Abstract
Recently, many different molecular datasets from cancers, such as genomic DNA copy number arrays, DNA methylation, exome sequencing, messenger RNA (mRNA), and microRNA (miRNA) have been generated. These datasets have provided many key insights into cancer subtypes, molecular heterogeneity, cancer pathogenesis and cancer progression. Although many datasets and tools have been developed for cancer analysis, there are still strong needs for more efficient computational methods.
Key words
molecular datasets, biological information, patients’ clinical information
In response, many researchers have developed more effective and robust tools that analyze these datasets. Until now, the main focus of the methodological development was to increase the efficiency for analyzing the individual dataset. However, recent methods being developed are capable of integrative analysis of datasets. In this short review, we provide an overview of three commonly used analysis of integration in cancer studies. The first case of commonly used tool is integrating different types of molecular datasets. The second tool is integrating one type of molecular datasets and biological information. The last case is integrating molecular datasets and clinical information.
In the case of integrating different types of molecular datasets, many methods have been published, such as miRNA and mRNA, mRNA and copy numbers, and more than two different molecular datasets. For the second case, the biological information source is the putative target gene of miRNA TargetScan (www.targetscan.org/) [1] or Miranda (www.microrna.org/) [2]. Pathway information is another good source of biological information. For example, integrating pathway information and microarray gene expression datasets is well known to improve the accuracy of the model than only using microarray gene expression datasets.
Lastly, compared to an individual analysis, an integrated analysis of molecular datasets containing clinical information also improves the prediction accuracy for cancer. Survival time is a good source for clinical information for cancer progression or cancer classification.
Integration of molecular datasets
Different types of molecular datasets can be obtained from the same cancer patient. Many studies proved that cancer analysis using integrative datasets performs better than analysis that just uses individual datasets. For examples, Lu et al. [3] and Peng et al. [4] distinguished eleven cancer types using miRNA and mRNA datasets. Lu et al. [3] presented that the classification (or prediction) accuracy based on mRNA is 5.9% while the accuracy of the model based on miRNA was 70.58%. However, Peng et al. [4] demonstrated that the performance of the analysis using only mRNA is better than that of only using miRNA. Even though the dataset was same, applying different tools produced completely opposite results. Regarding this, Kim et al. [5] presented the result of comparing three different types of datasets; miRNA alone, mRNA alone, and integrative datasets of miRNA and mRNA. For the research, two different cancer types, ovarian cancer (OV) and glioblastoma multiforme (GBM) were analyzed (Kim et al. [5]). The accuracies of OV are 84.04%, 75%, 63.64% using integrative miRNA-mRNA profiles, miRNA profiles, and mRNA profile, respectively. The accuracies of GBM are 87.76%, 79.59%, 77.55% using integrative miRNA-mRNA profiles, miRNA profiles, and mRNA profiles, respectively.
Integration of molecular dataset with biological information
Fu et al. [6] proposed an integrative method using miRNA and mRNA expression datasets, and miRNA and its target mRNA information from both TargetScan [1] and miRanda [2]. The method identified the robust target mRNA of miRNAs. Integrating pathway information and microarray gene expression datasets also provided more accurate results than just using mRNA expression profiles.
The classification based on pathway presented comparable or even better performance than the gene-based classification method. For example, Guo et al. [7] suggested a method to infer module activity using mean or median gene expression values in gene ontology [8]. Su et al. [9] presented a method for classification based on the probabilistic inference of pathway activity. In addition, Lee et al. [10,11] proposed a classification method for cancer phenotypes using the core genes in pathways as the differentiators of the disease phenotypes. Kim et al. [12] proposed an integrative method using KEGG pathway [13] information extracted from gene set enrichment analysis (GSEA) [14] and mRNA expression datasets. In the study, features based on core genes from enrichment pathway performed better than the methods using only the pathway features or mRNA itself (Figure 1).
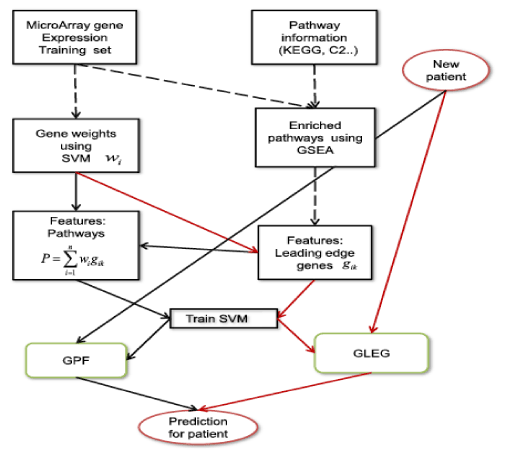
Figure 1. Overall flowchart for integrating pathway information and mRNA expression datasets. GLEG; genes of leading edge gene, GPF; pathway features, SVM; support vector machine [12]
Integration of molecular dataset with clinical information
Finally, integrating datasets is including not only the molecular datasets obtained from the laboratory, but also the patients’ clinical information. For example, intermediate survival times between long and short survival times can also be a good source for the data analysis.
Kim et al. [5] suggested a method for extensive datasets using intermediate survival times, integrative miRNA, and mRNA datasets with putative target gene information for classifying cancer survival times.
Figure 2 shows an overall flowchart for integrating datasets that include miRNA, mRNA, and intermediate survival times [15]. The results of using integrative datasets are more accurate than just using single profiles.
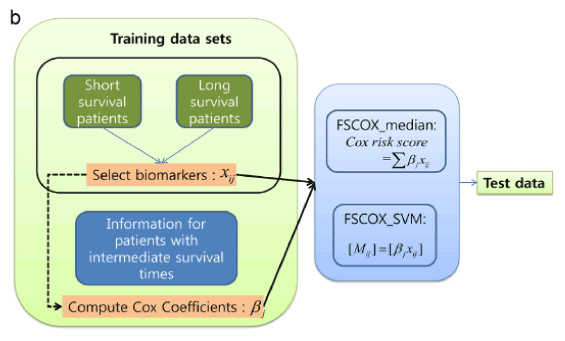
Figure 2. Overall flowchart for integrating datasets including miRNA/mRNA expression datasets and intermediate survival times [15]
Conclusions
There are many ways to integrate molecular datasets and extend the types of datasets. Here we shortly reviewed three ways of integrating the different types of data. Compared to using only individual data sets, integrating datasets that use molecular datasets with biological information or patients clinical information improves the accuracies of cancer analysis. Moreover, the analysis of integrating datasets could lead on to systemic insights of cancer progression and/or pathogenesis in the future.
Acknowledgement
This work was supported by the Bio-Synergy Research Project (2013M3A9C4078158) of the Ministry of Science, ICT and Future Planning through the National Research Foundation (NRF) Korea to TP and by Basic Science Research Program through the NRF funded by the Ministry of Education (NRF-2015R1D1A1A01060287) Korea to SK.
References
- Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15-20. [Crossref]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, et al. (2004) Human MicroRNA targets. PLoS Biol 2: e363. [Crossref]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, et al. (2005) MicroRNA expression profiles classify human cancers. Nature 435: 834-838. [Crossref]
- Peng S, Zeng X, Li X, Peng X, Chen L (2009) Multi-class cancer classification through gene expression profiles: microRNA versus mRNA. J Genet Genomics 36: 409-416. [Crossref]
- Kim S, Park T, Kon M (2014) Cancer survival classification using integrated data sets and intermediate information. Artif Intell Med 62: 23-31. [Crossref]
- Fu J, Tang W, Du P, Wang G, Chen W, et al. (2012) Identifying microRNA-mRNA regulatory network in colorectal cancer by a combination of expression profile and bioinformatics analysis. BMC Syst Biol 6: 68. [Crossref]
- Guo Z, Zhang T, Li X, Wang Q, Xu J, et al. (2005) Towards precise classification of cancers based on robust gene functional expression profiles. BMC Bioinformatics 6: 58. [Crossref]
- Ashburner M, Ball C2021 Copyright OAT. All rights reserv al. (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25: 25. [Crossref]
- Su J, Yoon BJ, Dougherty ER (2009) Accurate and reliable cancer classification based on probabilistic inference of pathway activity. PLoS One 4: e8161. [Crossref]
- Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, et al. (2010) Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 26: i237-245. [Crossref]
- Lee E, Chuang HY, Kim JW, Ideker T, Lee D (2008) Inferring pathway activity toward precise disease classification. PLoS Comput Biol 4: e1000217. [Crossref]
- Kim S, Kon M, DeLisi C (2012) Pathway-based classification of cancer subtypes. Biol Direct 7: 21. [Crossref]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, et al. (1999) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27: 29-34. [Crossref]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545-15550. [Crossref]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, et al. (2004) Human MicroRNA targets. PLoS Biol 2: e363. [Crossref]