Abstract
Among the thirteen types of water channel proteins, aquaporins (AQPs), which play various essential roles in human physiology, AQP4 is richly expressed in cells of the central nervous system and implicated in pathological conditions such as brain edema. Therefore, researchers have been looking for ways to inhibit AQP4’s water-conducting function. Many small molecules have been investigated for their interactions with the residues that form the AQP4 channel entry vestibule on the extracellular side and their interruption of waters entering into the conducting pore. Conducting all-atom simulations on the basis of CHARMM 36 force field, we study one such inhibitor, 5-acetamido-1,3,4-thiadiazole-2-sulfonamide (AZM), to achieve quantitative agreement between the computed and the experimentally measured values of AZM-AQP4 binding affinity. Using the same method, we examine the possibility of plugging up the AQP4 channel around the Asn-Pro-Ala motifs located near the channel center because a small molecule bound there would totally occlude water conduction through AQP4. We compute the binding affinities of 1,2-ethanediol (EDO) and1,3-propanediol (PDO) inside the AQP4 conducting pore and identify the specificities of the interactions. The EDO-AQP4 interaction is weak with a dissociation constant of 80 mM.The PDO-AQP4 interactionis rather strong with a dissociation constant of 328 µM, which indicates thatPDO is an efficacious AQP4 inhibitor with sufficiently high potency.Considering the fact that PDO is classified by the US Food and Drug Administration as generally safe, we predict that 1,3-propanediol could be an effective drug for brain edema and other AQP4-correlated neurological conditions.
Key words
ligand-protein interaction, aquaporin inhibitor, molecular dynamics
Abbreviations
1D: one-dimensional; 3D: three-dimensional; AQP: aquaporin; AZM: 5-acetamido-1:3:4-thiadiazole-2-sulfonamide; ar/R: aromatic/arginine; EDO: 1:2-ethanediol;hSMD: hybrid steered molecular dynamics; IC50: half maximal inhibitory concentration; MD: molecular dynamics; NOAEL:no observed adverse effect level; NPA: asparagine-proline-alanine; PDB: protein data bank; PDO: 1:3-propanediol; PMF: potential of mean force; SMD: steered molecular dynamics; vdW: van der Waals.
Introduction
The family of water channel proteins (aquaporins, AQPs) [1-16], consists of aquaporins that conduct water, but not glycerol, and aquaglyceroporins that conduct, in addition to water, glycerol and some other polar solutes. They are present in all living systems, constituting the cell’s “plumbing system”. In humans, 13 AQPs (AQP0–AQP12), expressed in various cells from head to toe, govern a wide spectrum of physiological functions with broad clinical importance [15,17-25].In particular, AQP4water channels are an essential component in a number of physiological processes including water movement into and out of the brain, neuro-excitation, as well as astrocyte migration toward injury sites, all of which are important functions in the central nervous system (CNS) [15]. These key roles of AQP4 suggest that inhibitors of this particular water channel could potentially treat (cytotoxic) brain edemaby reducing intracranial pressure and water content, epilepsy by increasing the seizure threshold, and CNS injuries (e.g. trauma or stroke) by accelerating neuronal regeneration via reducing the formation of glial scars, respectively [26-34].As such, AQP4 is an appealing drug target and the search for its inhibitors is being actively pursued by medical scientists [8,15,35-38]. However, finding an efficacious and potent AQP inhibitor has proven to be challenging [15].While virtual screening seems to be a good method due to the widely available structural information for AQP, identifying bona fide inhibitors via computational studieshasyettobearfruit.The virtually identified binding ofsmall molecules does not necessarily translate to channel blocking activity;thisis particularly true inthecaseof AQPs due to their small size and narrow aqueous channel.For example,via virtual docking and oocyte functional assays, some of the epilepsy drugswere identified to bind strongly at the entry vestibule of the channel and inhibit AQP4 water conduction at low µM half maximal inhibitory concentrations (IC50s) [36] but structural experiments and proteoliposome assays of AQP4 in the presence of mM amounts of these drugs showed otherwise [8].The lack of crystal structures of AQP4-drug complexes in conjunction with the disagreement about IC50s indeed highlights the inaccuracy in binding affinity estimations inherent in the current virtual screening techniques and the need for accurate computation of binding affinities that still takes too long, even on today’s high performance supercomputers.
In this paper, we employ our newly developed hybrid steered molecular dynamics (hSMD) method [39,40], which is fast (somewhat brute force) and accurate, to investigate one of the epilepsy drugs and twoother small molecules shown in Figure 1 by conducting all-atom simulations (model systems illustrated in Figure 2) on the basis of CHARMM 36 force field [41,42].On 5-acetamido-1,3,4-thiadiazole-2-sulfonamide (AZM), an epilepsy drug tested on AQP4 inhibition [8,36], we achieve quantitative agreement between the computed and the experimentally measured [8] values of AZM-AQP4 binding affinity. Using the same method, we examine the possibility of plugging up the AQP4 channel around the NPA motifs located near the channel center because a small molecule bound there would totally occlude water conduction through AQP4. We compute the binding affinities of 1,2-ethanediol (EDO) and 1,3-propanediol (PDO) inside the AQP4 conducting pore and identify the specificities of the interactions. The toxic EDO binds inside the AQP4 channel with a rather weak affinity (dissociation constant 80 mM). The nontoxic PDO-AQP4 interaction is rather strong with a dissociation constant of 328 µM, which leads us to predictthat PDO is an efficacious AQP4 inhibitor with sufficiently high potency.
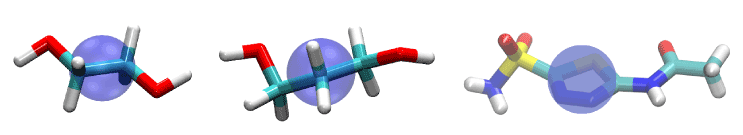
Figure 1.Ligands of this study: EDO (left), PDO (center), and AZM (right). They are represented as licorices colored by atom names: hydrogen, white; carbon, cyan; oxygen, red; nitrogen, blue; sulfur, yellow. The purple bubbles indicate the pulling center of each ligand.
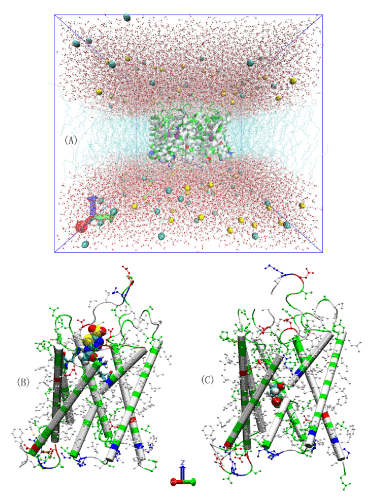
Figure 2.All-atom model systems.(A) The AQP4-PDO in silico system. Na+ and Cl- ions are represented as large spheres colored in yellow and cyan, respectively; waters are represented as balls-and-stickscolored in red for oxygen and white for hydrogen; protein is represented as a cartoon colored by residue types; andPDO is represented as large spheres colored in purple. (B) AZM bound to AQP4. The protein backbone is represented as cartoons and its H-less side chains as balls-and-sticks, both colored according to residue types. The ar/R residues (His 201 and Arg 216) are represented as licorices and AZM is represented as large spheres, both colored by atom names (hydrogen, white; carbon, cyan; oxygen, red; nitrogen, blue; sulfur, yellow). (C) PDO bound to AQP4. PDO is represented as large spheres colored by atom names (carbon, cyan; oxygen, red; hydrogen, white).Protein is represented in ways identical to (B).
Results
Binding AZMto the AQP4channelentry vestibule
AZM has low solubility in water (4.4 mMat30°C) [43].Oocyte functional assay studies suggested high potency of AQP4 inhibition by AZM with a half maximal inhibitory concentration (IC50) in the submicromolar range [36,37].However, AZM was not found bound to the protein in experiments ofcocrystallizing AQP4 with 5 mM of AZM and the IC50 measured in proteoliposome experiments was approximately 3 mM [8]. Our all-atom model study shows that AZM does bind to AQP4 in the channel entry vestibule with the sulfonamide group pointing away from the channel (Figure 3) but in a pose essentially opposite to what was found in the virtual docking studies which had the sulfonamide group plugging into the channel entry [36,37].We conducted a 50 ns MD run for each of the two poses and found that AZM in the latter pose fluctuates away from the protein by itself without being pulled. The pose illustrated in Figure 3 was found to be stable. The end state of the 50 ns run was chosen to be the one initial state for SMD runs of pulling AZM from the bound state to the dissociated state in the extracellular bulk. The fluctuations of the center of mass of AZM in this bound state (the last 10 ns, shown in SI, Figure S1) were used to compute the partial partition in Equation (7). The numerical results are:
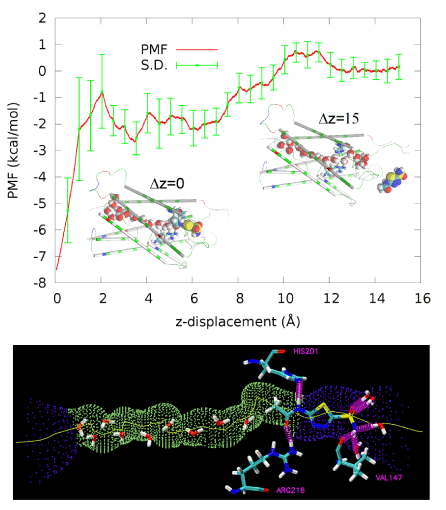
Figure 3.PMF along the dissociation path of pulling AZM from the binding site in the channel entry vestibule to the extracellular bulk.Inset Δz=0 (also drawn in the right panel) shows AZM at the binding site. Inset Δz=15 shows AZM in the extracellular bulk away from the protein. Protein is represented as cartoons colored by residue types. Waters inside the AQP4 conducting channel and AZM are shown as large spheres colored by atom names (hydrogen, white; oxygen, red; carbon, cyan; nitrogen, blue; sulfur, yellow). In the bottom panel, the AQP4 channel is illustrated as the purple-green-purple dotted pipe threaded with a yellow line. Three residues, His 201, Arg 216, and Val 147, form three hydrogen bonds with AZM. Also shown are waters inside the channel or hydrogen-bonded to AZM. All these waters, AZM, and the three residues are represented in licorices colored by atom names.
The PMF along the pulling path was computed from the work curves (shown in SI, Figure S2) using the Brownian dynamics fluctuation-dissipation theorem [44]. The PMF curve is shown in Figure 3 from which we obtained the PMF difference
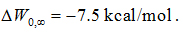
Plugging the values of equations (1) and (2) into Equation (7) and then into Equation (6), we obtained the dissociation constant of AZM as 1.8 mM, which is slightly lower than the experimentally measured IC50 at 3 mM [8]. Considering the fact that AZM fluctuates at the channel entry and thus does not totally plug up the water channel, the IC50 value should be higher than the dissociation constant. This nearly perfect agreement between our all-atom computation and the proteoliposome functional assay experiments [8] validates our hSMD method that was also validated in other protein-ligand complexes [39].
Binding PDO inside the AQP4 channel
PDO is a linear chain of three hydrocarbon groups terminated with a hydroxyl on each end (Figure 1). The fluctuations of PDO are non-Gaussian along the channel axis (z-axis). On the xy-plane perpendicular to the channel axis, the fluctuations can still be well approximated as Gaussian (shown in SI, Figure S3). Therefore, we conducted SMD runs of pulling the z-coordinate of PDO’s center of mass while allowing the xy-coordinates to fluctuate inside the AQP4 channel between z= −7.33Å and z= 8.67Å (Figure 4). We chose z= −7.33Å as the interface separating the single-file channel region(z>−7.33Å) from the non-single-file channel orifice to the cytoplasmic bulk region.On the xy-plane of z= −7.33Å, we recorded the fluctuations of PDO for 10 ns (shown in SI, Figure S3).We chose the end state of the 10 ns trajectory as the one initial state from which we conducted SMD runs of pulling PDO along a straight line from z= −7.33Å to the cytoplasmic bulk z<= −15.33Å disallowing xy-fluctuations (Figure 4). From the fluctuations on the interface, we obtained
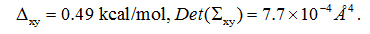
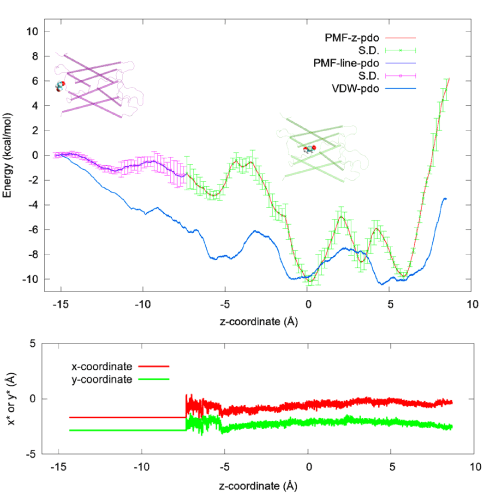
Figure 4.PDO-AQP4 binding characteristics. Top: The 1D PMF of PDO (red line with green error bars) as function of the z-coordinate of its center of mass vs the 3D PMF of PDO (blue line with purple error bars) along a line leading from the one chosen state to the corresponding dissociated state in the cytoplasmic bulk.Two insets of PDO (spheres)-AQP4 (cartoons) illustrate PDO bound inside the channel and PDO outside the channel respectively. Bottom: The x- and y-coordinates of the PDO center of mass along the pulling path. The x- and y-coordinates fluctuate freely inside the channel region where the 1D PMF was computed but they are not allowed to fluctuate along the partial path on which the 3D PMF was determined. Also plotted in the top panel is the vdW energy (blue line without error bars) between PDO and AQP4 as a function of the z-coordinate of the PDO center of mass, which was the result of time-coarse-graining a PDO trajectory.
From the work curves (shown in SI, Figure S4), we obtained the 1D PMF along the pulling path through part of the AQP4 channel and the 3D PMF from z= −7.33Åto the cytoplasmic bulk (shown in Figure 4). From the PMF curves, after integrating the 1D PMF in the channel region, we obtained
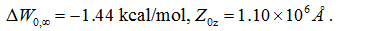
Plugging the numerical results in equations (3) and (4) into equation (11) and then into equation (6), we obtained the dissociation constant of PDO as 328 µM.
PDO vs EDO
In a procedure parallel to the above, we computed the dissociation constant of the highly toxic EDO as 80 mM, which is close to its lethal dosage. This rules out the medical use of EDO as an AQP4 inhibitor. However, it is interesting to note that the nontoxic PDO binds to AQP4 stronger than EDO by a factor of 244. This huge difference stems from the fact that a PDO molecule in a bulk of water outside the AQP4 channel disrupts, on average, 3.8 more water-water hydrogen bonds than an EDO molecule. Additionally, PDO has one more hydrocarbon group than EDO and thus has a stronger vdW interaction with the AQP4 channel.
Discussion
Efficacy of PDO as an AQP4 inhibitor
Illustrated in Figure 5 are the geometric characteristics of the AQP4 water channel. The water pore has a diameter ranging from 1.5 Å to 4.2 Å and thus allows only single-file lining of waters throughout the channel. No two waters can occupy the same z-coordinate inside the channel. However, near the NPA motifs (between z= 0Å and z= 5Å), the pore is wide enough to host a PDO molecule (Figure 5, top right panel). Pulling PDO away from this site either way, to the cytoplasmic or the extracellular side, actually causes distortions to the pore-lining sidechains of AQP4 because the channel is narrower on both sides of the binding site. When a PDO molecule binds there, it totally occludes the channel from water conduction. And its fluctuations inside the channel will not give space for water to squeeze by. (Another illustration is given inFigure 6.) Therefore, we conclude that PDO is an efficacious inhibitor of AQP4 water conduction.
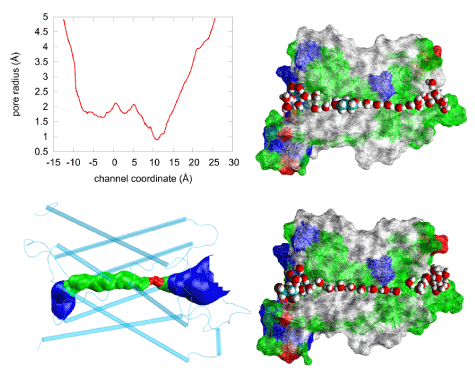
Figure 5.Steric fitness of PDO inside AQP4. The pore radius (top left panel) of the human AQP4 channel (PDB: 3GD8) obtained using the HOLE program [59].The narrowest point, with a diameter of ~1.5Å, corresponds to the ar/R (aromatic/arginine) selectivity filter region shown as red in the bottom left panel where the water-conducting channel is shown as the blue-green-red-green-blue pipe inside the protein monomer represented as cartoons colored in cyan. The two right panels illustrate the water channel being plugged up by PDO (top) or left open for water conduction. Approximately half of the protein residues are illustrated as wire-frame surfaces colored by residue types, selected waters are represented as large red (oxygen) and white (hydrogen) spheres. PDO inside (top) or outside (bottom) the channel is represented as large spheres colored in white (hydrogen), red (oxygen), and cyan (carbon).
Potency of PDO as an AQP4 inhibitor
Since a PDO bound to AQP4 totally occludes the water channel, the potency of PDO should be equal to the dissociation constant, IC50= kD= 328 µM. Considering that our PMF estimation has an error of ±1.2 kcal/mol, the IC50 of PDO should be less than 2.4 mM which indicates that PDO as an AQP4 inhibitor has sufficiently high potency. This is particularly true in light of the knowledge that PDO is nontoxic.
Interactions responsible for binding
In the bound state inside the AQP4 channel, PDO displaces two or three waters out of their places, which can be seen by comparing the top and the bottom panels of Figure 6. The three displaced waters, if not displaced, would form seven hydrogen bonds with the channel residues (three bonds) and with waters (four). In their place, PDO forms one or two hydrogen bonds with the channel residuesand two hydrogen bonds with two waters by its two hydroxyl groups. See Figure 6 (top panel) for a snap shot of PDO hydrogen-bonded to His 95 and Asn 97. Altogether, PDO in the bound state disrupts four hydrogen bonds on the average. In the dissociated state, when it is away from the protein, PDO forms four hydrogen bonds with waters. In terms of the hydrogen bonds PDO can form, there is no significant difference between its bound state and its dissociated state. However, in the dissociated state, PDO displaces four waters and disrupts 10 hydrogen bonds if we consider that each water forms 3.5 hydrogen bonds with other waters on the average. Therefore, the system of PDO-protein-waters has six more hydrogen bonds with PDO in the bound state than in the dissociated state. Additionally, the van der Waals interaction between PDO and the protein is also favorable for PDO to reside at the binding site (Figure 4, top panel) because it sterically fits in the channel near the NPA motifs. The van der Waals attraction between PDO and AQP4 and the hydrophobic effect (breaking more hydrogen bonds in the aqueous bulk than inside the protein) together are significantly stronger than the cost in conformational energies (SI, Figures S5 and S6). They are also much greater than the term of entropy loss due to binding (estimated in SI to be 4.4 kcal/mol). Therefore, we conclude that the van der Waals and the hydrophobic interactions are the relevant forces in the PDO-AQP4 system. The overall free energy of binding PDO inside AQP4 is approximately = –4.8 kcal/mol in correspondence to 328 µM.
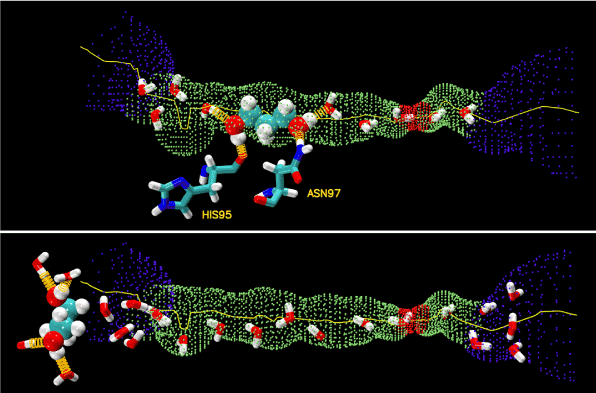
Figure 6.Hydrogen bonds.PDO inside (top panel) and outside (bottom panel) the AQP4 water channel. The channel is illustrated as the blue-green-red-green-blue pipe. PDO is shown as large spheres. Waters and residues His 95 and Asn 97 are shown as licorices. Colors: oxygen in red, carbon in cyan, nitrogen in blue, and hydrogen in white. The hydrogen bonds between PDO and protein residues/waters are shown as yellow springs.
Considering that the binding site of PDO is near the NPA motifs which are conserved across most aquaporins (human or other forms of life), we are tempted to speculate that PDO possibly binds inside other AQP channels as well. In Figure 7, we show the pore radii of human aquaporins whose coordinates are available from the PDB. The similarities between AQPs 2, 4, and 5 suggest that PDO may inhibit these three human aquaporins. However, we should not speculate that PDO would not inhibit AQP1 on the basis of the dissimilarities between AQPs 1 and 4 because the resolution is not sufficiently high for any of the three structures of AQP1. For any one of the aquaporins, we need to conduct detailed computations (currently underway) similar to this current work on AQP4.
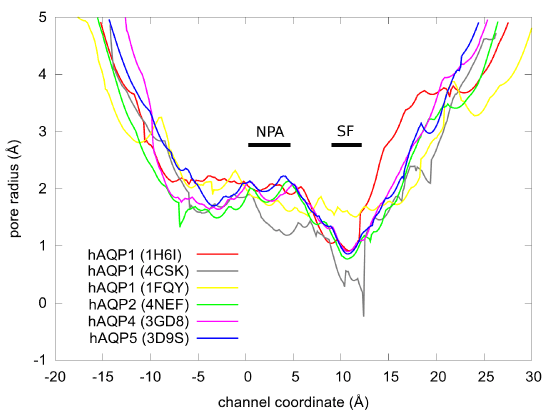
Figure 7.The pore radii of the human AQPs obtained from the PDB coordinates (PDB code: 1H6I, 4CSK, 1FQY, 4NEF, 3GD8, and 3D9S) using the HOLE program [59]. The ar/R selectivity filter and the NPA motifs are marked by SF and NPA bars respectively.
Possible medical use of 1,3-propanediol
A number of toxicity studies on PDO have been done to determine the potential harm that PDO may pose to humans. Under the toxicological profile for PDO in the Federal Register (June 12, 2013) [45], a 14-day inhalation toxicity study, a 90-day oral toxicity study, and a developmental toxicity study showed no evidence of any clinical signs of systemic toxicity at the limit dose of PDO. The mutagenic potential of PDO was also evaluated in both in vitro and in vivo experiments, and the results indicate that PDO is not considered mutagenic. Furthermore, the results from these studies indicate that there is no concern for the carcinogenicity of PDO or for the immunotoxicity or neurotoxicity of PDO. As such, the Environmental Protection Agency has concluded that there is a reasonable certainty that aggregate exposure to PDO presents no harm to the general population, including infants and children. The reported no-observed-adverse-effect-levels (NOAELs) are, respectively, 1.8 mg/L in the 14-day inhalation toxicity study; 1,000 mg/kg bw/day in the 90-day oral toxicity study; and 1,000 mg/kg bw/day in the developmental toxicity study.
In the FDA Agency Response Letter GRAS Notice No. GRN 000302 (March 2010) [46], the following is stated: “Based on the information provided by DuPont Tate & Lyle, as well as other information available to FDA, the agency has no questions at this time regarding DuPont Tate & Lyle's conclusion that PDO is GRAS (generally regarded as safe) under the intended conditions of use [in food products]”at a level of 34 mg/kg bw/day.
It is good to point out, however,that there has been one report of human death supposedly involving PDO in the cause of death in 2008 [47]: Two containers of antifreeze were found close to the decedent's body, and via gas chromatography–flame-ionization detection, the concentration of PDO in the decedent's body fluid was found to be 445 mg/dL (i.e. 58.5 mM).
Considering all the afore-stated data and our result that the IC50 of AQP4 inhibition by PDO is most probably around 328 µM and less than 2.4 mM, which is far less than the NOAELs, it should be safe to use 1,3-propanediol as a drug for brain edema and other AQP-correlated pathological conditions.
Conclusions
Using CHARMM 36 all-atom force fieldson the basis of the rigorous PMF formalism, we conducted hSMD simulations to compute the binding affinities of three AQP4 inhibitors: (1) AZM, an epilepsy drug, that binds to the entry vestibule of AQP4 channel and thus prevents waters from entering into the channel; (2) EDO, a toxic compound, that binds inside the channel and thus totally occludes it; and (3) PDO, a nontoxic food additive, that also binds inside the water-conducting pore and totally occludes water conduction. Our computed AZM-AQP4 dissociation constant agrees closely with the in vitro experimental measurement, which along with other tests on ligand-protein complexes shows the validity of our approach and the accuracy of the force field parameters. The weak binding affinity of EDO in conjunction with its toxicity precludes its medical use as an AQP4 inhibitor. In contrast, the reasonably strong affinity of the nontoxic PDO makes it a valid drug candidate. It is worth noting again that the location of the PDO bound state inside the single-file channel of AQP4 renders PDO’s efficacy so that the IC50 is simply equal to the dissociation constant kD. The computed dissociation constant of 328 µM indicates its potency as an AQP4 inhibitor. Taking into account the error bars of the computation, we have 44 µM<kD<2.4 mM (in accordance with the binding free energy = –4.8±1.2 kcal/mol). Therefore, we conclude with high confidence that 1,3-propanediol is an efficacious and potent AQP4 inhibitor, which could be a drug needed for neurotoxic brain edema and other AQP-correlated pathological conditions.
Methods
Parameters and procedures
We used the all-atom force field CHARMM36 [41,42] to represent all interactions. The van der Waals (vdW) cut-off distance was 10 Å with a switching distance of 9 Å. The electrostatic interactions were computed through the Particle-Mesh Ewald (PME) method, and periodic boundary conditions were applied in every direction. The temperature was kept at 298 K and the pressure at 1.0 bar. The time-step was 1.0 fs for the short range interactions and 2.0 fs for the long range interactions, and the Langevin damping coefficient was 5.0 ps-1. We used NAMD [48] to perform the simulations, and VMD [49]to set up simulation systems, analyze simulation results, and rendermolecular graphics.
We took the high-resolution crystal structure of AQP4 (PDB code: 3GD8) [8], embedded the tetramer in a 120 x 120 Å2patch of POPE bilayer, added a 30 Å-thick layer of waters to each side of the membrane patch, and salinated the system to an NaCl concentration of 150 mM. In this way, we obtained a pure AQP4 tetramer-membrane system consisting of 150,951 atoms without any ligands that can bind to AQP4. After the initial optimization and 50 ns equilibrium molecular dynamics (MD) run, the system settles down to dimensions of 115.3×112.6×110.6 Å3. This all-atom model system is similar to typical MD studies of AQPs in the current literature [50-55] pioneered in Ref. 4. In all MD production runs, we fixed the alpha carbons on the helices that are within the ±10 Å range from the central plane of the membrane to fully respect the crystal structure. Figure 1(A) illustrates the equilibrated system with one PDO in each AQP4 monomer.
Into the entry vestibule of an AQP4 monomer, we placed a 5-acetamido-1,3,4-thiadiazole-2-sulfonamide (AZM) molecule and deleted the waters occupying the site. We conducted a 10 ns MD run for AZM and waters to settle down followed by a 10 ns MD run to sample the fluctuations of AZM at the binding site. The equilibrated AZM-AQP4 structure is illustrated in Figure 1(B). From the bound state ensemble, we chose one state and conducted SMD runs of pulling AZM from the binding site to the extracellular bulk to compute the potential of mean force (PMF) along the unbinding path. We combined the PMF difference and the fluctuations to compute the binding affinity. In this case, we chose the center of mass of the thiadiazole ring as the pulling center (Figure 2). In hSMD runs for binding affinity computation, we used one AZM-AQP4 monomer complex in a 150 mMNaCl saline box of 80×80×109.7 Å3without the lipid bilayer. This reduction in system size speeds up simulations considerably but does not introduce additional errors because all the molecules reduced are beyond the cut-off distance from the ligand; the ligand is not charged; and the alpha carbons on the central parts of the helices are fixed as in the case of the full tetramer-membrane system.
Into the binding site near the NPA motifs of each AQP4 monomer, we placed an EDO/PDO molecule and deleted waters at or near the site to obtain the EDO/PDO-AQP4-membrane systems. After initial optimization and equilibration, we ran a 10 ns equilibrium MD run to sample EDO/PDO fluctuations. The equilibrated PDO-AQP4 complex is shown in Figure 1(C). Then we chose one state of the bound state ensemble and conducted SMD runs to compute the PMF along the path of pulling EDO/PDO from the channel to the cytoplasmic bulk and along the path of pulling EDO/PDO through the part of the channel around the NPA motifs. We combined the fluctuations and the PMF differences to compute the binding affinity. In this case we chose the center of mass of the entire ligand molecule as the pulling center (shown in Figure 2).In hSMD runs for binding affinity computation, we used one EDO/PDO-AQP4 monomer complex in a 150 mMNaCl saline box of 80×80×109.7Å3without the lipid bilayer. Again, this reduction in system size does not introduce additional errors because all the molecules reduced are beyond the cut-off distance from the ligand; EDO/PDO is not charged; and the alpha carbons on the central parts of the helices are fixed as in the case of the full tetramer-membrane system.
Computing binding affinities with hSMDmethod
The formulas and validation of hSMD method can be found in Refs. [39-40]. In this study, we only employ the case of one pulling center, n= 1, and briefly outline the hSMD formulation below.Following the standard literature [56,57], the binding affinity at one binding site is
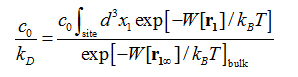
where is the standard concentration. For clarity and for convenience of unit conversion, we use two different but equivalent forms, on the left hand side and on the right hand side of the equation. kB is the Boltzmann constant and T is the absolute temperature. The three-dimensional (3D) integrations are over the x-, y-, and z-coordinates ofthe ligand’s position that can be chosen as the center of mass of one segment or of the whole ligand. (In the case of AZM, is chosen as the center of mass of the thiadiazole ring. In the case of PDO, is chosen as the center of mass of the entire ligand.) The integral has the units of Å3 that renders the right hand side dimensionless as it should be. is the 3D PMF. The subscripts “site” and “bulk” indicate that is at the binding site (near the PMF minimum) and in the bulk region far away from the protein, respectively. Expressed in terms of the PMF difference and the partial partition of the bound state , we have
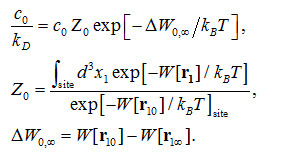
Here is the one bound state chosen from the bound state ensemble from which the ligand is pulled to the corresponding one state 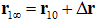 in the dissociated state ensemble.
in the dissociated state ensemble. 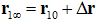 is the ligand displacement along the dissociation path from the binding site to the bulk region. The 3D PMF difference
is the ligand displacement along the dissociation path from the binding site to the bulk region. The 3D PMF difference  is computed from SMD runs following the multi-sectional scheme described in Ref. [58]. The partial partition
is computed from SMD runs following the multi-sectional scheme described in Ref. [58]. The partial partition  is computed in ways according to the nature of the ligand’s fluctuations at the binding site(described in the subsequent two subsections).
is computed in ways according to the nature of the ligand’s fluctuations at the binding site(described in the subsequent two subsections).
When the Ligand Fluctuations Can Be Approximated as 3D Gaussian, as in the case of AZM, all we have to do is to evaluate the fluctuations around the binding site. In this case, we have the dissociation constant, within the Gaussian approximation of the fluctuations at the binding site,

Here the dimensionless quantity  gives a measure of how far , the initial state chosen for SMD, is from the PMF minimum
gives a measure of how far , the initial state chosen for SMD, is from the PMF minimum  that is equal to the average vector under
that is equal to the average vector under the Gaussian approximation,
the Gaussian approximation,

The brackets stand for statistical average and the superscript T transposes a single-row matrix into a single-column matrix. represents the determinant. is the 3×3 matrix of the fluctuations/deviations of the pulling center coordinates etc.in the bound state ensemble,
, (9)
which can be accurately evaluated by running equilibrium MD in the bound state of the ligand-protein complex. is the inverse matrix.
When the ligand fluctuations cannot be approximated as 3D gaussian, as in the case of EDO/PDO, however, one would have to evaluate the partial partition differently. Considering integration on the xy-plane (parallel to the membrane and perpendicular to the AQP4 channel axis) separately from the integration along the channel axis (z-axis), we can factor the partial partition into two ratios:
. (10)
The first ratio can be well approximated as Gaussian when the SMD initial state is chosen as one inside the channel so that
. (11)
The second ratio can be expressed integration along the z-axis ( is constant in the integral) and the integrand is simply related to the 1D PMF difference,
(12)
The 1D PMF difference is computed by conducting SMD runs in which only the z-coordinate is steered while the x- and y-coordinates are free to fluctuate inside the channel [59]. The fluctuations on the xy-plane are measured to compute the deviation of the initial state from the average state on the xy-plane
(13)
and the 2×2 fluctuation matrix
. (14)
Slightly different from the case of AZM, the brackets in this case represent statistical average on the xy-plane.
Supplementary material
In the online supplementary information (SI.pdf), we present the fluctuation and work data that were used to obtain the potential of mean force (PMF) and the partial partition functions in the main text.
Conflict of interest 2021 Copyright OAT. All rights reserv
The authors filed a US patent application of the medical use of 1,3- propanediol.
Acknowledgements
The authors acknowledge support from the NIH (GM 084834) and the computing resources provided by the Texas Advanced Computing Center at University of Texas at Austin. LY acknowledges the Jiangsu Overseas Research & Training Program for University Prominent Young & Middle-aged Teachers and Presidents.ODV acknowledges a graduate scholarship from the Mexican National Council of Science and Technology (CONACYT Grant 533262).
References
- Agre P, Bonhivers M, Borgnia MJ (1998) The aquaporins, blueprints for cellular plumbing systems. J Biol Chem 273: 14659-14662. [Crossref]
- Heymann JB, Engel A (1999) Aquaporins: Phylogeny, Structure, and Physiology of Water Channels. News Physiol Sci 14: 187-193. [Crossref]
- Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, et al. (2000) Structural determinants of water permeation through aquaporin-1. Nature 407: 599-605. [Crossref]
- de Groot BL, Grubmüller H (2001) Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science 294: 2353-2357. [Crossref]
- Engel A, Stahlberg H. Aquaglyceroporins: Channel proteins with a conserved core, multiple functions, and variable surfaces. In: Thomas Zeuthen WDS, Ed. (2002) International Review of Cytology. Academic Press 75-104.
- Gonen T, Walz T (2006) The structure of aquaporins. Q Rev Biophys 39: 361-396. [Crossref]
- Carbrey JM, Agre P (2009) Discovery of the aquaporins and development of the field. Handb Exp Pharmacol: 3-28. [Crossref]
- Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WE, et al. (2009) Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc Natl Acad Sci U S A 106: 7437-7442. [Crossref]
- Branden M, Tabaei SR, Fischer G, Neutze R, Hook F (2010) Refractive-index-based screening of membrane-protein-mediated transfer across biological membranes. Biophys J 99: 124-133. [Crossref]
- Benga G (2012) On the definition, nomenclature and classification of water channel proteins (aquaporins and relatives). Mol Aspects Med 33: 514-517. [Crossref]
- Abramson J, Vartanian AS (2013) Biochemistry. Watch water flow. Science 340: 1294-1295. [Crossref]
- Gravelle S, Joly L, Detcheverry F, Ybert C, Cottin-Bizonne C, et al. (2013) Optimizing water permeability through the hourglass shape of aquaporins. Proceedings of the National Academy of Sciences 110: 16367-16372.
- Kosinska Eriksson U, Fischer G, Friemann R, Enkavi G, Tajkhorshid E, et al. (2013) Subangstrom resolution X-ray structure details aquaporin-water interactions. Science 340: 1346-1349. [Crossref]
- Oberg F, Hedfalk K (2013) Recombinant production of the human aquaporins in the yeast Pichia pastoris (Invited Review). Mol Membr Biol 30: 15-31. [Crossref]
- Verkman AS, Anderson MO2, Papadopoulos MC3 (2014) Aquaporins: important but elusive drug targets. Nat Rev Drug Discov 13: 259-277. [Crossref]
- Horner A, Zocher F, Preiner J2, Ollinger N, Siligan C, et al. (2015) The mobility of single-file water molecules is governed by the number of H-bonds they may form with channel-lining residues. Sci Adv 1: e1400083. [Crossref]
- King LS, Kozono D, Agre P (2004) From structure to disease: the evolving tale of aquaporin biology. Nat Rev Mol Cell Biol 5: 687-698. [Crossref]
- Krane CM, Goldstein DL (2007) Comparative functional analysis of aquaporins/glyceroporins in mammals and anurans. Mamm Genome 18: 452-462. [Crossref]
- Verkman AS, Mitra AK (2000) Structure and function of aquaporin water channels. Am J Physiol Renal Physiol 278: F13-28. [Crossref]
- Verkman AS (2002) Aquaporin water channels and endothelial cell function. J Anat 200: 617-627. [Crossref]
- Törnroth-Horsefield S, Hedfalk K, Fischer G, Lindkvist-Petersson K, Neutze R (2010) Structural insights into eukaryotic aquaporin regulation. FEBS Lett 584: 2580-2588. [Crossref]
- Delporte C, Steinfeld S (2006) Distribution and roles of aquaporins in salivary glands. Biochim Biophys Acta 1758: 1061-1070. [Crossref]
- Larsen HS, Ruus AK, Schreurs O, Galtung HK (2010) Aquaporin 11 in the developing mouse submandibular gland. Eur J Oral Sci 118: 9-13. [Crossref]
- Noda Y, Sohara E, Ohta E, Sasaki S (2010) Aquaporins in kidney pathophysiology. Nat Rev Nephrol 6: 168-178. [Crossref]
- Agre P, Preston GM, Smith BL, Jung JS, Raina S, et al. (1993) Aquaporin CHIP: the archetypal molecular water channel. Am J Physiol 265: F463-476. [Crossref]
- Assentoft M, Larsen BR, Olesen ETB, Fenton RA, MacAulay N (2014) AQP4 plasma membrane trafficking or channel gating is not significantly modulated by phosphorylation at COOH-terminal serine residues. Am J Physiol Cell Physiol 307: C957-C965. [Crossref]
- Hasegawa H, Ma T, Skach W, Matthay MA, Verkman AS (1994) Molecular cloning of a mercurial-insensitive water channel expressed in selected water-transporting tissues. J Biol Chem 269: 5497-5500. [Crossref]
- Jung JS, Bhat RV, Preston GM, Guggino WB, Baraban JM, Agre P (1994) Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc Natl Acad Sci U S A 91:13052-13056. [Crossref]
- Nagelhus EA, Ottersen OP (2013) Physiological roles of aquaporin-4 in brain. Physiol Rev 93: 1543-1562. [Crossref]
- Papadopoulos MC, Verkman AS (2005) Aquaporin-4 gene disruption in mice reduces brain swelling and mortality in pneumococcal meningitis. J Biol Chem 280: 13906-13912. [Crossref]
- Smith AJ, Jin BJ, Ratelade J, Verkman AS (2014) Aggregation state determines the localization and function of M1- and M23-aquaporin-4 in astrocytes. J Cell Biol 204: 559-573. [Crossref]
- Vella J, Zammit C, Di Giovanni G, Muscat R, Valentino M (2015) The central role of aquaporins in the pathophysiology of ischemic stroke. Front Cell Neurosci 9: 108. [Crossref]
- Badaut J, Fukuda AM, Jullienne A, Petry KG (2014) Aquaporin and brain diseases. Biochim Biophys Acta 1840: 1554-1565. [Crossref]
- Papadopoulos MC, Verkman AS (2013) Aquaporin water channels in the nervous system. Nat Rev Neurosci 14: 265-277. [Crossref]
- Papadopoulos MC, Verkman AS (2008) Potential utility of aquaporin modulators for therapy of brain disorders. Prog Brain Res 170: 589-601. [Crossref]
- Huber VJ, Tsujita M, Kwee IL, Nakada T (2009) Inhibition of aquaporin 4 by antiepileptic drugs. Bioorg Med Chem 17: 418-424. [Crossref]
- Huber VJ, Tsujita M, Yamazaki M, Sakimura K, Nakada T (2007) Identification of arylsulfonamides as Aquaporin 4 inhibitors. Bioorg Med Chem Lett 17: 1270-1273. [Crossref]
- Kato J, Hayashi MK, Aizu S, Yukutake Y, Takeda J, et al. (2013) A general anaestheticpropofol inhibits aquaporin-4 in the presence of Zn2+. Biochem J 454: 275-282. [Crossref]
- Chen LY (2015) Hybrid Steered Molecular Dynamics Approach to Computing Absolute Binding Free Energy of Ligand-Protein Complexes: A Brute Force Approach That Is Fast and Accurate. J Chem Theory Comput 11: 1928-1938. [Crossref]
- Rodriguez RA, Yu L, Chen LY (2015) Computing Protein-Protein Association Affinity with Hybrid Steered Molecular Dynamics. J Chem Theory Comput 11: 4427-4438. [Crossref]
- Brooks BR, Brooks CL 3rd, Mackerell AD Jr, Nilsson L, Petrella RJ, et al. (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30: 1545-1614. [Crossref]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, et al. (2010) CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31: 671-690. [Crossref]
- Yalkowsky SH, Dannenfelser RM (1992) Aquasol database of aqueous solubility. College of Pharmacy, University of Arizona, Tucson, AZ.
- Chen LY (2011) Exploring the free-energy landscapes of biological systems with steered molecular dynamics. Phys Chem Chem Phys 13: 6176-6183. [Crossref]
- 1,3-Propanediol; Exemptions From the Requirement of a Tolerance: A Rule by the Environmental Protection Agency on 06/12/2013, The Federal Register, The Daily Journal of the United States Government: 2013.
- The Food and Drug Administration Agency Response Letter GRAS Notice No. GRN 000302: 2010.
- Garg U, Frazee CC 3rd, Kiscoan M, Scott D, Peterson B, et al. (2008) A fatality involving ,3-propanediol and its implications in measurement of other glycols. J Anal Toxicol 32: 324-326. [Crossref]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, et al. (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26: 1781-1802. [Crossref]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33-38, 27-8. [Crossref]
- Jensen MØ, Park S, Tajkhorshid E, Schulten K (2002) Energetics of glycerol conduction through aquaglyceroporin GlpF. Proc Natl Acad Sci U S A 99: 6731-6736. [Crossref]
- Jensen MØ, Mouritsen OG (2006) Single-channel water permeabilities of Escherichia coli aquaporins AqpZ and GlpF. Biophys J 90: 2270-2284. [Crossref]
- Hub JS, de Groot BL (2008) Mechanism of selectivity in aquaporins and aquaglyceroporins. Proc Natl Acad Sci U S A 105: 1198-1203. [Crossref]
- Alberga D, Nicolotti O, Lattanzi G, Nicchia GP, Frigeri A, et al. (2014) A new gating site in human aquaporin-4: Insights from molecular dynamics simulations. Biochim Biophys Acta 1838: 3052-3060. [Crossref]
- Aponte-Santamaria C, Hub JS, de Groot BL (2010) Dynamics and energetics of solute permeation through the Plasmodium falciparum aquaglyceroporin. Phys Chem Chem Phys 12: 10246-10254. [Crossref]
- Cui Y, Bastien DA (2011) Water transport in human aquaporin-4: molecular dynamics (MD) simulations. Biochem Biophys Res Commun 412: 654-659. [Crossref]
- Zhou HX, Gilson MK (2009) Theory of free energy and entropy in noncovalent binding. Chem Rev 109: 4092-4107. [Crossref]
- Woo HJ, Roux B (2005) Calculation of absolute protein-ligand binding free energy from computer simulations. Proc Natl Acad Sci U S A 102: 6825-6830. [Crossref]
- Chen LY (2013) Glycerol modulates water permeation through Escherichia coli aquaglyceroporin GlpF. Biochim Biophys Acta 1828: 1786-1793. [Crossref]
- Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MS (1996) HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph 14: 354-360, 376. [Crossref]