about 1.5 million, of which 750 000 vertebral fractures, 250,000 hip fractures, 250,000 fractures in the wrist fracture and 250 000 other locations. According to a recent World Health Organization report, osteoporosis has become a global health problem with a disease incidence and mortality rate similar to that of cardiovascular diseases, diabetes and cancer [3,4]. Osteoporosisis is widely recognized as a major public health threat.
A high-fat diet appears to consistently induce impaired glucose tolerance with glucose levels reaching approximately 200 mg/dL [30-32]. Impaired glucose tolerance in C57BL/6J, which this mouse is the best-studied model of diet-induced obesity [30], appears to result from poor insulin secretion [33], but insulin resistance also develops with increasing age and concomitant exposure to a high-fat diet. Impaired glucose tolerance is more severe in males than females [33]. The C57BL/6J mouse appears to have adverse effects on trabecular bone components [34], and recent evidence suggests that the effect of the high-fat diet is more pronounced in skeletally immature compared to skeletally mature mice [35-37]. Lower trabecular bone volumes are primary the result of increased osteoclast resorption as indicated by serum biomarkers and histomorphotometry [35-37], though bone formation is also decreased [38]. Interestingly, recent data indicate that whereas body weight and fasting glucose levels can return to normal after a return to a normal diet, trabecular bone loss prior to skeletal maturity may not be recoverable without some other types of intervention [34]. The potential implications of those results are obvious considering the childhood obesity problem in the United States and other countries [30].
The spontaneous obese ob/ob mouse is a model of severe obesity resulting from a spontaneous inactivating mutation in the leptin gene [39,40]. ob/ob mice are used as a general model for insulin resistance with obesity. There is also a lack of complete β-cell failure in the model, indicating that diabetes is not very severe [41]. Weight of ob/ob mice increases rapidly starting at 2 weeks of age, and can reach up to three fold the weight of wild-type controls [41]. Hyperphagia, insulin resistance and hyperinsulinemia are all evident at 3 to 4 weeks of age, with obesity evident by 4 weeks [40]. Even after obvious hyperglycemia detected at 4 weeks, blood glucose levels continue to increase until reaching a peak at 12 to 20 weeks of age with glucose levels reaching up to 400 mg/dL [41]. Because leptin directly influences bone metabolism, the skeletal phenotype of the ob/ob mouse is not attributed solely to the observed metabolic changes. Moreover, many studies showed a complex skeletal phenotype with ob/ob having higher trabecular bone mass in the lumbar vertebrate, but lower trabecular and cortical bone mass in the long bones [42-44], as well as shorter femurs compared to control. Furthermore, ob/ob mice have decreased biomechanical properties (eg, maximum load) as determined by three-point bending of the femur [42-44]. This mouse also have lower bone mass and markedly reduced bone formation in vertebrate and long bones compared to wild-type controls, and this deficiency can be corrected with leptin treatment [30, 42-45].
The TallyHo mouse is a polygenic model of early onset, naturally occurring type 2 diabetes mellitus and obesity [30,46]. The genetic background is mixed, but is 86% homologous with SWR mice. TallyHo mice are fatter and weigh more than controls by 4 weeks of age [47,48]. Body weight continues to increase with age, but obesity in these mice is less severe than some other diabetic models [49]. Increased plasma triglycerides, cholesterol, and fatty acid levels are noticeable from 4 to 8 weeks of age [49]. Diabetes is present only in male mice [47]. Other characteristics of the TallyHo mouse include enlarged pancreatic islets, hyperleptinemia, and poor wound healing [48,49]. Investigations of the skeletal phenotype in TallyHo mice have utilized two different control strains [47,50]. Won et al. used C57BL/6J mice to show that male TallyHo have reduced BMD at 8 and 12 weeks of age [47]. TallyHo also exhibited reduced serum osteocalcin, and lower osteoblastogenic and higher osteoclastogenic gene expression patterns in bone marrow cells compared to C57BL/6J [50]. Devlin et al., using SWR mice as controls, showed that despite their greater body weight male TallyHo mice have reduced total body BMD as well as lowest trabecular bone volume at the distal femur. Femoral three-point bending showed that male TallyHo mice have a higher maximum load but less postyield deformation than SWR control [50].
As described above, there is growing evidence that abnormality and bone loss are induced in animal models for obesity and type 2 diabetes. Thus, osteoporosis is implicated with obesity and diabetes.
Anti-resorptive agents have been used the preferred standard of clinical care for the amelioration of bone loss in osteoporosis. However, clinical compounds that stimulate bone formation are under development. Novel analogues, which are synthesized from bioactive chemicals derived from food factors, may be developed as new drugs that reveal potent-osteogenic effects for the treatment of osteolysis, which is induced in various diseases including inflammation, obesity and diabetes. Development of osteogenic factors, which target the differentiation of bone marrow mesenchymal stem cells, may be importance in biomedical osteoporosis treatment. We found novel osteogenic factors including zinc compound, Satsuma mandarin orange (Citrus unshiu MARC.) β-cryptoxanthin, and plant component p-hydroxycinnamic acid as shown in Figure 1. These factors were found to reveal preventive effects on diabetic condition and bone loss that are induced in animal models for type 1 diabetes.
Effect of nutritional zinc: Nutritional zinc has been demonstrated to stimulate osteoblastic bone for mation and suppress osteoclasti bone resorption in vitro and in vivo [51]. We found that zinc acexamate reveals a potential anabolic effect on bone and that prevents bone loss in type 1 diabetic model rats induced by streptozotocin (STZ) [52,53]. Rats received a single subcutaneous administration of STZ (60 mg/kg body weight), and the animals were orally administered once daily for 7, 14 and 21 days with zinc acexamate (25, 50 or 100 mg/kg). Bone loss in the femoral- metaphyseal tissues induced by STZ administration is shown in Figure 2. Administration of STZ caused a significant increase in serum glucose, triglyceride and calcium concentrations and a significant decrease in body weight, serum zinc and inorganic phosphorus concentrations [52,53]. Moreover, calcium content, alkaline phosphatase activity and deoxyribonucleic acid (DNA) content in the femoral-diaphyseal and -metaphyseal tissues were significantly reduced in diabetic rats [52,53]. The change in these serum and bone components of STZ-diabetic rats was found to improve after the oral administration of zinc acexamate. This preventive effect was also observed by the administration of zinc acexamate for 7 days [53]. This study demonstrates that the oral administration of zinc acexamate has preventive effects on diabetic condition and bone loss in type 1 diabetes in vivo.
Effects of β-cryptoxanthin: β-cryptoxanthin, a carotenoid, has been demonstrated to stimulate osteoblastic bone for mation and suppress osteoclasti bone resorption in vitro and in vivo [54]. β-Cryptoxanthin was found to prevent bone loss in animal models for type 1 diabetes using STZ-treated rats [55]. Rats received a single subcutaneous administration of STZ (60 mg/kg body weight), and then the animals were orally administered of β-cryptoxanthin (50 or 100 μg /kg body weight) once daily for 7 or 14 days. Administration of STZ caused a significant decrease in body weight and a significant increase in serum glucose, triglyceride, and calcium concentrations, indicating a diabetic state [55]. These alterations were prevented by the administration of β-cryptoxanthin for 14 days. Administration of β-cryptoxanthin to normal rats for 14 days did not have a significant effect on body weight or on serum glucose, triglyceride, and calcium concentrations [55]. Calcium content, alkaline phosphatase activity, and DNA content in the femoral-diaphyseal and -metaphyseal tissues were significantly decreased in STZ-diabetic rats [55]. These decreases were significantly prevented by the administration of β-cryptoxanthin (50 or 100 μg/kg) for 14 days. Administration of β-cryptoxanthin to normal rats for 14 days caused a significant increase in calcium content, alkaline phosphatase activity, and DNA content in the femoral-diaphyseal and -metaphyseal tissues [55]. Thus, β-cryptoxanthin was found to reveal preventive effects on diabetic condition and bone loss in type 1 diabetic model rats.
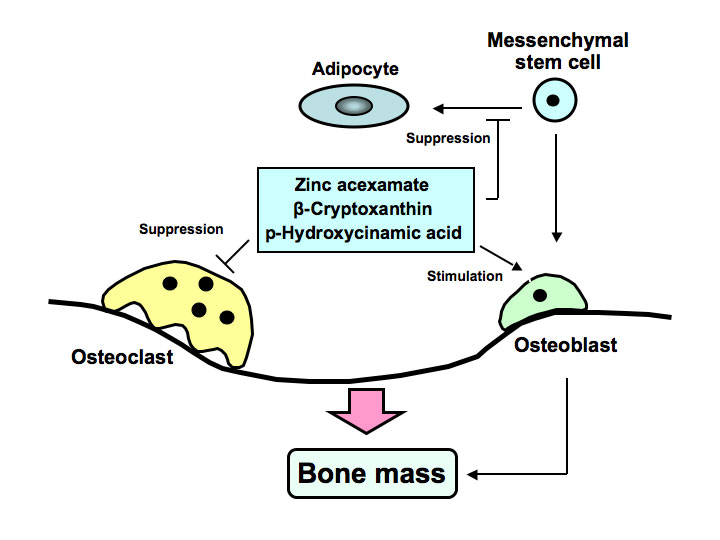
Figure 1. Zinc acexamate, β-cryptoxanthin and p-hydroxycinnamic acid (HCA) stimulates osteoblastic bone formation and suppresses osteoclastin bone resorption, thereby increasing bone mass. These factors were found to prevent bone loss induced in type 1 diabetic animal model. HCA was found to suppress adipogenesis from bone marrow mesenchymal stem cells, suggesting its preventive effects on obesity.
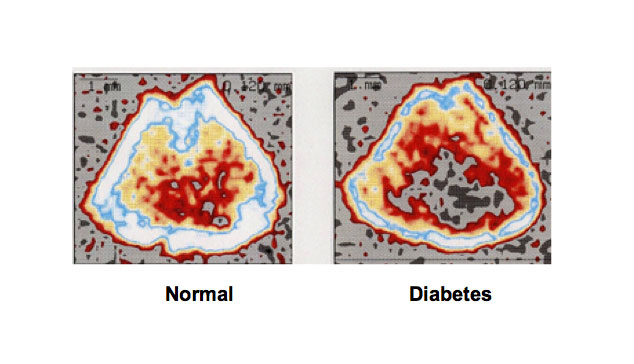
Figure 2. Bone loss is induced in type 1 diabetic animal models. Rats received a single subcutaneous administration of STZ (60 mg/kg body weight), which induces type 1 diabetes, and 21 days later the animals were killed. Bone mineral density was dramatically decreased in the femoral-metaphyseal tissues of rats treated with streptozotocin that causes type 1 diabetes. Bone mineral density was analyzed using a peripheral quantitative computed tomography.
Effects of p-hydroxycinnamic acid: Cinnamic acid, a flavonoid, is present in many plant and fruits. The flavonoid p-hydroxycinnamic acid is an intermediate-metabolic substance in plant and fruits, and it is synthesized from tyrosine. Among cinnamic acid and its related compounds (cinnamic acid, p-hydroxycinnamic acid, ferulic acid, caffeic acid and 3, 4-dimethoxycinnamic acid), p-hydroxycinnamic acid has been shown to have specific-anabolic effects on bone in vitro [56]. p-hydroxycinnamic acid was found to stimulate osteoblastic bone formation and to inhibit osteoclastic bone resorption in vitro [57,58]. Oral administration of p-hydroxycinnamic acid was found to reveal preventive and restorative effects on hyperglycemia, hyperlipidemia and bone loss in type 1 diabetic rats induced by treatment with STZ in vivo [59], demonstrating that this factor reveals preventive and restorative effects on diabetic states. Rats received a single subcutaneous administration of STZ (60 mg/kg body weight), and then the animals were orally administered p-hydroxycinnamic acid (2.5, 5, or 10 mg/kg body weight) once daily for 14 days [59]. STZ administration caused a significant decrease in body weight and a significant increase in serum glucose, triglyceride, and calcium levels, indicating a diabetic state. These alterations were prevented by the administration of p-hydroxycinnamic acid [59]. Calcium content in the femoral-diaphyseal and -metaphyseal tissues was significantly decreased in STZ-diabetic rats [59]. This decrease was prevented after the administration of p-hydroxycinnamic acid [59]. Alkaline phosphatase activity and DNA content in the diaphyseal and metaphyseal tissues was decreased in STZ-diabetic rats [59]. These decreases in STZ-diabetic rats were prevented after the administration of p-hydroxycinnamic acid [59]. Thus, intake of p-hydroxycinnamic acid was found to reveal preventive effects on bone loss in type 1 diabetic model animals and that reveals partial restrative effects on serum biochemical findings in the diabetic state.
Interestingly, p-hydroxycinnamic acid was found to stimulate osteoblastogenesis and suppress adipogenesis using culture systems with mouse bone marrow cells and mouse 3T3-L1 preadipocytes in vitro [60]. Bioactive flavonoid p-hydroxycinnamic acid, a novel osteogenic factor, may reveal preventive and therapeutic effects on osteoporosis associated with obese type 2 diabetes.
Obesity and diabetes induce secondary diseases with various pathophysiologic states, which are important in clinical aspects including cardiovascular disease, neural disturbance, kidney disease, cancer and osteoporosis. Diabetes is frequent in the elderly and therefore frequently coexists with osteoporosis. Drugs, which improve osteoporosis implicated with obesity and diabetes, have been poorly understood. Moreover, clinical compounds that stimulate bone formation are under development. Novel analogues, which are synthesized from bioactive chemicals derived from food factors, may be developed as new drugs that reveal potent-osteogenic effects for the treatment of osteolysis, which is induced in various diseases including inflammation, obesity and diabetes. Development of osteogenic factors, which target the differentiation of bone marrow mesenchymal stem cells, may be importance in treatment of obesity, diabetes and osteoporosis.
We found that diabetic states and bone loss in type 1 diabetes were improved by the intake of zinc compound, β-cryptoxanthin and p-hydroxycinnamic acid, which is novel osteogenic factors. These factors may be a useful tool in the prevention and improvement of diabetic osteoporosis, and development as new drugs will be expected in clinical aspects.
- 1. Leslie WD, Rubin MR, Schwartz AV, Kanis JA (2012) Type 2 diabetes and bone. J Bone Miner Res 27: 2231-2237. [Crossref]
- 2. Nielson CM, Srikanth P, Orwoll ES (2012) Obesity and fracture in men and women: an epidemiologic perspective. J Bone Miner Res 27: 1-10. [Crossref]
- 3. Walker-Bone K (2012) Recognizing and treating secondary osteoporosis. Nat Rev Rheumatol. 28: 480-492. [Crossref]
- 4. Weilbaecher KN, Guise TA, McCauley LK (2011) Cancer to bone: a fatal attraction. Nat Rev Cancer 11: 411-425. [Crossref]
- 5. Minguell JJ, Erices A, Conget P (2001) Mesenchymal stem cells. Exp Biol Med (Maywood) 226: 507-520. [Crossref]
- 6. Muruganandan S, Roman AA, Sinal CJ (2009) Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenic program. Cell Mol Life Sci 66: 236-253. [Crossref]
- 7. Laudes M (2011) Role of WNT signalling in the determination of human mesenchymal stem cells into preadipocytes. J Mol Endocrinol 46: R65-72. [Crossref]
- 8. Gharibi B, Abraham AA, Ham J, Evans BA (2011) Adenosine receptor subtype expression and activation influence the differentiation of mesenchymal stem cells to osteoblasts and adipocytes. J Bone Miner Res 26: 2112-2124. [Crossref]
- 9. Kawai M, Rosen CJ (2010) PPARγ: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol 6: 629-636. [Crossref]
- 10. Wu L, Cai X, Dong H, Jing W, Huang Y, et al. (2010) Serum regulates adipogenesis of mesenchymal stem cells via MEK/ERK-dependent PPARgamma expression and phosphorylation. J Cell Mol Med 14: 922-932. [Crossref]
- 11. Laudes M (2011) Role of WNT signalling in the determination of human mesenchymal stem cells into preadipocytes. J Mol Endocrinol 46: R65-72. [Crossref]
- 12. Gharibi B, Abraham AA, Ham J, Evans BA (2011) Adenosine receptor subtype expression and activation influence the differentiation of mesenchymal stem cells to osteoblasts and adipocytes. J Bone Miner Res 26: 2112-2124. [Crossref]
- 13. Kawai M, Rosen CJ (2010) PPARγ: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol 6: 629-636. [Crossref]
- 14. Wu L, Cai X, Dong H, Jing W, Huang Y, et al. (2010) Serum regulates adipogenesis of mesenchymal stem cells via MEK/ERK-dependent PPARgamma expression and phosphorylation. J Cell Mol Med 14: 922-932. [Crossref]
- 15. Parhami F, Tintut Y, Beamer WG, Gharavi N, Goodman W, et al. (2001) Atherogenic high-fat diet reduces bone mineralization in mice. J Bone Miner Res16: 182-188. [Crossref]
- 16. Pirih F, Lu J, Ye F, Bezouglaia O, Atti E, et al. (2012) Adverse effects of hyperlipidemia on bone regeneration and strength. J Bone Miner Res 27: 309-318. [Crossref]
- 17. Cortez M, Carmo LS, Rogero MM, Borelli P, Fock RA (2013) A high-fat diet increases IL-1, IL-6, and TNF-α production by increasing NF-κB and attenuating PPAR-γ expression in bone marrow mesenchymal stem cells. Inflammation 36: 379-386. [Crossref]
- 18. Li Y, Li A, Strait K, Zhang H, Nanes MS, et al. (2007) Endogenous TNFalpha lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-kappaB. J Bone Miner Res 22: 646-655. [Crossref]
- 19. Chang J, Wang Z, Tang E, Fan Z, McCauley L, et al. (2009) Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med 15: 682-689. [Crossref]
- 20. Weitzmann MN, Pacifici R (2006) Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest 116: 1186-1194. [Crossref]
- 21. Rossmeisl M, Rim JS, Koza RA, Kozak LP (2003) Variation in type 2 diabetes--related traits in mouse strains susceptible to diet-induced obesity. Diabetes 52: 1958-1966. [Crossref]
- 22. Ables GP, Perrone CE, Orentreich D, Orentreich N (2012) Methionine-restricted C57BL/6J mice are resistant to diet-induced obesity and insulin resistance but have low bone density. PLoS One 7: e51357. [Crossref]
- 23. Yamaguchi M (2010) Regucalcin and metabolic disorders: osteoporosis and hyperlipidemia are induced in regucalcin transgenic rats. Mol Cell Biochem 341: 119-133. [Crossref]
- 24. Yamaguchi M, Weitzmann MN, Baile CA, Murata T (2012) Exogenous regucalcin suppresses osteoblastogenesis and stimulates adipogenesis in mouse bone marrow culture. Integr Biol (Camb) 4: 1215-1222. [Crossref]
- 25. Raggatt LJ, Partridge NC (2010) Cellular and molecular mechanisms of bone remodeling. J Biol Chem 285: 25103-25108. [Crossref]
- 26. Chambers TJ, Fuller K (2011) How are osteoclasts induced to resorb bone? Ann N Y Acad Sci 1240: 1-6. [Crossref]
- 27. Chen G, Deng C, Li YP (2012) TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 8: 272-288. [Crossref]
- 28. Johnell O, Kanis JA (2006) An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int 17: 1726-1733. [Crossref]
- 29. Davidge Pitts CJ, Kearns AE (2011) Update on medications with adverse skeletal effects. Mayo Clin Proc 86: 338-343. [Crossref]
- 30. Fajardo RJ, Karim L, Calley VI, Bouxsein ML (2014) A review of rodent models of type 2 diabetic skeletal fragility. J Bone Miner Res 29: 1025-1040. [Crossref]
- 31. Rossmeisl M, Rim JS, Koza RA, Kozak LP (2003) Variation in type 2 diabetes--related traits in mouse strains susceptible to diet-induced obesity. Diabetes 52: 1958-1966. [Crossref]
- 32. Ables GP, Perrone2021 Copyright OAT. All rights reserv) Methionine-restricted C57BL/6J mice are resistant to diet-induced obesity and insulin resistance but have low bone density. PLoS One 7: e51357. [Crossref]
- 33. Ahrén B, Pacini G (2002) Insufficient islet compensation to insulin resistance vs. reduced glucose effectiveness in glucose-intolerant mice. Am J Physiol Endocrinol Metab 283: E738-744. [Crossref]
- 34. Ionova-Martin SS, Do SH, Barth HD, Szadkowska M, Porter AE, et al. (2010) Reduced size-independent mechanical properties of cortical bone in high-fat diet-induced obesity. Bone 46: 217-225. [Crossref]
- 35. Cao JJ, Gregoire BR, Gao H (2009) High-fat diet decreases cancellous bone mass but has no effect on cortical bone mass in the tibia in mice. Bone 44: 1097-1104. [Crossref]
- 36. Halade GV, Rahman MM, Williams PJ, Fernandes G (2010) High fat diet-induced animal model of age-associated obesity and osteoporosis. J Nutr Biochem 21: 1162-1169. [Crossref]
- 37. Patsch JM, Kiefer FW, Varga P, Pail P, Rauner M, et al. (2011) Increased bone resorption and impaired bone microarchitecture in short-term and extended high-fat diet-induced obesity. Metabolism 60: 243-249. [Crossref]
- 38. Baek K, Hwang HR, Park HJ, Kwon A, Qadir AS, et al. (2014) TNF-α upregulates sclerostin expression in obese mice fed a high-fat diet. J Cell Physiol 229: 640-650. [Crossref]
- 39. Castracane VD, Henson MC (2002) When did leptin become a reproductive hormone? Semin Reprod Med 20: 89-92. [Crossref]
- 40. Coleman DL (1978) Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 14: 141-148. [Crossref]
- 41. Lindström P (2007) The physiology of obese-hyperglycemic mice [ob/ob mice]. Scientific World Journal 7: 666-685. [Crossref]
- 42. Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, et al. (2000) Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell 100: 197-207. [Crossref]
- 43. Ealey KN, Fonseca D, Archer MC, Ward WE (2006) Bone abnormalities in adolescent leptin-deficient mice. Regul Pept 136: 9-13. [Crossref]
- 44. Hamrick MW, Pennington C, Newton D, Xie D, Isales C (2004) Leptin deficiency produces contrasting phenotypes in bones of the limb and spine. Bone 34: 376-383. [Crossref]
- 45. Turner RT, Kalra SP, Wong CP, Philbrick KA, Lindenmaier LB, et al. (2013) Peripheral leptin regulates bone formation. J Bone Miner Res 28: 22-34. [Crossref]
- 46. Kim JH, Stewart TP, Zhang W, Kim HY, Nishina PM, et al. (2005) Type 2 diabetes mouse model TallyHo carries an obesity gene on chromosome 6 that exaggerates dietary obesity. Physiol Genomics 22: 171-181. [Crossref]
- 47. Won HY, Lee JA, Park ZS, Song JS, Kim HY, et al. (2011) Prominent bone loss mediated by RANKL and IL-17 produced by CD4+ T cells in TallyHo/JngJ mice. PLoS One 6: e18168. [Crossref]
- 48. Kim JH, Saxton AM (2012) The TALLYHO mouse as a model of human type 2 diabetes. Methods Mol Biol 933: 75-87. [Crossref]
- 49. Kim JH, Stewart TP, Soltani-Bejnood M, Wang L, Fortuna JM, et al. (2006) Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. J Endocrinol 191: 437-446. [Crossref]
- 50. Devlin M, Vliet MV, Motyl K, Karim L, Brooks D, et al. (2014) Early onset Type 2 diabetes impairs skeletal acquisition in the male TALLYHO/JngJ mouse. Endocrinology. [Crossref]
- 51. Yamaguchi M (2010) Role of nutritional zinc in the prevention of osteoporosis. Mol Cell Biochem 338: 241-254. [Crossref]
- 52. Uchiyama S, Yamaguchi M (2003) Alteration in serum and bone component findings induced in streptozotocin-diabetic rats is restored by zinc acexamate. Int J Mol Med 12: 949-954. [Crossref]
- 53. Yamaguchi M, Uchiyama S (2003) Preventive effect of zinc acexamate administration in streptozotocin-diabetic rats: Restoration of bone loss. Int J Mol Med 12: 755-761. [Crossref]
- 54. Yamaguchi M (2012) Role of carotenoid β-cryptoxanthin in bone homeostasis. J Biomed Sci 19: 36. [Crossref]
- 55. Uchiyama S, Yamaguchi M (2005) Oral administration of beta-cryptoxanthin prevents bone loss in streptozotocin-diabetic rats in vivo. Biol Pharm Bull 28: 1766-1769. [Crossref]
- 56. Lai YL, Yamaguchi M (2006) Phytocomponent p-hydroxycinnamic acid stimulates bone formation and inhibits bone resorption in rat femoral tissues in vitro. Mol Cell Biochem 292: 45-52. [Crossref]
- 57. Yamaguchi M, Lai YL, Uchiyama S, Nakagawa T (2008) Phytocomponent p-hydroxycinnamic acid stimulates mineralization in osteoblastic MC3T3-E1 cells. Int J Mol Med 22: 287-291. [Crossref]
- 58. Lai YL, Yamaguchi M (2007) Phytocomponent p-hydroxycinnamic acid inhibits osteoclast-like cell formation in mouse bone marrow cultures. Int J Mol Med 19: 123-128. [Crossref]
- 59. Yamaguchi M, Uchiyama S, Lai YL (2007) Oral administration of phytocomponent p-hydroxycinnamic acid has a preventive effect on bone loss in streptozotocin-induced diabetic rats. Int J Mol Med 19: 803-807. [Crossref]
- 60. Yamaguchi M, Baile CA, Zhu S, Shoji M (2013) Bioactive flavonoid p-hydroxycinnamic acid stimulates osteoblastogenesis and suppresses adipogenesis in bone marrow culture. Cell Tissue Res 354: 743-750. [Crossref]