Abstract
Type 2 diabetes mellitus (T2DM), characterized by chronic hyperglycaemia and insulin resistance, is part of the metabolic syndrome and an independent risk factor for cardiovascular and cerebrovascular events, being particularly associated with carotid atheromatous disease. Arterial stiffness and reduced vasodilatation present in diabetes are induced by multiple mechanisms at molecular level. Chronic accumulation of advanced glycation end products (AGEs) and depletion of nitric oxide (NO) lead to oxidative stress that enhances inflammatory cytokine cascades. Concurrently, overproduction of vasoconstrictors promotes smooth muscle cell migration and endothelium impairment. Thus, chronic inflammation leads to cellular senescence through interaction with cellular regulatory systems. Consequently, vascular functionality is deteriorated, while vessel wall thickening and formation of atheromatous plaques are precipitated, resulting in carotid disease. So, effective glycaemic control and pharmaceutical modification of cell regulatory systems may be able to prevent diabetic vascular complications. This review summarizes current evidence on the issue aiming to a deeper understanding of these pathogenic mechanisms that will contribute to the development of targeted diagnostic and treatment approaches.
Keywords
diabetes mellitus, carotid disease, senescence, vascular dysfunction, inflammation
Introduction
Diabetes mellitus (DM) is a heterogeneous pathological entity characterized by an absolute or relative deficiency of insulin secretion and action that results in chronic hyperglycaemia, if untreated [1]. Its spectrum of pathogenic disorders ranges from the autoimmune destruction of beta- pancreatic cells and subsequent pancreatic inability to produce insulin (type 1 DM) to inadequate insulin secretion and impaired response of target cells to it (type 2 DM) [2]. Most prevalent in general population worldwide though is type 2 DM. This condition is strongly related with the metabolic syndrome, which is defined by the alteration of at least three out of the five following parameters: abdominal obesity, high triglycerides, low high-density lipoprotein (HDL) cholesterol levels, elevated blood pressure, and elevated fasting glucose [3].
The aforementioned dysmetabolic status of chronic hyperglycaemia and insulin resistance seen in type 2 DM, entails a number of complications remaining a primary cause of mortality and morbidity especially in older ages. Diabetic long-term consequences include end-organ dysfunction, such as peripheral and autonomic (gastrointestinal and genitourinary) neuropathy as well as microvascular (nephropathy and retinopathy) and macrovascular disorders (coronary and cerebrovascular disease) [2].
Thus, DM has been identified as an independent risk factor for cardiovascular events, including carotid artery atherosclerosis and stenosis [4]. Carotid disease clinically manifested as stroke, transient ischemic attack (TIA) or syncope due to cerebral hypoperfusion or arterial embolism, is characterized by vascular wall thickening as well as by the formation of atheromatous plaques vulnerable to rupture. These structural and functional changes define a dynamic procedure known as vascular ageing, which is different from normal ageing as an entity, beginning at a young age under the impact of risk factors such as obesity, dyslipidaemia and hyperglycaemia (components of metabolic syndrome) [5].
Taking into account the high prevalence of carotid disease among diabetic patients and its clinical significance for healthcare, we are convinced that the elucidation of underlying pathology could contribute to a thorough understanding of the disease.
At this point, it is worth mentioning that diabetes mellitus is a major public health problem estimated to be affecting more than 550 million people within the next decade [2,4]. Current data support that its complications from large and small vessels share the same pathological background [4]. Carotid atheromatous disease, an important clinical issue among diabetic patients, has been studied and described in the context of cardiovascular disease and a large body of evidence underlines the role of macroscopic and morphological changes of vascular wall in the progression of atheromatous disease [5].
This review aims to address pathogenic mechanisms leading to structural and functional alterations in carotid atherosclerosis in particular. It underlines the relation of cellular pathways with the pathology of this critical large vessel. This mechanistic approach will allow a deeper comprehension of carotid atheromatous disease as a separate entity.
Moreover, the article aims to highlight the perspective of new therapies and prevention methods with the help of biomedical sciences. It focuses on the role of senescence, a recently recognized cellular process involved in the disease and a very interesting new field in biomedical sciences.
Thus, we have performed a comprehensive search online and collected data on pathogenic mechanisms at intracellular and molecular level to summarize current evidence in literature.
Association of metabolic syndrome and vascular disease
Multiple studies have reported the robust relation between metabolic syndrome, carotid disease and confirmed relevant alterations in blood flow [1-6]. This demonstrates a higher pulse wave velocity in carotid arteries of diabetic patients along with age [6-9]. Especially two of the metabolic syndrome components-hypertension and hyperglycaemia - not obesity or hypertriglyceridemia- were associated with subclinical carotid atherosclerosis, suggesting that efficient metabolic control can prevent occurrence of carotid disease [10]. Similarly, HbA1c (glycated haemoglobin), a common marker of glycaemic control in type 2 DM, has been considered to be a useful marker for the prediction of carotid atheromatous disease, being associated independently with elevated carotid intima media thickness (cIMT) but not with the development of plaques [11]. In addition, daily glucose fluctuations are related with cIMT in a linear way and associated with progression of atherosclerosis in older diabetics with a long duration of diabetes. Hence, proper glycaemic control can also delay the progression of carotid disease [12]. Finally, other studies indicate that the carotid plaque score (independently) but not cIMT alone can be a useful tool for the prediction of diabetic microvascular complications. This underlines that asymptomatic diabetic patients with a high carotid score are in need of special care in order to avoid further vascular complications [11].
The main underlying mechanism of the above clinical outcomes is arterial stiffness and reduced vasodilatation because of abnormalities in endothelial and vascular smooth muscle cell function [13]. These conditions are independently associated with cardiovascular disease [14] (Homeostasis Model Assessment) precipitated by the dysfunction of endothelial cells, on which insulin resistance and hyperglycaemia in type 2 DM have a deteriorating effect [13-18]. Atherosclerosis’ consequences though are further exacerbated by vascular calcification and cartilaginous metaplasia according to preclinical studies on mutant mice [19,20]. Hyperglycaemia, hyperlipidaemia and subsequent insulin resistance intensify the formation of calcium lesions and the osteochondrogenic differentiation of smooth muscle cells through a process mediated by advanced-glycation-end products and their receptor [21]. At cellular level, inflammation, Reactive Oxygen Species (ROS) generation and subsequent senescence have been identified as the major factors triggering vascular injury in DM. For example, Vpo1, a recently discovered peroxidase with a significant role in induction of senescence on endothelial cells has been associated with vascular complications of DM according to preclinical studies [22].
Endothelial cells, constituting a layer located strategically between circulating blood and the vascular wall, have an active and regulatory role concerning vascular function, structure and homeostasis. They release numerous substances in order to adjust blood flow and nutrient supply, and to prevent thrombosis and leukocyte diapedesis. As mentioned above, DM alters the functions of multiple systems, including vessels, being associated with cardiovascular and cerebrovascular disease and other microvascular or macrovascular complications [22,23].
In fact, diabetic milieu, as characterized by hyperglycaemia, free fatty acid excess and insulin resistance, promotes oxidative stress, inflammation and subsequent cellular senescence via numerous intracellular pathways that affect endothelial and smooth muscle cells. Endothelial dysfunction has been detected in various studies on arteries of diabetic subjects in vivo and ex vivo [23-29]. Vascular changes in DM are quite similar to those of vascular ageing, resulting in impaired vessel functionality (Flowchart).
A number of mechanisms contributing to vascular dysfunction related with diabetes mellitus are described below in order to show their impact on the progression of carotid disease (Figure 1 and Table 1).
Cytokine cascades |
Transcription factors |
miRNAs |
Increase |
TGF-β1 |
FOXO1 |
miR-155 |
IL-6 |
FOXO3a |
miR-146a |
ICAM |
NF-κΒ |
miR-4448 |
ROS |
PGC-1a |
miR-338-3p |
AGEs |
|
miR-190a-5p |
TNFa |
|
miR-485-5p |
INF-γ |
|
miR-9-5p |
IL-1β |
|
|
NF-κΒ |
|
|
CRP |
|
|
TXA2 |
|
|
PGs |
|
|
Decrease |
NO |
Nrf2 |
miR-34a |
IL-10 |
Sirt1 |
miR-27a |
| |
Nampt |
|
Table 1. Molecules involved in inflammation and senescence inducing carotid disease in diabetic patients
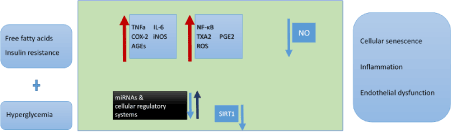
Figure 1. Metabolic derangements in type 2 diabetes mellitus affect vascular function through multiple mechanisms
Overproduction of cytokines such as TNF-a (tumor necrosis factor-a) and IL6 (interleukin 6) accelerate inflammation, production of ROS (reactive oxygen species) and oxidative stress. Especially NF-κΒ (nuclear factor kappa-B) is an inflammatory cytokine that regulates multiple cellular pathways. Hyperglycaemia, in particular, is a major stress stimulus that leads to generation of AGEs (advanced glycation end products) that in turn enhance inflammation and senescence. In addition, hyperglycaemia affects microRNAs and other cellular regulatory systems (SIRT1 (sirtuin), FOXO1/2(forkhead box protein O1/2), Nampt (Nicotinamide phosphoribosyltransferase), Nrf2 (Nuclear factor erythroid 2-related factor 2)) inducing senescence. At the same time, diabetic milieu causes upregulation of COX2 (cyclooxygenase 2) and iNOS (inducible nitric oxide synthase), resulting in generation of vasoconstrictors (thromboxane and prostaglandins). Thus, an overall depletion of NO (nitric oxide) levels contribute to endothelial dysfunction and vascular disease.
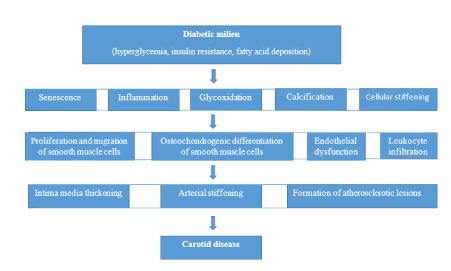
Flowchart. Impaired vessel functionality
Diabetic vascular inflammation and subsequent cellular senescence
First of all, inflammation and DM are reciprocally related. DM induces inflammation while chronic inflammation –related with cellular senescence- stimulates onset of diabetes. Experimental evidence shows that high leukocyte count, neutrophilia in particular, is associated with insulin resistance and occurrence of diabetes, demonstrating the value of inflammatory biomarkers in the assessment and prediction of incidence not only of diabetes, but also of its complications [30]. According to data from the Diabetes Control and Complications Trial (DCCT) [31] as well as from other studies [32-34], higher plasma levels of CRP (C-reactive protein) and other inflammatory markers are associated with diabetic vascular dysfunction, especially cardiovascular disease risk. CRP, in particular, contributes to exacerbation of osteochondrogenic trans differentiation and calcification of primary human aortic smooth muscle cells (HAoSMCs) through promotion of oxidative stress [35].
NO (nitric oxide) depletion induces endothelial dysfunction
The aforementioned metabolic derangements mediate abnormalities in endothelial cell function by affecting the synthesis or degradation of NO. An extensively studied condition in fact is the endothelium dysfunction deriving from NO depletion. It is known that NO generated by endothelial NO synthase is responsible for vascular wall relaxation and for the protection of the vessel from endogenous injury. It controls signalling pathways that inhibit platelet and leukocyte adhesion as well as vascular smooth muscle cell proliferation and migration into the intima while it lessens their tone. Any decrease of NO levels (due to decreased production or increased degradation) leads to augmented activation of inflammatory cascades, overexpression of pro-inflammatory cytokines, leukocyte and monocyte interaction with vessel wall, vascular smooth muscle cell migration and formation of macrophage foam cells. These procedures already known to promote atherosclerosis have been reported in type 1 and 2 diabetic patients, highlighting its atherogenic impact [28,36-38].
Hyperglycaemia triggers inflammatory pathways
So, hyperglycaemia seems to induce a series of cellular reactions that increase the production of reactive oxygen species (superoxide anion) that inactivate NO into peroxynitrite [39,40]. Peroxynitrite uncouples NOS (NO synthase) triggering further superoxide anion production. Superoxide anion then promotes a cascade of endothelial processes that enhance oxygen-derived free radicals’ generation. For example, activation of protein kinase C (PKC), by glucose is implicated in the modulation of membrane-associated NAD(P)H (Nicotinamide adenine dinucleotide phosphate)-dependent oxidases activity for the production of superoxide anion. Hence, this chain of events causes overabundance of superoxide anion rather than NO [36-39].
Apart from NO, numerous circulating cytokines have been identified in diabetes underlying its inflammatory potential [40,41]. Overexpression of pro-inflammatory molecules, such as IL-1β (interleukin-1β) and IL-6 (interleukin-6), TNF-α (tumor necrosis factor- a), NF-кB, (nuclear factor- kappa B) myeloperoxidase and ICAM (intercellular adhesion molecule), has been correlated with vascular dysfunction in diabetic patients [42-44]. Higher levels of the inflammatory adipokine, leptin, are related with the development of type 2 diabetes [45]. However, prevalence of the anti-inflammatory adipokine, adiponectin diminishes risk for DM onset but also protects diabetic patients from cardiovascular disease according to findings from experimental animal studies [46,47].
Similarly, up-regulation of anti-inflammatory cytokine IL-10 gene in diabetic rats seems to prevent endothelial dysfunction [48]. Hyperglycaemia-induced ROS (reactive oxygen species) formation in endothelial cells triggers NF-кB transcription that initiates signalling of inflammatory pathways involving VEGF (vascular endothelial growth factor), TNF-α, IL-1β, which through a positive feedback loop, enhance transcription of this factor [49,50]. Multiple interactions of the aforementioned molecules and triggering of relevant cytokine cascades promote inflammation leading to atherosclerosis.
Increased synthesis of vasoconstrictors leads to vascular dysfunction in diabetes mellitus
Impaired vasodilatation in DM is a consequence not only of decreased NO production but also of increased synthesis of vasoconstricting prostanoids and endothelin [51-54], mostly due to the prevalent hyperglycaemic status. Vasoconstrictors, especially endothelin, are responsible for diabetes’ effects on vascular smooth muscle cells by promoting their migration into the intima, elevating their tone and contributing to inflammation (increased PKC activity, NF-kB production, and oxygen free radicals) ending up to the progression of atherosclerosis [51-57]. In atherosclerotic plaques, vascular smooth muscle cells produce then a matrix of metalloproteinases and undergo apoptosis making the plaque more vulnerable to rupture. In particular, hyperglycaemia seems to increase the expression of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2) and vasoconstricting prostanoids in both endothelial and vascular smooth muscle cells according to findings of in vitro and in vivo studies (human cell cultures and animal models [58-60]. Other experimental studies on mice have demonstrated that deletion of the iNOS gene and thus conservation of NO preserves endothelial vasorelaxation in carotid arteries as well as the animals’ cerebral arteriolar vasomotor function [38,61]. Up-regulation of COX-2 in atheromatous plaques of diabetic subjects [62] has been associated with elevated levels of thromboxane A2 (TXA2), prostaglandin E2 (PGE2), prostaglandin I2 (PGI2) as well as elevated arterial vascular smooth muscle tone [63-65]. Finally, hyperglycaemia-induced oxidative stress may increase levels of asymmetric dimethylarginine, a competitive antagonist of NOS, through blocking its degradation by dimethylaminohydrolase [66].
MicroRNAs involved in inflammation and cellular senescence in diabetes mellitus
This inflammatory phenotype occurring in DM and similar conditions, such as ageing, seems to be regulated by a number of microRNAs. These microRNAs are already known to be inducers or suppressors of senescence. Although data is still scarce, the role of these macromolecules has been identified in several studies and experimental animal models. Firstly, hyperglycaemia related down-regulation of miR-155 and miR-146a was reported in peripheral blood mononuclear cells from patients with type 2 diabetes [67]. Reduction of miR-146a reinforced inflammation in human aortic endothelial cells [68]. On the contrary, high glucose levels led to overexpression of miR-34a [69]. Similar dysregulations of miR-27a has been detected in tissues of diabetic rats [70] and in diabetic patients in correlation with fasting glucose. Furthermore, miR-4448, miR-338-3p, miR-190a-5p, miR-485-5p, and miR-9-5p were found to be implicated in diabetic retinopathy in human subjects, regulating 55 target genes related with NAD metabolism, sirtuin, and aging [71,72].
Regulatory systems of inflammation and senescence in DM
Additional regulatory systems such as Nampt (Nicotinamide phosphoribosyl transferase), SIRT1 (sirtuin 1), FOXO1 (forkhead box protein O1), and PGC-1α (Peroxisome proliferator-activated receptor gamma coactivator 1-alpha), related with cellular senescence, endothelial cell survival, redox states, vascular inflammation and bioenergetics have been reported to be implicated in diabetes. SIRT1, a member of sirtuins family considered to have anti-inflammatory effects [73], is reduced in endothelial and vascular smooth muscle cells of diabetic subjects in rats [74] as well as in endothelial cell cultures incubated in high glucose or hydrogen peroxide (H2O2) concentrations that induced their premature senescence [75-77]. Low SIRT1 levels were associated with p53 and FOXO1 activation and NO depletion [76-79]. Nrf2 (Nuclear factor erythroid 2-related factor 2) another modulatory molecule when inactivated by hyperglycaemia leads to endothelial dysfunction [80]. Moreover Nampt (senescence suppressor) overexpression enhances glycolysis and reduces ROS in high glucose-treated endothelial cells. Its rescue effect was dependent on SIRT1 activity and was inhibited by active FOXO1 [81-83].
In a high glucose concentration environment nuclear FOXO1 and FOXO3a activation in endothelial cells (- in vitro studies- of human aortic and rat brain/retinal cells) lead to production of peroxynitrite, consecutively to endothelial NO synthase (eNOS) dysfunction, and cell apoptosis [84-88]. Depletion of FOXO1/3/4 improved endothelial tissue insulin sensitivity and conserved normal angiogenesis in HFD (high in fat diet) -fed mice. 88 Similarly, ROS generation leads to an increase in PGC-1α levels in endothelial cells of diabetic mice, in cultures at high glucose environment, and in endothelial progenitor cells of diabetic patients [89]. Induction of PGC-1α seems to promote endothelial cell migration and angiogenesis through activation of Notch pathway and inhibition of Akt (Protein kinase B)/eNOS (endothelial NO synthase) signaling cascade, while its overexpression in experimental models (diabetic mice) leads endothelial dysfunction. On the other hand, partial genetic silencing of endothelial cells PGC-1α could have a protective effect in postischemic blood flow recovery in Type 1 and Type 2 diabetic mice and wound healing [89].
Role of mitochondrial metabolism in vascular dysfunction in DM
Endothelial cell metabolism though depends primarily on anaerobic glycolysis for baseline needs. For this reason, ECs express GLUT1 (glucose transporter) that allows for higher glucose levels in ECs in a hyperglycaemic environment. However, endothelial cell survival and response to stress conditions is based on aerobic reaction and energy production from cell mitochondria, whose metabolism is impaired in hyperglycaemic conditions mediating exacerbation of oxidative stress [90,91].
Oxidative stress in DM
As a stress stimulus, long term hyperglycaemia slows the pentose phosphate pathway (PPP) flux through inhibition of glucose-6-phosphate dehydrogenase (G6PD), diminishing NADPH (antioxidant) production [92]. At the same time, increased xanthine or NADPH (Nicotinamide adenine dinucleotide phosphate) oxidases activity produces superoxide anions that consume NO to peroxynitrite (ONOO–).
What is more, chronic hyperglycaemia, ROS and RNS (reactive nitric species) accumulation cause DNA damage activating the enzyme poly-ADP-ribose polymerase 1 (PARP1), which inactivates the glycolytic enzyme GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) by ADP (Adenosine diphosphate)-ribosylation [92-96].
Advanced glycation end products and endothelial dysfunction
In addition, slowing of the glycolytic flux causes glycolytic intermediates to accumulate directing metabolism into three glycolysis branch pathways that end up to the formation of advanced glycation end-products: 1) the hexosamine biosynthetic pathway 2) the glycation pathway that involves angiogenic capacity under hyperglycaemia. 3) the polyol pathway (glucose converts to sorbitol to fructose 3- deoxyglucosone, a highly reactive α-oxo-aldehyde that non-enzymatically generates toxic advanced glycation end-products (AGEs) (Maillard reaction) furtherly consuming NADPH and increasing ROS [92,97-99].
In particular, the AGES, substances of diverse structures with high reactivity and through multiple interactions, especially by binding to their receptor (RAGE) on endothelial cells, contribute to inflammation, leakage and ROS production. They activate arginase, blocking NO synthase and enhancing superoxide anion production, subsequent senescence and vascular dysfunction accelerate the progression of diabetic atherosclerosis and vascular calcification [100-104]. AGEs also enhance mitochondrial production of superoxide anion, which activates the hexosamine pathway, diminishing NOS activation by protein kinase Akt [105-115]. These processes promote oxidative stress by extracellular xanthine oxidase. As a result, endothelial vessel wall cells express pro-inflammatory phenotypes developing atherosclerosis [106,107].
Various AGEs have been studied in multiple studies, in fact. For example, AGE CML (Advanced glycation end-product Nε-carboxymethyl-Lysine) has been shown to be correlated with vascular calcification and progression of atherosclerosis, especially asymptomatic carotid disease, in diabetic patients [100].
Insulin resistance- induced-oxidative stress promoting senescence
Finally, it is worth mentioning the role of insulin resistance, which is a main feature of type 2 DM as well as a characteristic of the metabolic syndrome and a significant condition leading to cardiovascular diseases, including atherosclerosis. At cellular level, there is a clear differentiation between insulin resistance on EC bioenergetics and hyperglycaemia [108]. According to experimental data on animal models, insulin resistance is related with augmented release of free fatty acid (FFA) from adipose tissues. Then free fatty acids inducing oxidative stress, NO depletion and production of ROS exacerbate endothelial dysfunction [109-111]. The most important reason is the inhibition of the phosphatidylinositol-3 kinase pathway [112-114]. Production of the lipid second messenger diacylglycerol causes the membrane translocation and activation of PKC that inhibits the activity of the phosphatidylinositol 3 kinase pathway, causing limiting NO synthesis. Experimental studies have shown that diminished endothelium-dependent relaxation of rabbit aorta exposed to elevated glucose levels is restored by PKC inhibition, while infusion of free fatty acids reduces endothelium- dependent vasodilation in animal models and humans in vivo [115-117].
Therapeutic Implications and Future Perspectives
An effective inhibition of inflammatory pathways can restore glycaemic control and prevent diabetic vascular complications [118]. Such anti-inflammatory interventions blocking IL-1β are likely to improve glycaemic status of in type 2 diabetic patients according to various studies [118-121]. Other approaches aiming to blockage of NF-κB, have similar effects on glycaemic status [122-126]. In the same context, medications with pleiotropic actions and anti-inflammatory effects, such as rosiglitazone and atorvastatin, could delay diabetic vascular dysfunction [127,128]. Moreover, restoration of NO and mitochondrial superoxide levels as well as restriction of ROS generation and oxidative stress could have beneficial effects on endothelial function [129]. Similarly, the inhibition of PKC on healthy subjects seem to rescue normal vessel relaxation despite prevalence of hyperglycemia [130,131].
Another promising approach is the modulation of cellular regulatory systems, including microRNAs and other genes. Overexpression of miR-146a in rat aorta has been correlated with lower NF-κB levels and suppression of inflammation and senescence, according to preliminary evidence [132]. In general, bioenergetics (glycolysis, mitochondrial oxidative phosphorylation, oxidative stress) regulate cell cycle, survival, proliferation, differentiation and death [133-135]. Any intervention on them can alter cell fate. Finally, as insulin resistance and free fatty acid excess are primary therapeutical targets, drugs improving insulin sensitivity, such as metformin and thiazolidinediones, enhance endothelium-dependent vasodilation [134-139] through phosphatidylinositol-3 kinase pathway, and seem to have anti-senescent effects [55-57,115-117]. Consistently, findings from several studies suggest that incretin treatment with GLP1 (glucagon-like peptide-1) analogues or DPP4 (dipeptidyl peptidase 4) inhibitors as well as with SGLT2 (sodium-glucose transport protein 2) inhibitors delays the evolution of atheromatous disease in diabetic patients through specific anti-inflammatory pathways [136-139]. There is still much to learn concerning pathogenesis of vascular disease in diabetes mellitus. The emersion of the field of senescence has offered a new perspective in the elucidation of pathogenic pathways and the development of innovative methods for prevention as well as for the diagnosis and treatment of diabetic vascular complications (Table 2).
Senolytic factors (Death of senescent cells) |
GLP-1 analogues |
SGLT-2 inhibitors |
DPP-4 inhibitors |
Metformin |
Glyburide |
Anti-inflammatory factors (Cascades suppression- increased NO bioavailability) |
Atorvastatin (pleiotropic benefits) |
Kanacinuma (Il-1β Inhibitors) |
Rosiglitasone (pleiotropic benefits) |
NF-κB inhibitors |
Salsalates |
Modulators of regulatory pathways (Inhibition of senescence and inflammation) |
Resveratrol (Sirt1 activator) |
miRNAs inhibitors or activators |
Tetrahydro-biopterin |
|
|
Modification of risk factors (Lifestyle changes preventing metabolic syndrome) |
Exercise |
Caloric (fat and sugar) restriction |
Smoking cessation |
|
|
Table 2. Therapeutic approach of carotid disease in diabetic patients
In conclusion, carotid disease is a significant vascular complication occurring in diabetic patients. Development of atheromatous plaques and exacerbation of arterial stiffness are precipitated by metabolic disorders present in diabetes mellitus. A number of intracellular pathways seem to be implicated in the development of carotid disease. Underlying mechanisms involve chronic inflammation and cellular senescence that affect endothelial and smooth muscle cells [140]. In this review, we have demonstrated that chronic hyperglycaemia is the primary stress stimulus that alters cell metabolism and regulatory pathways that lead to vascular dysfunction and arterial stiffening, while free fatty acid accumulation and insulin resistance have a strong atherogenic potential. Thus, efficient glycemic control and antisenescent medications constitute promising therapeutic approaches [136-141].
Highlights
- Carotid atheromatous disease is a major clinical issue among diabetic patients.
- Diabetic milieu promotes oxidative stress, inflammation and subsequent cellular senescence leading to endothelial dysfunction.
- Inflammation and diabetes are reciprocally related.
- The senescent phenotype occurring in diabetes is regulated by microRNAs and other cellular systems.
- Antidiabetic medications with anti-senescent effects seem to delay the evolution of atheromatous disease in diabetic patients.
Conflict of interest
The authors declare that no conflict of interest exists.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
References
- Alam U, Asghar O, Azmi S, Malik RA (2014) General aspects of diabetes mellitus. Handb Clin Neurol 126: 211-222. [Crossref]
- American Diabetes Association (2014) Diagnosis and classification of diabetes mellitus. Diabetes Care 37: S81-S90. [Crossref]
- Cuspidi C, Sala C, Provenzano F, Tadic M, Gherbesi E, et al. (2018) Metabolic syndrome and subclinical carotid damage: a meta-analysis from population-based studies. J Hypertens 36: 23-30. [Crossref]
- Bartman W, Nabrdalik K, Kwiendacz H, Sawczyn T, Tomasik A, et al. (2017) Association between carotid plaque score and microvascular complications of type 2 diabetes. Pol Arch Intern Med 127: 418-422. [Crossref]
- Scuteri A, Cunha PG, Rosei EA, Badariere J, Bekaert S, et al. (2014) Arterial Stiffness and Influences of the Metabolic Syndrome: A Cross-Countries Study. Atherosclerosis 233: 654-660. [Crossref]
- Cruickshank K, Riste L, Anderson SG, Wright JS, Dunn G, et al. (2002) Aortic pulse-wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation 106: 2085-2090. [Crossref]
- Lukich E, Matas Z, Boaz M, Shargorodsky M (2010) Increasing derangement of glucose homeostasis is associated with increased arterial stiffness in patients with diabetes, impaired fasting glucose and normal controls. Diabetes Metab Res Rev. 26: 365-370. [Crossref]
- Zhang M, Bai Y, Ye P, Luo I, Xiao W, et al. (2011) Type 2 diabetes is associated with increased pulse wave velocity measured at different sites of the arterial system but not augmentation index in a Chinese population. Clin Cardiol 34: 622-627. [Crossref]
- Naka KK, Papathanassiou K, Bechlioulis A, Kazakos N, Pappas K, et al. (2012) Determinants of vascular function in patients with type 2 diabetes. Cardiovasc Diabetol. 11: 127. [Crossref]
- Yan Z, Liang Y, Jiang H, Cai C, Sun B, et al. (2014) Metabolic syndrome and subclinical carotid atherosclerosis among Chinese elderly people living in a rural community. Metab Syndr Relat Disord 12: 269-276. [Crossref]
- Nazish S, Zafar A, Shahid R, Albakr A, Alkhamis FA, et al. (2018) Relationship Between Glycated Haemoglobin and Carotid Atherosclerotic Disease Among Patients with Acute Ischaemic Stroke. Sultan Qaboos University Med J. 18: e311-e317. [Crossref]
- Chen X, Zhang Y, Shen X, Huang Q, Ma H, et al. (2010) Correlation between glucose fluctuations and carotid intima-media thickness in type 2 diabetes. Diabetes Res Clin Pract 90: 95-99. [Crossref]
- Prenner SB, Chirinos JA (2014) Arterial stiffness in diabetes mellitus. Atherosclerosis. 238: 370-379. [Crossref]
- Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, et al. (2007) Insulin resistance as estimated by Homeostasis Model Assessment predicts incident symptomatic cardiovascular disease in caucasian subjects from the general population: the Bruneck study. Diabetes Care 30: 318-324. [Crossref]
- Jin SM, Noh CI, Yang SW, Bae EJ, Shin CH, et al. (2008) Endothelial dysfunction and microvascular complications in type 1 diabetes mellitus. J Korean Med Sci 23: 77-82. [Crossref]
- Naylor LH, Green DJ, Jones TW, Kalic RJ, Suriano KL, et al. (2011) Endothelial function and carotid intima-medial thickness in adolescents with type 2 diabetes mellitus. J Pediatr 159: 971-974. [Crossref]
- Avogaro A, de Kreutzenberg SV, Federici M, Fadini GP (2013) The endothelium abridges insulin resistance to premature aging. J Am Heart Assoc 2: e000262. [Crossref]
- van Slotten TT, Henry RM, Dekker JM, Nijpels G, Unger T, et al. (2014) Endothelial dysfunction plays a key role in increasing cardiovascular risk in type 2 diabetes: the Hoorn study. Hypertension. 64: 1299-1305. [Crossref]
- Nguyen N, Naik V, Speer MY (2013) Diabetes Mellitus Accelerates Cartilaginous Metaplasia and Calcification in Atherosclerotic Vessels of LDLr Mutant Mice. Cardiovasc Pathol 22: 167-75. [Crossref]
- Sawada N, Arany Z (2017) Metabolic Regulation of Angiogenesis in Diabetes and Aging. Physiology (Bethesda) 32: 290-307. [Crossref]
- Wang Z, Jiang Y, Liu N, Ren L, Zhu Y, et al. (2012) Advanced glycation end-product Nε-carboxymethyl-Lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis 221: 387-396. [Crossref]
- Liu SY, Yuan Q, Li XH, Hu CP, Hu R, et al. (2015) Role of vascular peroxidase 1 in senescence of endothelial cells in diabetes rats. Int J Cardiol 197: 182-191. [Crossref]
- Rodríguez- Mañas L, Angulo J, Peiro C, Llergo JL, Sanchez-Ferrer A, et al. (1998) Endothelial dysfunction and metabolic control in streptozotocin-induced diabetic rats. Br J Pharmacol 123: 1495-1502. [Crossref]
- Leo CH, Hart JL, Woodman OL (2011) Impairment of both nitric oxide-mediated and EDHF-type relaxation in small mesenteric arteries from rats with streptozotocin-induced diabetes. Br J Pharmacol 162: 365-377. [Crossref]
- Kim YK, Lee MS, Son SM, Kim IJ, Lee WS, et al. (2002) Vascular NADH oxidase is involved in impaired endothelium-dependent vasodilation in OLETF rats, a model of type 2 diabetes. Diabetes. 51: 522-527. [Crossref]
- Rodríguez-Mañas L, López-Dóriga P, Petidier R, Neira M, Solís J, et al. (2003) Effect of glycaemic control on the vascular nitric oxide system in patients with type 1 diabetes. J Hypertens. 21: 1137-1143. [Crossref]
- Angulo J, Cuevas P, Fernández A, Gabancho S, Allona A, et al. (2003) Diabetes impairs endothelium-dependent relaxation of human penile vascular tissues mediated by NO and EDHF. Biochem Biophys Res Commun 312: 1202-1208. [Crossref]
- Molnar J, Yu S, Mzhavia N, Pau C, Chereshnev I, et al. Diabetes induces endothelial dysfunction but does not increase neointimal formation in high-fat diet fed C57BL/6J mice. Circ Res 96: 1178-1184. [Crossref]
- Schjørring O, Kun A, Flyvbjerg A, Kirkeby HJ, Jensen JB, et al. (2012) Flow-evoked vasodilation is blunted in penile arteries from Zucker diabetic fatty rats. J Sex Med 9: 1789-1800. [Crossref]
- Vozarova B, Weyer C, Lindsay RS, Pratley RE, Bogardus C, et al. (2002) High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 51:455-461. [Crossref]
- Lin J, Glynn RJ, Rifai N, Manson JE, Ridker PM, et al. (2008) Inflammation and progressive nephropathy in type 1 diabetes in the Diabetes Control and Complications Trial. Diabetes Care 31: 2338-2343. [Crossref]
- Schulze MB, Rimm EB, Li T, Rifai N, Stampfer MJ, et al. (2004) C-reactive protein and incident cardiovascular events among men with diabetes. Diabetes Care 27: 889-894. [Crossref]
- Friedman AN, Hunsicker LG, Selhub J, Bostom AG, Collaborative Study Group (2005) C-reactive protein as a predictor of total arteriosclerotic outcomes in type 2 diabetic nephropathy. Kidney Int 68:773-778. [Crossref]
- Krzyzanowska K, Mittermayer F, Wolzt M, Schernthaner G (2007) Asymmetric dimethylarginine predicts cardiovascular events in patients with type 2 diabetes. Diabetes Care 30: 1834-1839. [Crossref]
- Henze LA, Luong TTD, Boehme B, Masyout J, Schneider MP, et al. (2019) Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging (Albany NY) 11: 5445-5462. [Crossref]
- Sedeek M, Callera G, Montezano A, Gutsol A, Heitz F, et al. (2010) Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Ren Physiol 299: F1348-F1358. [Crossref]
- Förstermann U, Sessa WC (2012) Nitric oxide synthases: regulation and function. Eur Heart J. 33: 829-37, 837a-837d. [Crossref]
- Gunnett CA, Heistad DD, Faraci FM (2003) Gene-targeted mice reveal a critical role for inducible nitric oxide synthase in vascular dysfunction during diabetes. Stroke 34: 2970-2974. [Crossref]
- Su Y, Qadri SM, Hossain M, Wu L, Liu L (2013) Uncoupling of eNOS contributes to redox-sensitive leukocyte recruitment and microvascular leakage elicited by methylglyoxal. Biochem Pharmacol 86: 1762-1764. [Crossref]
- Schinzari F, Tesauro M, Bertoli A, Valentini A, Veneziani A, et al. Calcification biomarkers and vascular dysfunction in obesity and type 2 diabetes: influence of oral hypoglycemic agents. Am J Physiol Endocrinol Metab 317: E658-E666. [Crossref]
- Zhang C (2008) The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol 103: 398-406. [Crossref]
- Corada M, Nyqvist D, Orsenigo F, Caprini A, Giampietro C, et al. (2010) The Wnt/beta- catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev Cell 18: 938-949. [Crossref]
- Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, et al. (2003) Inflammatory cytokines and the risk to develop type 2 diabetes. Results of the prospective populatioon-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Postdam Study. Diabetes 52: 812-817. [Crossref]
- Natali A, Toschi E, Baldeweg S, Ciociaro D, Favilla S, et al. (2006) Clustering of insulin resistance with vascular dysfunction and low-grade inflammation in type 2 diabetes. Diabetes 55: 1133-1140. [Crossref]
- Julia C, Czernichow S, Charnaux N, Ahluwalia N, Adreeva V, et al. (2014) Relationships between adipokines, biomarkers of endothelial function and inflammation and risk of type 2 diabetes. Diabetes Res Clin Pract 105: 231-238. [Crossref]
- Lindberg S, Jensen JS, Bjerre M, Pedersen SH, Frystyk J, et al. (2015) Adiponectin, type 2 diabetes and cardiovascular risk. Eur J Prev Cardiol 22: 276-283. [Crossref]
- Bitar MS, Ayed AK, Abdel-Halim SM, Isenovic ER, Al-Mulla F (2010) Inflammation and apoptosis in aortic tissues of aged type II diabetes: amelioration with α-lipoic acid through phosphatidylinositol 3-kinase/Akt-dependent mechanism. Life Sci 86: 844-853. [Crossref]
- Gunnett CA, Heistad DD, Faraci FM (2002) Interleukin-10 protects nitric oxide-dependent relaxation during diabetes: role of superoxide. Diabetes 51: 1931-1937. [Crossref]
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM (2002) Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23: 599-622. [Crossref]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, et al. (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404: 787-790. [Crossref]
- Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, et al. (2003) Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med 9: 294-299. [Crossref]
- Harjes U, Bridges E, McIntyre A, Fielding BA, Harris AL (2014) Fatty acid-binding protein 4, a point of convergence for angiogenic and metabolic signaling pathways in endothelial cells. J Biol Chem. 289: 23168-23176. [Crossref]
- Hazarika S, Dokun AO, Li Y, Popel AS, Kontos CD, et al. (2007) Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor. Circ Res 101: 948-956. [Crossref]
- Pechlivani N, Ajjan RA (2018) Thrombosis and Vascular Inflammation in Diabetes: Mechanisms and Potential Therapeutic Targets. Front Cardiovasc Med 5: 1. [Crossref]
- Herranz D, Muñoz-Martin M, Cañamero M, Mulero F, Martinez-Pastor B, et al. (2010) Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun 1: 3. [Crossref]
- Yang J, Park Y, Zhang H, Gao X, Wilson E, et al. (2009) Role of MCP-1 in tumor necrosis factor-alpha-induced endothelial dysfunction in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 297: H1208–H1216. [Crossref]
- Jang C, Arany Z (2013) Metabolism: Sweet enticements to move. Nature 500: 409-411. [Crossref]
- Cosentino F, Eto M, De Paolis P, van der Loo B, Bachsmid M, et al. (2003) High glucose causes upregulation of cyclooxygenase 2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation 107: 1017-1023. [Crossref]
- Gordillo-Moscoso A, Ruiz E, Carnero M, Reguillo F, Rodriguez E, et al. (2013) Relationship between serum levels of triglycerides and vascular inflammation, measured as COX-2, in arteries from diabetic patients: a translational study. Lipids Health Dis. 12: 62. [Crossref]
- Ahmad M, Turkseven S, Mingone CJ, Gupte SA, Wolin MS, et al. (2005) Heme oxygenase-1 gene expression increases vascular relaxation and decreases inducible nitric oxide synthase in diabetic rats. Cell Mol Biol (Noisy-le-grand) 30; 51: 371-376. [Crossref]
- Kitayama J, Faraci FM, Gunnett CA, Heistad DD (2006) Impairment of dilator responses of cerebral arterioles during diabetes mellitus: role of inducible NO synthase. Stroke 37: 2129-2133. [Crossref]
- Baldan A, Ferronato S, Olivato S, Malerba G, Scuro A, et al. (2014) Cyclooxygenase 2, toll-like receptor 4 and interleukin 1β mRNA expression in atherosclerotic plaques of type 2 diabetic patients. Inflamm Res 63: 851-858. [Crossref]
- Bagi Z, Erdei N, Toth A, Li W, Hintze TH, et al. (2005) Type 2 diabetic mice have increased arteriolar tone and blood pressure. Enhanced release of COX-2-derived constrictor prostaglandins. Arterioscler Thromb Vasc Biol 25: 1610-1616. [Crossref]
- Szerafin T, Erdei N, Fulop T, Pasztor ET, Edes I, et al. (2006) Increased cyclooxygenase-2 expression and prostaglandin-mediated dilation in coronary arterioles of patients with diabetes mellitus. Circ Res. 99: e12-e17. [Crossref]
- Chen SS, Jenkins AJ, Majewski H (2009) Elevated plasma prostaglandins and acetylated histone in monocytes in type 1 diabetic patients. Diabet Med 26: 182-186. [Crossref]
- Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, et al. (2002) Impaired Nitric Oxide Synthase Pathway in Diabetes Mellitus. Role of Asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation 106: 987-992. [Crossref]
- Corral-Fernandez NE, Salgado-Bustamante M, Martınez-Leija ME, Cortez-Espinosa N, Garcıa-Hernandez MH, et al. (2013) Dysregulated miR-155 expression in peripheral blood mononuclear cells from patients with type 2 diabetes. Exp Clin Endocrinol Diabetes 121: 347-353. [Crossref]
- Wang Q, Bozack SN, Yan Y, Boulton ME, Grant MB, e al. (2014) Regulation of retinal inflammation by ryhthmic expression of miR-146a in diabetic retina. Invest Ophthalmol Vis Sci 55: 3986-3994. [Crossref]
- Zhang L, He S, Guo S, Xie W, Xin R, et al. (2014) Down-regulation of mir-34a alleviates mesangial proliferation in vitro and glomerular hypertrophy in early diabetic nephropathy mice by targeting GAS1. J Diabetes Complications 28: 259-264. [Crossref]
- Herrera BM, Lockstone HE, Taylor JM, Ria M, Barrett A, et al. (2010) Global microRNA expression profiles in insulin target tissues in a spontaneous rat model of type 2 diabetes. Diabetologia 53: 1099-1109. [Crossref]
- Karolina DS, Tavintharan S, Armugam A, Sepramaniam S, Pek SL, et al. (2012) Circulating miRNA profiles in patients with metabolic syndrome. J Clin Endocrinol Metab 97: E2271-E2276. [Crossref]
- Li Z, Dong Y, He C, Pan X, Liu D, et al. (2019) RNA-Seq Revealed Novel Non-proliferative Retinopathy Specific Circulating MiRNAs in T2DM Patients. Front Genet 10: 531. [Crossref]
- Winnik S, Stein S, Matter CM (2012) SIRT1 – an anti-inflammatory pathway at the crossroads between metabolic diseases and atherosclerosis. Curr Vasc Pharmacol 10: 693-696. [Crossref]
- Toniolo A, Warden EA, Nassi A, Cignarella A, Bolego C (2013) Regulation of SIRT1 in vascular smooth muscle cells from streptozotocin-diabetic rats. PLoS One 8: e65666. [Crossref]
- Chen H, Wan Y, Zhou S, Lu Y, Zhang Z, et al. (2012) Endothelium-specific SIRT1 overexpression inhibits hyperglycemia-induced upregulation of vascular cell senescence. Sci China Life Sci 55: 467-473. [Crossref]
- de Kreutzenberg SV, Ceolotto G, Papparella I, Bortoluzzi A, Semplicini A, et al. (2010) Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes 59: 1006-1015. [Crossref]
- Guclu A, Erdur FM, Turkmen K (2016) The Emerging Role of Sirtuin 1 in Cellular Metabolism, Diabetes Mellitus, Diabetic Kidney Disease and Hypertension. Exp Clin Endocrinol Diabetes 124: 131-139. [Crossref]
- Orimo M, Minamino T, Miyauchi H, Tateno K, Okada S, et al. (2009) Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol 29: 889-894. [Crossref]
- Ota H, Akishita M, Eto M, Iijima K, Kaneki M, et al. (2007) Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol 43: 571-579. [Crossref]
- Wang Y, Zhang Z, Sun W, Tan Y, Liu Y, et al. (2014) Sulforaphane attenuation of type 2 diabetes-induced aortic damage was associated with the upregulation of Nrf2 expression and function. Oxid Med Cell Longev 2014: 123963. [Crossref]
- Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, et al. (2008) Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res 80: 191-199. [Crossref]
- Zhou S, Chen HZ, Wan YZ, Zhang QJ, Wei YS, et al. (2011) Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res 109: 639-648. [Crossref]
- Cheng X, Siow RC, Mann GE (2011) Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid Redox Signal 14: 469-487. [Crossref]
- Borradaile NM, Pickering JG (2009) Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 8: 100-112. [Crossref]
- Behl Y, Krothapalli P, Desta T, Roy S, Graves DT (2009) FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes 58: 917-925. [Crossref]
- Hou J, Chong ZZ, Shang YC, Maiese K (2010) FOXO3a governs early and late apoptotic endothelial programs during elevated glucose through mitochondrial and caspase signaling. Mol Cell Endocrinol 321: 194-206. [Crossref]
- Tanaka J, Qiang L, Banks AS, Welch CL, Matsumoto M, et al. (2009) Foxo1 links hyperglycemia to LDL oxidation and endothelial nitric oxide synthase dysfunction in vascular endothelial cells. Diabetes 58: 2344-2354. [Crossref]
- Nwadozi E, Roudier E, Rullman E, Tharmalingam S, Liu HY, et al. (2016) Endothelial FoxO proteins impair insulin sensitivity and restrain muscle angiogenesis in response to a high-fat diet. FASEB J. 30: 3039-3052. [Crossref]
- Sawada N, Jiang A, Takizawa F, Safdar A, Manika A, et al. (2014) Endothelial PGC-1α mediates vascular dysfunction in diabetes. Cell Metab 19: 246-258. [Crossref]
- Davidson SM, Duchen MR (2007) Endothelial mitochondria: contributing to vascular function and disease. Circ Res 100: 1128-1141. [Crossref]
- Victor VM, Rocha M, Herance R, Hernandez-Mijares A (2011) Oxidative stress and mitochondrial dysfunction in type 2 diabetes. Curr Pharm Des 17: 3947-3958. [Crossref]
- Zhang Z, Apse K, Pang J, Stanton RC (2000) High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J Biol Chem 275: 40042-40047. [Crossref]
- Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54: 1615-1625. [Crossref]
- Giacco F, Brownlee M (2010) Oxidative stress and diabetic complications. Circ Res 107: 1058-1070. [Crossref]
- Liu R, Liu H, Ha Y, Tilton RG, Zhang W (2014) Oxidative stress induces endothelial cell senescence via downregulation of Sirt6. Biomed Res Int 2014: 902842. [Crossref]
- Dymkowska D, Drabarek B, Podszywałow-Bartnicka P, Szczepanowska J, Zabłocki K. Hyperglycaemia modifies energy metabolism and reactive oxygen species formation in endothelial cells in vitro. Arch Biochem Biophys 542: 7-13. [Crossref]
- Wautier JL, Schmidt AM (2004) Protein glycation: a firm link to endothelial cell dysfunction. Circ Res 95: 233-238. [Crossref]
- Du X, Matsumura T, Edelstein D, Rossetti L, Zsengellér Z, et al. (2003) Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 112: 1049-1057. [Crossref]
- De Bock K, Georgiadou M, Carmeliet P (2013) Role of endothelial cell metabolism in vessel sprouting. Cell Metab 18: 634-647. [Crossref]
- Wang Z, Jiang Y, Liu N, Ren L, Zhu Y, et al. Advanced glycation end-product Nε-carboxymethyl-Lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis 221: 387-396. [Crossref]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, et al. (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154: 651-663. [Crossref]
- Manigrasso MB, Juranek J, Ramasamy R, Schmidt AM (2014) Unlocking the biology of RAGE in diabetic microvascular complications. Trends Endocrinol Metab 25: 15-22. [Crossref]
- Goveia J, Stapor P, Carmeliet P (2014) Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol Med 6: 1105-1120. [Crossref]
- Berrone E, Beltramo E, Solimine C, Ape AU, Porta M (2006) Regulation of intracellular glucose and polyol pathway by thiamine and benfotiamine in vascular cells cultured in high glucose. J Biol Chem 281: 9307-9313. [Crossref]
- Stefano GB, Challenger S, Kream RM (2016) Hyperglycemia-associated alterations in cellular signaling and dysregulated mitochondrial bioenergetics in human metabolic disorders. Eur J Nutr 55: 2339-2345. [Crossref]
- Goldin A, Beckman JA, Schmidt AM, Creager MA (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114: 597-605. [Crossref]
- Dias IK, Griffiths HR (2014) Oxidative stress in diabetes – circulating advanced glycation end products, lipid oxidation and vascular disease. Ann Clin Biochem. 51: 125-127. [Crossref]
- King GL, Loeken MR (2004) Hyperglycemia-induced oxidative stress in diabetic complications. Histochem Cell Biol 122: 333-338. [Crossref]
- Moldogazieva NT, Mokhosoev IM, Mel'nikova T, Porozov YB, Terentiev AA (2019) Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid Med Cell Longev 2019: 3085756. [Crossref]
- Ghosh A, Gao L, Thakur A, Siu PM, Lai CWK (2017) Role of free fatty acids in endothelial dysfunction. J Biomed Sci 24: 50. [Crossref]
- Hardy OT, Czech MP, Corvera S (2012) What causes the insulin resistance underlying obesity? Curr Opin Endocrinol Diabetes Obes 19: 81-87. [Crossref]
- Talior I, Yarkoni M, Bashan N, Eldar-Finkelman H. Increased glucose uptake promotes oxidative stress and PKC-δ activation in adipocytes of obese, insulin-resistant mice. Am J Physiol Endocrinol Metab 285: E295-302. [Crossref]
- Koya D, King GL (1998) Protein kinase C activation and the development of diabetic complications. Diabetes 47: 859-866. [Crossref]
- Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, et al. (1992) Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci USA 89: 11059-11063. [Crossref]
- Kim JA, Montagnani M, Koh KK, Quon MJ (2006) Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113: 1888-1904. [Crossref]
- Montagnani M, Golovchenko I, Kim I, Koh GY, Goalstone ML, et al (2002). Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem 277: 1794-1799. [Crossref]
- Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, et al. (2000) High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49: 1939-1945. [Crossref]
- Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444: 840-846. [Crossref]
- Agrawal NK, Kant S (2014) Targeting inflammation in diabetes: newer therapeutic options. World J Diabetes 5: 697-710. [Crossref]
- Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, et al. (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356: 1517-1526. [Crossref]
- Ridker PM, Howard CP, Walter V, Everett B, Libby P, et al. (2012) Effects of interleukin-1β inhibition with canakinumab on haemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo controlled trial. Circulation 126: 2739-2748. [Crossref]
- Hensen J, Howard CP, Walter V, Thuren T (2013) Impact of interleukin-1β antibody (canakinumab) on glycaemic indicators in patients with type 2 diabetes mellitus: results of secondary endpoints from a randomized, placebo-controlled trial. Diabetes Metab. 39: 524-531. [Crossref]
- Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, et al. (2010) The effects of salsalate on glycaemic control in patients with type 2 diabetes. Ann Intern Med 152: 346-357. [Crossref]
- Pieper GM, Siebeneich W, Olds CL, Felix CC, Del Soldato P (2002) Vascular protective actions of a nitric oxide aspirin analog in both in vitro and in vivo models of diabetes mellitus. Free Radic Biol Med 32: 1143-1156. [Crossref]
- Yang J, Park Y, Zhang H, Gao X, Wilson E, et al. (2009) Role of MCP-1 in tumor necrosis factor-α-induced endothelial dysfunction in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 297: H1208-H1216. [Crossref]
- Murthy SN, Desouza CV, Bost NW, Hilaire RC, Casey DB, et al. (2010) Effects of salsalate therapy on recovery from vascular injury in female Zucker fatty rats. Diabetes 59: 3240-3246. [Crossref]
- Bruder-Nascimento T, Callera GE, Montezano AC, He Y, Antunes TT, et al. (2015) Vascular injury in diabetic db/db mice is ameliorated by atorvastatin: role of Rac1/2-sensitive Nox-dependent pathways. Clin Sci 128: 411-423. [Crossref]
- Esposito K, Ciotola M, Carleo D, Schisano B, Saccomano F, et al. (2006) Effect of rosiglitazone on endothelial function and inflammatory markers in patients with the metabolic syndrome. Diabetes Care 29: 1071-1076. [Crossref]
- Konduracka E, Galicka-Latala D, Cieslik G, Rostoff P, Fedak D, et al. (2008) Effect of atorvastatin on endothelial function and inflammation in long-duration type 1 diabetic patients without coronary heart disease and essential hypertension. Diabetes Obes Metab 10: 719-725. [Crossref]
- Dodson M, Darley-Usmar V, Zhang J (2013) Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med. 63: 207-221. [Crossref]
- Eelen G, Cruys B, Welti J, De Bock K, Carmeliet P (2013) Control of vessel sprouting by genetic and metabolic determinants. Trends Endocrinol Metab 24: 589-596. [Crossref]
- Emadi SS, Soufi FG, Khamaneh AM, Alipour MR (2014) MicroRNA-146a expression and its intervention in NF-кB signaling in diabetic rat aorta. Endocr Regul 48: 103-108. [Crossref]
- Shah MS, Brownlee M (2016) Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res 118: 1808-1829. [Crossref]
- Mugoni V, Postel R, Catanzaro V, De Luca E, Turco E, et al. (2013) Ubiad1 is an antioxidant enzyme that regulates eNOS activity by CoQ10 synthesis. Cell 152: 504-518. [Crossref]
- Stroes E, Kastelein J, Cosentino F, Erkelens W, Wever R, et al. (1997) Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest 99: 41-46. [Crossref]
- Pancholia AK (2018) Sodium-glucose cotransporter-2 inhibition for the reduction of cardiovascular events in high-risk patients with diabetes mellitus. Indian Heart J 70: 915-921. [Crossref]
- Barbieri M, Marfella R, Esposito A, Rizzo MR, Angellotti E, et al. (2017) Incretin treatment and atherosclerotic plaque stability: Role of adiponectin/APPL1 signaling pathway. J Diabetes Complications 31: 295-303. [Crossref]
- Finkel T, Deng CX, Mostoslavsky R (2009) Recent progress in the biology and physiology of sirtuins. Nature 460: 587-591. [Crossref]
- Kao CL, Chen LK, Chang YL, Yung MC, Hsu CC, et al. (2010) Resveratrol protects human endothelium from H(2)O(2)-induced oxidative stress and senescence via SirT1 activation. J Atheroscler Thromb 17: 970-979. [Crossref]
- Guo Z, Su W, Allen S, Pang H, Dougherty A, et al. (2005) Up-regulation and smooth muscle contractile hyperreactivity in spontaneous diabetic db/db mice. Cardiovasc Res 67: 723-735. [Crossref]
- Carmeliet P, Wong BW, De Bock K (2012) Treating diabetes by blocking a vascular growth factor. Cell Metab 16: 553-555. [Crossref]