Absence of the second X chromosome in Turner syndrome is generally well tolerated, but in cases of small fragments or rings derived from the X chromosome, the incidence of mental retardation and other unusual congenital abnormalities can be significantly higher. This investigation focuses on a 9-year-old female who was referred for molecular analysis of the karyotype because of dysmorphic features uncharacteristic of Turner’s syndrome such as moderate psychomotor retardation, significant language delay and autism.
Karyotype analysis by Affymetrix CytoScan™ high-density (HD) array allowed to identify a small fragment derived from the X chromosome which was detected in 70% of cells. The fragment appeared to be made mainly of alphoid sequences with breakpoints in the juxtacentromeric region Xp11.1 and Xq13.2. The fragment did not include the XIST locus and might not, therefore, be subject to X-inactivation. Interestingly we also detected a 15q13.3 microdeletion of 1.84 Mb, including the genes: FAN1, TRPM1, MIR211, KLF13, OTUD7A, CHRNA7 and ARHGAP11A. The 15q13.3 microdeletion syndrome (OMIM#612001) is characterized by a wide range of phenotypic features, including intellectual disability, autism and psychiatric conditions.
15q13.3 microdeletion, aneuploidy, autism, CHRNA7, ring chromosome, Turner syndrome
Turner syndrome (TS) is defined as the total or partial absence of the second sex chromosome in women [1]. Its incidence is 1 in every 1,850 newborn girls (7th International Conference on Turner Syndrome, 2009) although it is higher at the moment of fertilization, since it is estimated that 3% of all human fertilizations are 45,X [2] but only 1% survive beyond 24 weeks gestation [3,4]. The Turner phenotype is quite variable, even among women with the same karyotype. However, there are some cardinal features: short stature (>99%), gonadal dysgenesis (>90%) and anatomic malformations such as Pterigium colli or cubitus valgus (>80%) [5]. In addition, there are other less frequent characteristics such us cardiovascular congenital defects, renal alterations, aorta anomalies, and also a specific neuropsychological profile, nevertheless autism and mental deficiency are not usual characteristics of TS. Only patients with fragments or rings derived from the X chromosome have been described with a more severe phenotype that depends on: origin, size, replication timing of the fragment (ring) chromosome, genes affected by copy number variations, level of mosaicism and status of the X inactivation [6].
Furthermore, the 15q13.3 microdeletion syndrome (OMIM#612001) is a disorder characterized by a high degree of variable expressivity. Individuals with this deletion often present a variety of phenotypic features including intellectual disability, seizures, autism, behavioural issues, and psychiatric conditions such as schizophrenia or bipolar disorder [7,8]. Most patients with 15q13.3 microdeletions have a typical deletion containing six genes (MRMR15, MTMR10, TRPM1, KLF13, OTUD7A, and CHRNA7); therefore, recently a series of probands was reported with smaller deletions encompassing only the CHRNA7 gene and neurodevelopmental symptoms similar to those observed with the larger 15q13.3 microdeletion [9,10]. Therefore, it was suggested that CHRNA7 may be the gene responsible for the abnormal phenotype in patients with 15q13.3 microdeletion syndrome.
Subject
The patient was a 9-year-old female diagnosed at the age of 8 months with TS with a no-mosaic karyotype 46,X,+mar(?) defined by G banding. Physical examination revealed a female phenotype without genital ambiguity, abundant body hair and short stature. She showed features suggestive of TS such as widely spaced nipples, Pterygium colli and coarctation of the aorta. Pelvic ultrasonography revealed a small uterus and rudimentary gonads that were surgically removed. Other features were deficient intrauterine growth, low birth weight (1.97 kg), psychomotor, language and mental delay, lumbar scoliosis, strabismus, hypothyroidism, celiac disease and autism.
Cell culture, cytogenetic and FISH analysis
The standard techniques for culturing lymphocytes from peripheral blood were used [11]. The X chromosome was studied by using the biotinylated probe DXZ1 (Oncor), which specifically hybridizes with the centromeric region of this chromosome. The hybridization procedure originally described by [12] was used in accordance with the manufacturer’s instructions (Oncor).
Genotype Assessment by High-Density (HD) Array
Genomic DNA was extracted using the DNeasy Blood & Tissue Kit from Qiagen. The genome-wide DNA copy number analyses were performed with Affymetrix CytoScan™ HD array in accordance with the manufacturer’s instructions. It has a high resolution (1 probe/880 bp; 2.6 million probes-1.9 million CNV and non-polymorphic, 750,000 single nucleotide polymorphism (SNP) probes, and it encompasses almost all known OMIM and RefSeq genes.
Copy number analysis was performed using Affymetrix® Chromosome Analysis Suite 2.0. The classification of CNV was obtained from information deposited in public international databases, such as GeneCards® http://www.genecards.org, MedGen http://www.ncbi.nlm.nih.gov/medgen, ClinVar http://www.ncbi.nlm.nih. gov/clinvar and Database of Genomic Variants http://dgv.tcag.ca.
The case presented common clinical characteristics of TS such as growth retardation, ovarian dysgenesis, widely spaced nipples, Pterygium colli and coarctation of the aorta, an in addition, unusual characteristics such as language and mental delay, and autism. The origin of the mar chromosome was determined by FISH using a probe for the centromeric region of the X chromosome (Fig. 1). Also a 70% level of mosaicism was determined by scoring FISH signals on metaphases and interphases. The karyotype analysis by Affymetrix CytoScan™ HD array identified the DNA material of the fragment, as mainly alphoid sequences with breakpoints in the juxtacentromeric region Xp11.1 and Xq13.2 (Table 1). The frag(X) has lost 58.17 Mb and 290 genes in the p arm and 82.24 Mb and 331 genes in q arm, including TSIX (300181) and XIST (314670).
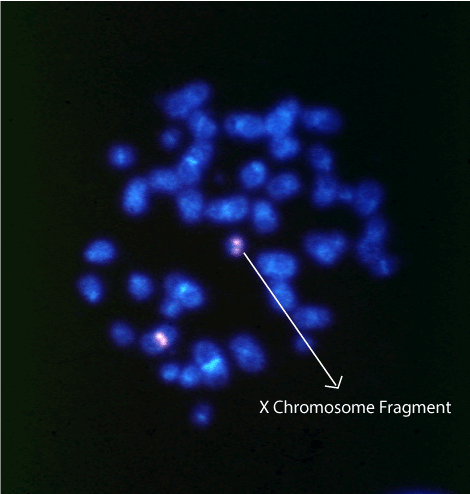
Figure 1. Characterization of X chromosomes by FISH. A metaphase spread hybridized with the X centromere DXZ1 probe showing two X chromosomes, one of which (arrow) is a small fragment derived from the X chromosome.
However, cytogenetic microarray analysis also revealed a 15q13.3 microdeletion of 1.84 Mb, in location chr15:31073735-32915593, including the genes FAN1 (613534), TRPM1 (603576), MIR211 (613753), KLF13 (605328), OTUD7A (612024), CHRNA7 (118511), and ARHGAP11A (610589) (Table 1; Fig 2). The deletion and some characteristics of the phenotype correspond to the 15q13.3 syndrome (OMIM#612001).
Table 1. Molecular analysis of the karyotype by Affymetrix CytoScan™ high-density (HD) arrays
CN State |
Type |
Chromosome |
Cytoband Start |
Size (Kbp) |
OMIN genes count |
OMIN genes |
Call |
Interpretation |
Microarray Nomenclature |
Full location |
1 |
Loss |
14 |
q11.2 |
454,056 |
0 |
- |
Benign |
-- |
arr[hg19] 14q11.2(22,505,306-22,959,362)x1 |
chr14:22505306-22959362 |
3 |
Gain |
14 |
q32.33 |
613,069 |
0 |
- |
Benign |
-- |
arr[hg19] 14q32.33(106,079,822-106,692,891)x3 |
chr14:106079822-106692891 |
1 |
Loss |
15 |
q13.2 |
1,841,858 |
7 |
FAN1 (613534), TRPM1 (603576), MIR211 (613753), KLF13 (605328), OTUD7A (612024), CHRNA7 (118511), ARHGAP11A (610589) |
Pathogenic |
15q13 syndrome |
arr[hg19] 15q13.2q13.3(31,073,735-32,915,593)x1 |
chr15:31073735-32915593 |
1 |
Loss |
X |
p22.33 |
58,170,085 |
290 |
290 genes |
Pathogenic |
Turner syndrome |
arr[hg19] Xp22.33p11.1(168,554-58,338,639)x1 |
chrX:168554-58338639 |
1 |
Loss |
X |
q13.2 |
82,244,168 |
331 |
TSIX (300181), XIST (314670), and others to 331 |
Pathogenic |
Turner syndrome |
arr[hg19] Xq13.2q28(72,989,563-155,233,731)x1 |
chrX:72989563-155233731 |
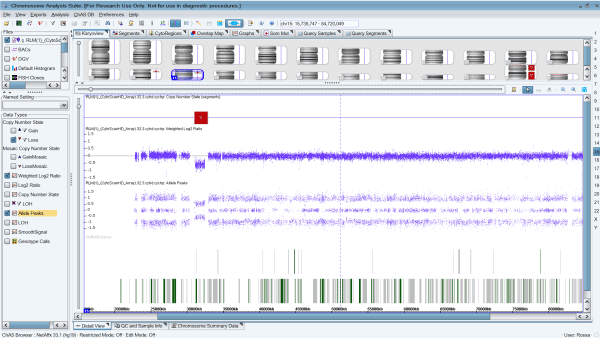
Figure 2. Cytogenetic microarray results representing the 15q13.3 microdeletion of 1.84 Mb arr[hg19] 15q13.2q13.3(31,073,735-32,915,593), including the genes FAN1, TRPM1, MIR211, KLF13, OTUD7A, CHRNA7, and ARHGAP11A, causing the 15q13.3 syndrome in the patient.
In this work, we describe a case of TS with 46,X + mar(?) karyotype without mosaicism defined by G-banding, and a mixed phenotype with frequent and atypical characteristics of TS that suggest another chromosome alteration besides the absence of the second sex chromosome.
Even with the same karyotype, it has been demonstrated that the TS phenotype is very variable [13,14,15]. But the presence of a severe phenotype is generally linked to the presence of small chromosome fragments, generally rings, derived from the X. This atypical severe phenotype depends on origin, size, replication timing of the fragment chromosome, genes affected by copy number variations and level of mosaicism [16], but the failure of the small fragment chromosome inactivation due to deletion of the X-inactive specific transcript (XIST) at Xq13, is what most influences a severe Turner phenotype [16, 17].
Here we describe a TS girl diagnosed at only 8 months due to insufficient intrauterine growth, low birth weight (1.97 kg), Pterygium colli and coarctation of the aorta. The G-banding karyotype analysis confirmed the diagnosis.
At the age of 9 years, the patient was referred for molecular analysis of the karyotype because of dysmorphic features uncharacteristic of TS such as moderate psychomotor retardation, significant language delay and autism. The FISH analysis confirmed the origin of the fragment and the level of mosaicism [45,X (30%)/46,X + frag(X)(70%)]. The material involved in the deletion was confirmed by Affymetrix CytoScan™ HD array. Cytogenetic microarray exactly mapped breakpoints in Xp11.1 and Xq13.2. The fragment appeared to be formed mainly of alphoid sequences and did not include the XIST locus, affirming the possible implication of the absence of the fragment inactivation in the severity of the phenotype.
But to our knowledge, the absence of the second sex chromosome in humans is not associated with an autistic spectrum disorder. In this case, applying the molecular analysis of the karyotype by HD array has given a possible explanation for such an unusual phenotype. In addition to the absence of practically all the second X chromosome, a 15q13.3 microdeletion of 1.84 Mb arr[hg19] 15q13.2q13.3(31,073,735-32,915,593), including the genes FAN1, TRPM1, MIR211, KLF13, OTUD7A, CHRNA7, and ARHGAP11A was revealed, causing the 15q13.3 syndrome [10,18] that could explain phenotype features such as autism and intellectual and language delay. The 15q13.3 microdeletion syndrome (OMIM#612001) is characterized by a wide range of phenotypic features, including intellectual disability, autism, and psychiatric conditions. It has been suggested that the gene responsible for the 15q13.3 microdeletion syndrome is CHRNA7[10,18] and this gene is deleted in our patient.
As confirmed in this report and in our previous investigations [13,14,15], the TS phenotype is very variable and generally well tolerated. But in very specific cases, the severity of the phenotype indicates the presence of other genetic alterations, only detectable at molecular level.
Patients who carry a fragment or a ring derived from the X chromosome are an interesting group who provide the opportunity to evaluate genotype/phenotype correlation in TS. Thus, the application of HD arrays could help us to better define the TS phenotype.