Abstract
Majority of the HCV infections are caused by HCV genotype 1 having high resistance to anti-HCV therapy among the Western population. Standard interferon (IFN) along with pegylated-interferon (pegIFN) α 2a or 2b combination with or without ribavirin is currently the approved therapy for HCV infection. Several factors are related with antiviral reaction and stratified according to factors related with virus, host, and on-treatment. The objectives of this review included exploration of phenomenon of interaction between HCV and IFN-α signalling pathway in patients receiving treatment with RBV and IFN-α as a combination therapy for chronic HCV infection (CHC). Another important objective of this review was to demonstrate mutations associated with ISDR region to develop understanding of the controversies and the correlations in resistance through chronic HCV-G1; in comparison to Japanese and other studies. Changes in SNPs occurring within IL28B were also evaluated along with the examination of relation between drugs and mutation. A literature review was performed commencing after literature search. Pertinent literature was searched through electronic databases PubMed, Science Direct, ProQuest, and MEDLINE during the period 2010 to 2014. Manual searching of the grey literature and various websites was conducted to obtain epidemiological data of HCV. The standard treatment for HCV infection is RBV and pegIFN combination therapy. However, SVR is induced in 50% of the HCV genotype 1 infection patients. The results of IL28B genotyping studies revealed that there appear to be differences between the distributions of the polymorphisms in different populations infected with HCV. NS5A mutations to be connected with decreased reaction to IFN treatment among HCV patients with genotype 1. PegIFN can be administered along with RBV regardless of the genotype conferring polymorphism and mutations for the production of interferon. Conclusion: Standard treatment for HCV infection is combination therapy of RBV and pegIFN- α2a or -α2b. Mutations in the IL28B have to be considered prior to the administration of RBV and pegIFN therapy for treating HCV-G1 infection in patients. Clinical trials should evaluate the efficacy of RBV and pegIFN combination therapy for HCV- G1 infection.
Key words
Hepatitis C virus (HCV), interferon sensitivity determining region (ISDR), sustained virological response (SVR), Interferons (ifns)
Introduction
A major cause of hepatitis C is hepatitis C virus (HCV) which is globally the leading source of chronic liver disease. Diverse epidemiologic profiles and responses to antiviral treatment have discovered that there is a difference of 10-30% in the genome sequence of the six genotypes. HCV poses the risk of mortality and morbidity to approximately 170 million individuals throughout the world [1]. Wiessing et al. [2] contends that majority of the HCV infections are caused by HCV genotype 1 among the Western population which has high resistance to anti-HCV therapy. HCV infection is a major public health concern because there is a risk of long-term sequelae, including hepatocellular carcinoma and cirrhosis of liver. HCV is the chief reason of primary liver carcinoma and cirrhosis in the Europe. Conversely, Ditah et al. [3] presented the current HCV prevalence in the USA providing verification pertaining to a decreasing trend. A decline from 1.9% during 2001-2002 to 1.3% in 2005-2006 was reported with sustainability till 2010. Chronic HCV infection was observed among 2.3 million individuals in the US.
Kanda et al. assert that the availability of pegylated interferon since 2001 has enabled effective HCV infection treatment [1]. Standard interferon (IFN) along with the combination of pegylated-interferon (pegIFN) α 2a or 2b with or without ribavirin is currently the approved therapy for HCV infection. According to Chak et al. ameliorations in the natural history and patient outcomes is presented through treatment with interferon- based therapy. However, several factors are related with antiviral response, and stratified according to host, viral, and on-treatment factors. Achievement of sustained virologic response (SVR) can be predicted by the genetic polymorphisms in the interleukin (IL) 28b gene. Lowered rates of SVR to combination therapy for HCV have been presented among the African American population in comparison to the Caucasians contributing to the increased prevalence of HCV genotype 1.
The objective of antiviral therapy for HCV is enduring eradication of the virus following the completion of treatment. Mortality and morbidity associated with liver disease caused by HCV can be decreased as a consequence of eradication of HCV with antiretroviral therapy. There are evidences regarding the long-term clearance of HCV resulting in ameliorations in liver necrosis, inflammation and fibrosis, and decline in the incidence of liver failure compared to patients without SVR [5]. Thus, the basic goal of treatment is to achieve SVR which is defined as undetectable HCV RNA at week 24 after treatment completion. The primary endpoint for the evaluation of efficacy of treatment with various anti-HCV therapies n clinical trials is SVR. Discontinuation with treatment is considered in patients with detectable HCV RNA at 24th week of treatment [6].
First therapy accepted for the HCV treatment was interferon alpha which was permitted in 1991 by the US FDA. The initially approved dose was 3 MU 3 times per week with treatment extended over a period of 6 months. In 1998, a dramatic improvement in the efficacy of anti-HCV therapy by addition of RBV to IFN therapy was attained. The degree of success of HCV treatment is proportionate to the adherence. The gold standard for the treatment of HCV infection is 80% pegylated interferon, 80% ribavirin, and 80% duration of treatment [7]. Several factors are associated with the effectiveness of treatment for instance socio-demographic factors. Ghany et al. predicted the incidence of HCV in people aged less than 40 years, not being African American, and weighing less than 75 kilograms as one the important factor contributing to efficacious treatment of HCV [8]. Tolerance among patients for two adequate doses of the two available pegIFN was also considered as an important factor for successful treatment with pegIFN along with an appropriate dose of ribavirin. However, the most important predictors of success were genotypes of HCV and the pretreatment viral load.
Primary HCV infection leads to persistent viremia in 80-85% of the cases, with chronicity developed in approximately 60% of the cases. Meanwhile, 20% of these reach the End stage liver disease, cirrhosis and /or hepatocellular carcinoma (HCC) is caused in 20% of these cases varying according to the genotypes and subtypes of virus. The Non-responsiveness to IFNs is fairly common with varying rates in HCV infection. Response to IFN-α is observed in nearly 40-50% of patients with HCV infection, with improvement in rates by the addition of Ribavirin [9].
Most of the studies related to IFN and HCV demonstrate continual antagonism between the virus and antiviral drugs associated primarily with the manner in which patient responds to mono or combination therapy. Various factors such as patient immunity, sex, age, personal habits, ancestry, and geographical region are the areas in which considerable role is played by the drug doses. These factors appear separate entities but they are closely linked to each other. Interferon (IFN), ribavirin (RBV), and Viraferon (VER) or any other derivatives of recombinant interferon drugs or even the newly approved protease inhibitors (PI) usually concede as an antiviral therapy in the treatment of HCV [10].
The objectives of this review included exploration of phenomenon of interaction between HCV and IFN-α signalling pathway in patients receiving treatment with RBV and IFN-α as a combination therapy for chronic HCV infection (CHC). Another important objective of this review was to demonstrate mutations associated with ISDR region to develop understanding of the controversies and the correlations in resistance through chronic HCV-G1; comparing with the Japanese studies, as well as other studies. Changes in SNPs occurring within IL28B were also evaluated along with the examination of relation between drugs and mutation.
Method
Studies for this review were retrieved by searching relevant literature stipulating a variety of key words such as ‘Hepatitis C virus’, ‘interferon sensitivity determining region’, ‘sustained virologic response’, and ‘Interferons’. Search for the pertinent literature was performed through electronic databases PubMed, Science Direct, ProQuest, and MEDLINE during the period 2010 to 2014 to access the current literature. Manual searching of the grey literature and various websites was conducted to obtain epidemiological data of HCV. The inclusion and exclusion criteria of the research assured that only those studies are included in the review that have been published between 2010 and 2014. Studies published in other languages were excluded from the review. Studies were also excluded if their full-text was not available.
Literature review
Identification of interferon occurred over 50 years ago by Lindenmann and Isaacs in the year 1957 while studying the viral interference phenomenon. This capability of virus is based on the capacity of activated or inactivated virus to obstruct the development of disparate virus. At present, approximately 10 IFN species belonging to mammals and several subspecies have been discovered, with each having their own distinctive properties but possessing antiviral activity. Currently, classification of these species has been done in three groups namely: IFNs type I, II and III. IFNβ, IFNκ, IFNαs, IFNω, IFNε, and IFNν are included in the type I IFNs. There are 12 different IFNαs in humans and a single IFNβ. Viral response is generated by cells to a variety of viruses through the production of IFNαs, IFNβ and IFNλs [11]. Host-derived components produce the structure of viruses distinct from fungi and bacteria which poses microbe-specific constructs discernible from structures of host cells. Cellular receptors for detecting viruses have evolved due to the lack of virus specific lipids and proteins. Hence, they are able to identify the existence of viral genome constituted by nucleic acids. Discovery and characterisation of two significant pathways detecting viral genomes and inducing IFNs type I and III has occurred in the recent years. These pathways are the cytosolic pathway and the toll-like receptor dependent pathway activated by viral RNA binding with the melanoma differentiation antigen 5 (MDA5) and RNA helicases retinoic acid inducible gene-I (RIG-I) [12].
IFN-α is a multi-gene family consisting over 20 types synthesised by leukocytes; while, synthesis of IFN-β occurs in majority cell types but particularly in fibroblasts. Reaction to cytokines, IL-18 and IL-12 along with the stimulation of NK cell or T-cell antigen receptors by activated T lymphocytes and NK cells, causes the synthesis of type II IFNs consisting INF-gamma (IFN-γ) [13]. A cytokine class with activity similar to IFN constitutes the third type called IFN-λ or interleukin 28s/29. Alpha (α) and Beta (β) IFNs bind to cell surface receptors comprising two major sub-units, IFNαR1 and IFNαR2. On the contrary, gamma interferon binds to IFNγR1 and IFNγR2 acting through two distinct but related pathways [14]. The binding of IFN-α to its receptor initiates the Janus Tyrosine kinase JAK–signal transducer and activator of the transcription STAT pathway (Figure 1). In humans, four JAKs (JAK 1-3 and Tyk2) and seven STATs (STAT 1-7) have been identified so far. The ligand-receptor binding causes a conformational change in the cytoplasmic part of the receptor, which in turn activates the receptor associated kinases Tyk 2 and JAK1. Tyk2 phosphorylates the tyrosine at amino acid 466 on the IFNαR1 to create a docking site for STAT2. STAT2 phosphorylates Tyk at tyrosine 690 serving as a platform for the recruitment of the STAT1. STAT2 becomes phosphorylated at tyrosine 701 [15].
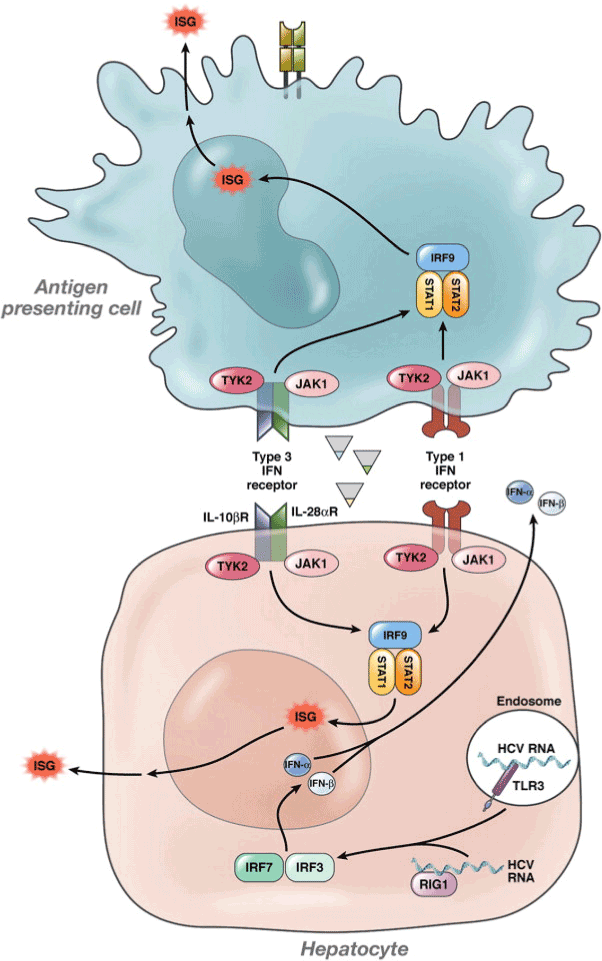
Figure 1. IFN-λ, IFN-α, and IFN-β signalling in response to HCV
Human IFNs compete for binding with receptors on cell composed of major sub-units for interferon activator receptors (IFNARs) to start the signaling. Member of the protein family carries the signal serving as transcriptional activators, known as STAT for Jak and STAT pathway. Through the SH2 domain, Tyk2 then phosphorylates the tyrosine (Try) at specific position to create a docking site for the STAT. [13,20]
Antiviral effects
Recognition of virus is mediated by a group of receptors located either at the surface of endosomes or in the cytoplasm. These mediators lead to the production of interferons and enable the cell to detect viral invasion.MDA-5, RIG-1, and TLRs 3, 7, 8, and 9 are the receptors. Inhibition of these cell intrinsic viral sensors is performed specifically by the evolved viral proteins [16]. An antiviral state is generated in the cell through regulation of several genes induced by type I IFN. Antiviral state confers protection to the cell against virus. Functions of numerous genes involved in this process have not yet been recognised. Translation of the viral genome is prohibited by the binding of IFIT1 and IFIT2 (the tetratricopeptide repeats) and members of the interferon-induced protein with eIF3. Another important antiviral effector is 2’-5’ oligoadenylate synthetase (OAS) which involves the gene transcription and the regulation of activity of enzyme [17].
Evading the immune response of host, both adaptive and innate, is necessary for establishing a persistent infection by HCV. TLR-dependent pathways and the cytosolic pathways prompted by the binding of RNA helicases MDA5 and RIG-I with viral RNA enables the sensing of viruses. Inactivation of the components of both these pathways can be caused by HCV through NS3-4A (a non-structural protein) via its proteolytic activity [18]. In cell cultures and in vitro, NS3-4A has been proven competent in slicing MAVS and TRIF. Nevertheless, there is absence of inhibition of the IFN-inducing pathways in vivo. Significant initiation of hundreds of ISGs has been observed in the transcriptome analysis of liver homogenates of chimpanzees suffering from CHC [19].
The IFN-λ and IFN-α/β gene expression can be induced following infection of hepatocytes with HCV. It results in phosphorylation of STAT 1 and 2, causing the formation of heterodimers of STAT1 and 2. Binding of these dimers to IRF9 structure the complex of ISGF3, whereupon they migrate to the nucleus and binds to the elements of ISRE for facilitating ISGs transcription. According to Balagopal et al. IFN-λ binding receptors can create the complex required to stimulate the TYK2 and JAK1 consisting an intracellular domain of 270 amino acids [20]. Cross-phosphorylation of these two kinases of the IFN-λ activates both the kinases facilitating the phosphorylation of Tyr343, Tyr406 and Tyr517 which are three tyrosine residues located at IFNλ- R1’s intracellular region. A docking site is created by the Tyr343 and Tyr517 for the Src Homology 2 (SH2) domain of STAT2 transcription factor. Binding of IFN-λR1 to STAT activates JAK1 and TYK2, allowing the tyrosine residue to be delivered towards the STAT proteins at the C-terminal end. A quay site is supplied for the SH2 domains. Formation of ISGF3 complex occurs by the activation of STATs 1 and 2 enabling the fusion of IRF9. The chief entrance for IFN-λ stimulation is considered to be the ISGF3 complex which consecutively causes the activation of STAT 3 and STAT 5 [21].
Interleukin-28B (IL28B)
Another SNP referred to as rs12979860 is linked to the course and prognosis of chronic HCV near the IL28B gene. Interferon lambda is encoded by this SNP which is among the type III interferons and is induced by binding of antigens to the toll-like receptors. Activation of various cellular pathways including STAT/JAK occurs by the production of type III interferons. In vitro replication of HCV is inhibited by the type III interferon suggesting their non-specificity for particular viruses. Correlation between the presence of HCV RNA in cells and expression of ISG mRNA in the specimen of liver tissue suggested that ISG expression is induced by HCV [22]. Replication of a variety of different tumour cells is also inhibited by type III interferons [23].
Kurosaki et al. affirmed that combination therapy of RBV and pegIFN for HCV infection has been effective in 50% of the cases [24]. An association between treatment response and IL28B genotype has been observed and polymorphism of IL28B correlates with early virological reaction and predicts void and sustained virological reaction. Relapse and independency of SVR from IL28B has been predicted by mutations in the ISDR. Highest null virologic response and lowest SVR was observed by these investigators in patients with low platelet counts and minor IL28B allele. Contrarily, lowest NVR and highest SVR were observed in patients with ISDR mutations, major IL28B allele, or elevated counts of platelet. Thus, mutations in the ISDR and polymorphisms in IL28B of HCV predict pre-treatment response to RBV and pegIFN. Similarly, De Nicola et al. posits that the major baseline predictors of SVR to RBV and pegIFN in patients having HCV infection caused by genotype 1 are the single nucleotide polymorphisms (SNPs) located nearby the region of IL28B [25]. Analysis of patients with HCV genotype 1 infection revealed that SVR can be achieved in 50% of these cases.
Several genome-wide association studies (GWAS) have recently demonstrated that SNPs polymorphisms near the IL28B gene have a strong association with the response of HCV patient to RBV and pegIFN-α therapy [26,27]. Spontaneous viral elimination has also been demonstrated diverse populations. Thus, host factors can be presented by IL28B variants determining susceptibility to HCV treatment providing a genetic explanation for various genetic outcomes of HCV infection in a variety of hosts. Hayes et al. genotyped two SNPs located in the IL28B locus from 817 patients suffering from chronic HCV [28]. Analysis of the substitutions performed within the NS5A and at amino acids 70 and 91 of the HCV csore protein exposed substitutions at the core amino acid 70, viral load at baseline, age, and IL28B rs12979860 CC genotype as the independent predictor of SVR. Age and IL28B rs12979860 CC genotype were the independent predictors of non-virologic response. Pineda et al. also inferred that variations in IL28B are related with SVR to pegIFN and RBV in mono-infected patients of HCV genotype 1 [29]. The effect of this genotype can be attributed by the low-density lipoprotein receptor based on the evidence provided by the relation between plasma low-density lipoprotein cholesterol and rs12979860.
According to Lyoo et al. Korean patients frequently presented the genotypes rs8099917 TT and rs12979860 CC in comparison to other ethnicities [30]. Genetic characteristics of patients can be prognostic factors predicting antiviral response to peg-IFN therapy. Liu et al. points out significant improvement in the efficacy of IFN-α by the combination of RBV for use against HCV [31]. Suppression of replication of HCV in the hepatocytes has been suggested to be the underlying mechanism involved in this phenomenon. Cell cycle is arrested by RBV at G1 phase by the employment of HCV and HepG2 replicons. Stabilisation and activation of p53 related with the activity of RBV is also caused by this process. Increased activation of p53 and elevated HCV suppression results from combination therapy of IFN- α and RBV as compared to IFN- α therapy alone. There is a link between p53 activation and RBV signals through ERK1/2. Inhibitory effects of RBV on HCV replication are moderately alleviated by p53 and ERK1/2 knockdown. It suggests that ERK1/2 is involved in the anti-HCV effects of RBV.
Donnelly et al. determined the efficiency of IFN-λ as therapeutic agent as an alternative to IFN- α. IFN- λ is a type III interferon possessing the capacity to induce antiviral response in hepatocytes [32]. Less adverse reactions were induced by IFN- λ because its receptors are mainly restricted to cells originated from the epithelial region. IL28B was identified by these researchers as the SNPs related with enhanced SVR achievement by combination therapy of RBV and pegIFN- α in HCV infected patients. Coppola et al conducted a meta-analysis to evaluate the efficiency of RBV and pegIFN a-2a or a-2b administered as dual therapy in comparison to triple therapy [33]. Based on the analysis, triple therapy was reported to have better outcomes for HCV patients with the provision of significantly higher rate of SVR as compared to dual therapy. However, dual therapy was observed to have increased rate of SVR in RVR patients. The outcomes were obtained irrespective of the genotype IL28B and HCV sub-genotype. Thus, pegIFN can be used in conjuction with RBV regardless of the genotype conferring polymorphism and mutations for the production of interferon.
Kanda et al. analysed the safety and efficacy of combination treatment with pegIFN alfa-2a and RBV in Japanese patients with type 2 genotype HCV infection [34]. Sustained SVR was observed in 60-66.6% of the Japanese patients who have previously failed response to treatment with monotherapy of pegIFN and 69.9% in patients receiving RBV and pegIFN combination therapy. Similarly, Oze et al. conducted a study with Japanese patients to observe response to re-treatment after failure of response generation following the initial treatment with RBV and pegIFN [35]. Significant rate of SVR was reported among 56% patients infected with type 2 genotype as compared to 41% SVR rate among those with genotype 1 infection. Decreased levels of HCV RNA in serum were associated with increased SVR rate at re-treatment.
Considerable results were obtained regarding combination therapy of pegIFN, RBV and boceprevir by Sulkowski et al. during assessment of HCV patients with genotype 1 and HIV co-infection [36]. Double-blind RCT reported SVR at 24th week of follow up among 63% patients in the group receiving triple combination therapy as compared to 29% in the control group. Thus, boceprevir can be used in combination with pegIFN and RBV for treatment of HCV genotype 1 and HIV co-infection. Feuerstadt et al. posit that HCV patients belonging to minority groups when treated with pegIFN and RBV combination therapy demonstrated SVR among 14% of the genotype 1 patients [37]. The decreased level of SVR rate in the minority population of the US suggested the need for novel treatment regimens due to lack of responsiveness among these patients for the tested regimen. Kumthip et al. demonstrated NS5A mutations to be connected with decreased reaction to IFN treatment among HCV genotype 1 patients in Thailand. Mutations in amino acids in the centre and NS5A proteins of HCV 1b, 3b, a, 3a, and 6f were observed to be correlated with response to pegIFN and RBV therapy. Mutations in C-terminus, 3rd variable region and flanking region of HCV-1b NS5A protein, and IFN sensitivity determining region were significant in the responder group of the treatment in comparison to the failure group. A correlation was observed between mutations in the amino acid residues of NS5A of HCV-1b, 1a, and 6f, and reaction to combination therapy of RBV and pegIFN.
Discussion
Several studies demonstrate that HCV infection is not only serious rather it is life threatening. A limited range of treatment options were available until recently. The pathogenicity of HCV is conferred primarily by infecting hepatocytes; therefore, HCV has globally become a leading source of liver disease [38]. Asselah et al. suggested that there has been a considerable evolution in HCV infection over the past few years. The standard treatment therapy for HCV infection is combination of RBV and pegIFN [39]. However, SVR is induced in only half of the patients with HCV genotype 1 infection. Non-response has been related with several factors including insulin resistance, obesity, genotype, male gender, age, steatosis, and ethnicity. Relatively less information is available in the literature pertaining to genotype 3 and 4 infections. There is even lesser information about the molecular level factors of the virus that could explain the differences observed in treatment response [40].
New developments in HCV treatment have emerged as a result of limitations in current treatment for viral hepatitis C. Two new drugs boceprevir and telaprevir that target the NS3 protease have now been licensed against HCV [41-44]. Furthermore, clinical trials of other compounds directed against the NS5B RNA polymerase are also being conducted. Fernández-Montero et al. introduced Telaprevir (NS3/4A serine-protease inhibitors) and Filibuvir (a non-nucleoside NS5B RNA polymerase inhibitor) in their study [45]. These two compounds are awaiting clinical evaluation. It is anticipated that these new drugs will be utilised along with the present standard therapy of pegIFN and RBV. In a has also shown High antiviral activity was observed in another trial of monotherapy with protease inhibitors but the frequent selection of resistant HCV variants created difficulty [45]. The deployment of these new drugs and other specific antiviral inhibitors suggests that the use of PEG-IFN-α2b + RBV might be superseded in the forthcoming years, thus minimising the interest in ISDR from a prognostic perspective.
Systematic GWAS were reported in 2009 in the HCV infection and antiviral control context. Analysis of >500,000 single nucleotide polymorphisms (SNPs) identified two polymorphisms in the IL28B gene (rs12979860C/T and rs8099917G/T) associated with a poor response to IFNα + RBV therapy and in the control of acute HCV infection [26,27,46]. Subsequently a number of studies have confirmed and extended these findings [47,48]. However, there is insufficiency of information about the distribution of these polymorphisms in Saudi Arabian populations and their effects on response to HCV therapy with IFN-based regimens. The results of IL28B genotyping studies revealed that there appear to be differences between the distributions of the polymorphisms in HCV infected Saudi patients compared to other published studies in Northern European and US populations. This observation could reflect the relatively stable genetic homogeneity of the Saudi population which is a predominantly tribal based system. However, it was also interesting that the distribution of IL28B genotypes differed between patients with HCV mono-infection and those with co-infection with HBV but not with HIV [49].
Conclusion and future recommendations
Currently, the care standard of treatment for HCV infection is combination therapy of RBV and pegIFN- α2a or -α2b and has been reported to have less adverse reactions. The functionality of IFN- α is in resemblance with that of the IFN- λ, particularly in terms of the anti-viral activity. However, signalling of IFN- λ through a unique receptor complex result in diminished hematopoietic toxicity and lesser side effects in comparison to IFN- α. Responsiveness of interferon remains a major clinical issue related to the eradication of HCV. Some activities are coded by the HCV proteins that limit and interfere with the signalling pathway of IFN. Therefore, use of novel antiviral drugs for instance, polymerase and protease inhibitors enabling the restoration of IFN signalling pathway and detecting non-responsiveness pertaining to several factors is expected to facilitate successful treatment of HCV. Mutations in the IL28B have to be considered prior to the administration of RBV and pegIFN therapy for treating HCV genotype 1 infection in patients. Studies based on RCTs are recommended to elucidate the existence of factors interfering with HCV-1 response to interferons. Clinical trials should evaluate the efficacy of pegIFN and RBV combination therapy for HCV genotype 1 infection and compare its effectiveness with that of other combination therapies of polymerase and protease inhibitors.
References
- Kanda T, Imazeki F, Yokosuka O (2010) New antiviral therapies for chronic hepatitis C. Hepatol Int 4: 548-561. [Crossref]
- Wiessing L, Ferri M, Grady B, Kantzanou M, Sperle I, et al. (2014) Hepatitis C virus infection epidemiology among people who inject drugs in Europe: A systematic review of data for scaling up treatment and prevention. PloS one 9: e103345. [Crossref]
- Ditah I, Ditah F, Devaki P, Ewelukwa O, Ditah C, et al. (2014) The changing epidemiology of hepatitis C virus infection in the United States: National health and nutrition examination survey 2001 through 2010. J Hepatol 60: 691-698. [Crossref]
- Chak E, Saab S (2010) Pegylated Interferon and Ribavirin Dosing Strategies to Enhance Sustained Virologic Response. Curr Hepat Rep 9: 147-154. [Crossref]
- Morisco F, Granata R, Stroffolini T, Guarino M, Donnarumma L, et al. (2013) Sustained virological response: a milestone in the treatment of chronic hepatitis C. World J Gastroenterol 19: 2793-2798.[Crossref]
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB (2011) An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatol 54: 1433-1444. [Crossref]
- Weiss JJ, Brau N, Stivala A, Swan T, Fishbein D (2009) Review article: Adherence to medication for chronic hepatitis C - building on the model of human immunodeficiency virus antiretroviral adherence research. Aliment Pharmacol Ther 30: 14-27 [Crossref]
- Ghany MG, Strader DB, Thomas DL, Seeff LB, American Association for the Study of Liver Diseases (2009) Diagnosis, management, and treatment of hepatitis C: an update. Hepatol 49: 1335-1374. [Crossref]
- Abbas Z, Tayyab GN, Qureshi M, Memon MS, Subhan A, et al. (2013) Consensus Interferon Plus Ribavirin for Hepatitis C Genotype 3 Patients Previously Treated With Pegylated Interferon Plus Ribavirin. Hepatitis Monthly 13: e14146. [Crossref]
- Foster GR, Serfaty LD (2012) Triple combination treatment for chronic hepatitis C with Protease Inhibitors, pegylated interferon and ribavirin:‘Lead-in or no lead-in’?. Liver Int 32: 61-63. [Crossref]
- George PM, Badiger R, Alazawi W, Foster GR, Mitchell JA (2012) Pharmacology and therapeutic potential of interferons. Pharmacol Ther 135: 44-53. [Crossref]
- Peng Y, Xu R, Zheng X (2014) HSCARG negatively regulates the cellular antiviral RIG-I like receptor signaling pathway by inhibiting TRAF3 ubiquitination via recruiting OTUB1. PLoS Pathog 10: e1004041.
- Lemon SM (2010) Induction and evasion of innate antiviral responses by hepatitis C virus. J Biol Chem 285: 22741-22747. [Crossref]
- Khanizadeh S, Ravanshad M, Mohebbi SR, Naghoosi H, Abrahim Tahaei M, et al. (2012) Polymorphisms within the Promoter Region of the Gamma Interferon (IFN-γ) Receptor1 Gene are Associated with the Susceptibility to Chronic HBV Infection in an Iranian Population. Hepat Mon 12: e7283. [Crossref]
- Konishi H, Okamoto K, Ohmori Y, Yoshino H, Ohmori H, et al. (2012) An orally available, small-molecule interferon inhibits viral replication. Sci Rep 2: 259. [Crossref]
- Devasthanam AS (2014) Mechanisms underlying the inhibition of interferon signaling by viruses. Virulence 5: 270-277. [Crossref]
- Ezelle HJ, Hassel BA (2012) Pathologic effects of RNase-L dysregulation in immunity and proliferative control. Front Biosci (Schol Ed) 4: 767-786. [Crossref]
- Doehle BP, Gale Jr M (2013) Innate Immune Evasion Strategies of HCV and HIV. Nucleic Acid Sensors and Antiviral Immunity 148.
- Dill MT, Makowska Z, Trincucci G, Gruber AJ, Vogt JE, et al. (2014) Pegylated IFN- α regulates hepatic gene expression through transient Jak/STAT activation. J Clin Invest 124: 1568-1581. [Crossref]
- Balagopal A, Thomas2021 Copyright OAT. All rights reservtrol of hepatitis C virus infection. Gastroenterology 139: 1865-1876. [Crossref]
- Kelly C, Klenerman P, Barnes E (2011) Interferon lambdas: the next cytokine storm. Gut 60: 1284-1293. [Crossref]
- Wieland S, Makowska Z, Campana B, Calabrese D, Dill MT, (2014) Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 59: 2121-2130. [Crossref]
- Abushahba W, Balan M, Castaneda I, Yuan Y, Reuhl K, et al. (2010) Antitumor activity of type I and type III interferons in BNL hepatoma model. Cancer Immunology, Immunotherapy 59: 1059-1071. [Crossref]
- Kurosaki M, Tanaka Y, Nishida N, Sakamoto N, Enomoto N, et al. (2011) Pre-treatment prediction of response to pegylated-interferon plus ribavirin for chronic hepatitis C using genetic polymorphism in IL28B and viral factors. J hepatol 54: 439-448. [Crossref]
- De Nicola S, Aghemo A, Grazia Rumi M, Galmozzi E, Valenti L, et al. (2012) Interleukin 28B polymorphism predicts pegylated interferon plus ribavirin treatment outcome in chronic hepatitis C genotype 4. Hepatology 55: 336-342. [Crossref]
- Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, et al. (2009) Genome-wide association of IL28B with response to pegylated interferon-a and ribavirin therapy for chronic hepatitis C. Nature Genetics 41: 1105-1109. [Crossref]
- Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, et al. (2009) Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461: 399-401. [Crossref]
- Hayes CN, Kobayashi M, Akuta N, Suzuki F, Kumada H (2011) HCV substitutions and IL28B polymorphisms on outcome of peg-interferon plus ribavirin combination therapy. Gut 60: 261-267. [Crossref]
- Pineda JA, Caruz A, Rivero A, Neukam K, Salas I, et al. (2010) Prediction of response to pegylated interferon plus ribavirin by IL28B gene variation in patients coinfected with HIV and hepatitis C virus. Clin Infect Dis 51: 788-795. [Crossref]
- Lyoo K, Song MJ, Hur W, Choi JE, Hong SW, et al. (2011) Polymorphism near the IL28B gene in Korean hepatitis C virus-infected patients treated with peg-interferon plus ribavirin. J Clin Virol 52(4): 363-366. [Crossref]
- Liu WL, Yang HC, Su WC, Wang CC, Chen HL, et al. (2012) Ribavirin enhances the action of interferon-α against hepatitis C virus by promoting the p53 activity through the ERK1/2 pathway. PLoS One 7: e43824. [Crossref]
- Donnelly RP, Dickensheets H, O'Brien TR (2011) Interferon-lambda and therapy for chronic hepatitis C virus infection. Trends Immunol 32: 443-450. [Crossref]
- Coppola N, Pisaturo M, Sagnelli C, Sagnelli E1, Angelillo IF3 (2014) Peg-interferon plus ribavirin with or without boceprevir or telaprevir for HCV genotype 1: a meta-analysis on the role of response predictors. PLoS One 9: e94542. [Crossref]
- Kanda T, Nakamoto S, Nishino T, Takada N, Tsubota A, et al. (2013) Peginterferon Alfa-2a plus ribavirin in Japanese patients infected with hepatitis C virus genotype 2 who failed previous interferon therapy. Int J Med Sci 10(1): 43. [Crossref]
- Oze T, Hiramatsu N,Yakushijin T, Mochizuki K, Oshita M, et al. (2011) Efficacy of re-treatment with pegylated interferon plus ribavirin combination therapy for patients with chronic hepatitis C in Japan. J gastroenterol 46(8): 1031-1037. [Crossref]
- Sulkowski M, Pol S, Mallolas J, Fainboim H, Cooper C, et al. (2013) Boceprevir versus placebo with pegylated interferon alfa-2b and ribavirin for treatment sof hepatitis C virus genotype 1 in patients with HIV: a randomised, double-blind, controlled phase 2 trial. Lancet Infect Dis 13(7): 597-605. [Crossref]
- Feuerstadt P, Bunim AL, Garcia H, Karlitz JJ, Massoumi H, et al. (2010) Effectiveness of hepatitis C treatment with pegylated interferon and ribavirin in urban minority patients. Hepatology 51: 1137-1143. [Crossref]
- Candice Marie Jackel-Cram (2009) Molecular Mechanisms Of Hepatitis C Virus Associated Steatosis. Phd Thesis, Department Of Veterinary Microbiology, University Of Saskatchewan, Saskatoon.
- Asselah T, Estrabaud E, Bieche I, Lapalus M, De Muynck S, et al. (2010) Hepatitis C: viral and host factors associated with non-response to pegylated interferon plus ribavirin. Liver Int 30: 1259-1269. [Crossref]
- Uhde-Holzem K, Schlösser V, Viazov S, Fischer R, Commandeur U (2010) Immunogenic properties of chimeric potato virus X particles displaying the hepatitis C virus hypervariable region I peptide R9. J virol methods 166(1):12-20. [Crossref]
- Poordad F, McCone J Jr, Bacon BR, Bruno S, Manns MP, et al. (2011) Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 364: 1195-1206. [Crossref]
- Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, et al. (2011) Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 364: 1207-1217. [Crossref]
- Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, et al. (2011) Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 364: 2405-2416. [Crossref]
- Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, et al. (2011) Telaprevir for retreatment of HCV infection. N Engl J Med 364: 2417-2428. [Crossref]
- Fernández-Montero JV, Soriano V (2011) [Future perspective in the treatment of chronic hepatitis C]. Rev Esp Sanid Penit 13: 21-29. [Crossref]
- Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, et al. (2009) IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 41: 1100-1104. [Crossref]
- Thompson AJ, Muir AJ, Sulkowski MS, Ge D, Fellay J, et al. (2010) Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterol 139(1): 120-129. [Crossref]
- O'Brien TR, Everhart JE, Morgan TR, Lok AS, Chung RT, et al. (2011) An IL28B genotype-based clinical prediction model for treatment of chronic hepatitis C. PLoS One 6: e20904. [Crossref]
- Qattan IT, Omer WH, AlOmer IA, Bereagesh SA, Kabbani KM, et al. (2012) Cross-sectional study of il28b polymorphisms amongst saudi arabian and british hcv patients. European Sci J 8(27): 1-24.