Breast cancer is a global burden with a woman’s lifetime risk of developing breast cancer at 1 in 8. Although breast cancer is a disease that affects mostly women, the lifetime risk in men is about 1 in 1000. Most cases of breast cancer are associated with somatic mutations in breast cells that are acquired during a person's lifetime. In this scenario, the mutations are not inherited and they do not cluster in families. In hereditary breast cancer, the specific genetic factors involved will determine the inherited cancer risk. Inherited mutations in the BRCA1 or BRCA2 genes have been the well-described, but mutations in ATM, CDH1, CHEK2, PALB2, PTEN, STK11, and TP53 also confer breast cancer risk. Understanding the functional significance of hereditary mutations has opened new paths for breast cancer prevention and is uncovering promising treatment strategies.
Breast cancer is the second most common cancer to affect women, but in rare cases it can also develop in men. There are three distinct types of breast cancer. Ductal carcinoma in situ (DCIS) arises in epithelial cells lining the breast ducts. Several studies suggest that at least one third of DCIS cases will progress to invasive cancer if left untreated [1]. Lobular carcinoma in situ (LCIS) develops in milk producing glands, and poses an increased risk for developing invasive cancer. The majority of breast cancers are invasive or infiltrating, and prognosis is dependent on the stage of the disease. Breast cancer is progressively becoming considered as a group of diseases distinguished by molecular subtypes, risk factors, clinical behaviors, and responses to treatment [2]. Biological markers are used to categorize breast cancer types into distinct classes for treatment. The factors include estrogen receptor status (ER+/ER-), progesterone receptor status (PR+/PR-), and human epidermal growth factor receptor 2 status (HER2+/HER2-). Transcriptional profiling of tumors has further led to a second, but related, classification system based on a PAM50 score, which utilizes the expression levels of 50 unique genes, and it is used for a standardizing subtype classification. The intrinsic subtypes of breast cancer are known as luminal A, luminal B, HER2-enriched, and basal-like. The PAM50 score has been providing relevant hints for biomarkers selection in treatment decisions, and it can be used as a predicative tool in cancer progression and patient survival [3].
Hereditary breast cancer accounts for only 5-10 percent of all breast cancers diagnosed in the U.S. In the mid-19th century, the famous French physician Pierre Paul Broca documented that in one family, in over four generations, 10 out of 24 women died from breast cancer. Though he speculated that some inherited factor might play a role, his questions came a hundred years before the scientific tools were available to test his hypothesis [4]. His initial finding, although supported by others [5], led to controversy in the field since some studies at that time concluded that inheritance did not play a role in breast cancer development [6].
Pharoah et al. combined data from 74 published studies conducted between 1935 to 1995 to investigate the incidence of hereditary breast cancer. The group determined that the relative risk for an individual is 2-fold increased if they have a first-degree relative who has been diagnosed with breast cancer [7]. Moreover, the relative risk increases to almost 4-fold if two first-degree relatives have been diagnosed with breast cancer, strongly suggesting that heredity may play a role in breast cancer. Later, Hall et al. identified that the chromosomal locus 17q21 was frequently mutated in individuals from 23 families suspected of having hereditary breast cancer [8]. They also concluded that breast cancer is not completely penetrant among susceptible individuals, and that gender, age and non-genetic risk factors also play important roles. Hall also proposed plausible genes localized in the 17q region that potentially could be critical in breast cancer: HER2 (oncogene), EDHB17 (estradiol-17β dehydrogenase), HOX2 (homeobox 2), NM23 (associated with metastasis), RARA (retinoic acid receptor α) and WNT3 (integration site of mouse mammary tumor virus). Follow-on studies confirmed that chromosome 17 contained a region that contributed specifically to families with an early onset of breast and ovarian cancer [9]. This region in the 17q chromosomal locus was further studied by analyzing haplotypes to identify the minimal genomic regions inherited in common by affected family members. Miki Y et al. demonstrated that the 17q21.3 region contained the BRCA1 gene using positional cloning methods [10]. Simultaneously, another team of scientists, focused on studying male breast cancer, mapped a second breast cancer related gene – BRCA2, and showed that male breast cancer is unlikely to be directly caused by BRCA1 mutation [11]. Additional studies, demonstrated that the BRCA2 gene was located within the chromosomal region 13q12-13 [12].
The BRCA1 gene is composed of 22 exons, encoding a 220kDa nuclear protein of 1863 amino acids [13]. BRCA1 is comprised of a zinc binding RING domain at the amino terminus region, and an acidic carboxyl terminus, which is conserved among species and throughout evolution (Figure 1). The BRCA1 gene is expressed in several tissues, such as breast and ovarian tissue. Initially, the mutations identified in the BRCA1 gene included an 11-base pair deletion, a 1-base pair insertion, a stop codon, a missense substitution, and an inferred regulatory mutation [10]. One year later, a collaborative study including 372 unrelated patients with breast or ovarian cancer selected from high-risk families, demonstrated that eighty patients had a BRCA1 mutation (21.5% of the cohort). Thirty-eight common mutations were recognized among sixty-three mutations identified in a complete screen of the BRCA1 gene. These distinct mutations occurred 8, 7 or 5 times each, and 86% of them predictively resulted in a truncated BRCA1 protein [14]. Currently, more than 1600 mutations have been identified in the BRCA1 gene, and the majority of them promote frameshifts resulting in miss-sense or non-functional protein. Generally, in individuals with a germline BRCA1 mutation, the wild-type allele is somatically mutated, which leads to the conclusion that BRCA1 is a tumor suppressor gene [15]. Women with BRCA1 mutations have an increased risk of developing ovarian cancer, while men have a higher risk, to a lesser extent, of developing prostate cancer [16].
The BRCA2 gene is larger than BRCA1, and it has a 10.3 kb open reading frame encoding a 384 kDa nuclear protein (Figure 1). BRCA2 does not share a high degree of sequence homology with other known genes, and the generated protein is comprised of regions with domains that are undefined [12]. However, the proteins encoded by BRCA1 and BRCA2 genes seem to share functional similarities that justify why mutations in these genes lead to a similar and specific hereditary predisposition to breast and ovarian cancer [15].
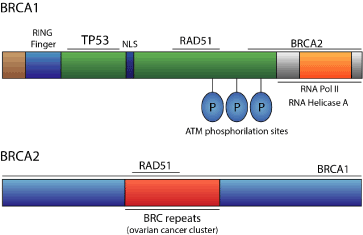
Figure 1. Schematic representation of BRCA1 and BRCA2 genes.
In early studies, BRCA2 was linked to six different germline mutations in breast cancer families, typically by causing disruption of the open reading frame of the transcriptional unit 17. These mutations were related to the interruption of protein translation, for example deletions and/or frameshifts leading to premature stop codons. Currently, more than 1800 mutations have been identified in BRCA2, that include frameshift deletions, insertions, or nonsense mutations that lead to premature truncation of proteins. These events are consistent with the loss of function that is expected in mutations subsequent to tumor suppressor genes [15]. Carriers of BRCA2 mutations also have a higher risk of gall bladder, bile duct, stomach cancer and melanoma [18].
Although only 5 to 10% of breast cancer cases are inherited, recent estimates suggest that 55 to 65% of BRCA1 mutation carriers, and approximately 45% of BRCA2 mutation carriers will develop breast cancer by age 70 [19,20]. Furthermore, the 10-year risk of developing ovarian cancer has been reported to be 12.7% and 6.8% for women carrying BRCA1 and BRCA2 mutations respectively [21]. A recent study of 21,401 families suspected of having a deleterious BRCA mutation showed that 24% of the families carried a pathogenic BRCA1 or BRCA2 mutation [22]. Because BRCA1 and BRCA2 are tumor suppressor genes, they are functionally recessive, and therefore, both copies of the allele must be mutated in the cell for breast cancer to develop (Figure 2).
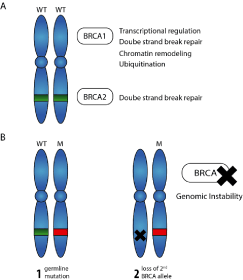
Figure 2. A) Schematic representation of BRCA1 and BRCA2 functions. B) Loss of second BRCA allele in a BRCA mutation carrier.
BRCA genes have a high density of repeated elements allowing for Alu-mediated genomic rearrangements – small recombination events unrecognized by conventional screening techniques can occur within BRCA1 and BRCA2. For example, 22 different genomic rearrangements were identified and ranged in size from less than 1 kb to greater than 170 kb, in high-risk families with negative (wild-type) genetic test results for BRCA1 and BRCA2 [23]. This suggests that germline BRCA mutations can be easily undetected. Several reviews summarize the genomic rearrangements that can occur in the BRCA1 and BRCA2 genes [24-26].
BRCA1 is a pleiotropic DNA damage response protein that operates in both checkpoint activation and DNA repair. BRCA2 is a mediator of homologous recombination [27,28]. The role of BRCA1 in tumorigenesis is related to several cellular processes, namely transcriptional regulation of DNA repair associated genes, heterochromatin formation on the X chromosome, double strand break repair, and ubiquitination [29]. BRCA1 binds to BRCA2, TP53, and RAD51 (repair of DNA double strand breaks), among other proteins associated with the cell cycle and DNA damage response pathways (Table 1). Cells lacking a functional BRCA1 protein are not capable of undergoing arrest in the G2 phase of the cell cycle following DNA damage, and are deficient in transcription-coupled repair [30]. Moreover, BRCA1 modifies chromatin structure to allow access of DNA repair proteins at sites of damage, by interacting with γH2AX [31]. Like BRCA1, the role of BRCA2 is associated with the maintenance of chromosome stability and recombination-mediated double strand break repair of DNA [32]. BRCA2 deficiency leads to deficits in chromosome segregation, and unexpected chromosomal abnormalities that develop after several divisions, namely double-stranded, tri-radials and quadri-radials [33].
The absence of an effective repair mechanism allows DNA damage to occur at many sites, including genes required for cell cycle checkpoint expression. For example, genetic mutations in the TP53 gene, which would prevent p21 expression, allow BRCA-deficient cells to escape apoptosis and perpetuate. Patients with BRCA1 or BRCA2 mutations frequently harbor TP53 mutations, and it is thought that several oncogenes undergo mutation as a result of BRCA insufficiency [34].
BRCA1 and BRCA2 are known to interact with many proteins (Table 1). They are localized in different pathways and play unique roles in recombination and DNA repair [35]. For instance, BRCA1 is localized to the sub-nuclear foci during the S and G2 phases of the cell cycle, where RAD51 is also present. The RAD51 protein is important for the repair of double-strand DNA breaks by binding single-stranded DNA to form a nucleoprotein filament that can penetrate into a homologous duplex DNA molecule [35]. The mechanism through which BRCA1 and BRCA2 promote DNA repair occurs by homologous recombination of DNA replication forks and double strand breaks. In this process, BRCA2 binds directly to RAD51 and guides it to the damaged DNA site. Simultaneously, BRCA1 controls the signaling involved in homologous recombination and it ensures that the double strand break is not resected before RAD51 protein formation [36]. In the case of BRCA mutation, the DNA is repaired in a non-conservative manner, in which the two DNA ends are united as they are, giving rise to new DNA mutations, particularly deletions [37]. If these new mutations affect cancer driver genes, tumorigenesis can occur.
Table 1. BRCA interacting proteins and their function
Reference |
Protein |
Function of interacting protein |
[39] |
RAD51 |
Double strand break repair |
[40] |
RAD50 |
Double strand break repair |
[41] |
BASC complex |
Mismatch repair |
[42] |
H2AX |
Signaling of DNA damage |
[43] |
TP53 |
Tumor suppressor gene – Transcription factor |
[44] |
pRB |
Tumor suppressor gene – Cell cycle regulator |
[45] |
c-myc |
Oncogene – Transcription factor |
[46] |
STAT1 |
Signal Transducer – Transcription factor |
[47] |
E2F |
Cell cycle regulator – Transcription factor |
[48] |
RNA Pol II |
Transcription |
[49] |
Estrogen Receptor |
Ligand responsive transcription factor |
[50] |
Androgen Receptor |
Ligand responsive transcription factor |
[51] |
SWI/SNF |
Chromatin remodeling complex |
[52] |
HDAC |
Histone deacetylation – chromatin remodeling |
[53] |
BRAP2 |
Cytoplasmic retention |
[54] |
PALB2 |
Double-strand break repair |
[55] |
BARD1 |
Ubiquitin ligase |
BRCA1 and BRCA2 also operate as transcriptional regulators of specific target genes. Interactions have been shown between BRCA proteins and specific transcription factors such as c-myc (BRCA1) and TP53 (BRCA1 and BRCA2) [38]. Candidates in the TP53 pathway have been identified as BRCA-target genes, such as p21 and GADD45. This suggests that BRCA1 function can be related to the expression of genes fundamental to checkpoint control or DNA repair.
In addition to BRCA1 and BRCA2, rare mutation susceptibility alleles exist with different penetrance levels, and account for a small fraction of hereditary breast cancer cases. For instance, STK11/LKB1 is a serine–threonine kinase and mutations in the STK11 gene can potentially cause Peutz-Jegher syndrome, which is characterized by hamartomatous polyps in the small bowel and pigmented macules, and is also linked with a relative risk for breast cancer of 20.3 compared with non-carriers [56]. Another highly penetrant mutation occurs in phosphatase and tensin homolog (PTEN) and is related to Cowden syndrome. This mutation is associated with an increase of 20 to 30% lifetime risk of breast cancer [57]. In patients with germline mutations in TP53, related with Li-Fraumeni syndrome, the breast cancer penetrance approaches 100% if the mutation carriers survive childhood [58].
Moderate penetrance genes have been more recently considered as having the status of hereditary breast cancer genes, and are often related to BRCA function. Carriers of mutations in the ATM gene (ataxia-telangiectasia) have an increased risk of breast cancer [59]. CHEK2, a cell cycle checkpoint kinase that is required in the DNA repair pathway involving BRCA1 and TP53, has pathogenic variants that result in a two-fold increase in the risk of developing breast cancer. However, it does not confer risk in BRCA mutation carriers [60]. Another example, the PALB2 gene, also known by the localizer of the BRCA2 gene, is related to the production of a functional protein that interacts with BRCA2 to repair damaged DNA. Fanconi anemia type N is a disease caused by the inheritance of two abnormal PALB2 genes and it is characterized by extremely low levels of red and white blood cells, and platelets. Recent work demonstrates that women with abnormal PALB2 levels have a 14% risk of developing cancer until 50 years old, and 35% risk until 70 years old [61]. Mutations in RAD51 have also been identified [62]. A recent study utilizing a focused panel of 25 genes sequenced in more than 35,000 women with breast cancer demonstrated pathogenic variants were present in 9.3% of the tested population. From these variants, 51.5% occurred in BRCA genes, 9.7% in ATM, 11.7% in CHECK2, and 9.3% in PALB2. The prevalence of pathogenic variants in BARD1 and RAD51 were statistically higher among women with triple-negative breast cancer [63]. It is important to recognize that BRCA1, BRCA2, PTEN, ATM, PALB2, CHEK2, RECQL, NBN as well as a large number of low penetrance variants together account for only ~50% of breast cancer susceptibility [64]. This finding demonstrates the polygenic nature of breast cancer risk and indicates that variants contributing to breast cancer risk remain to be discovered.
Advanced genomic studies using whole exome sequencing have the capability of revealing unique hereditary mutations [65]. In a study with 4398 breast cancer cases and 4316 controls, followed by a second phase to test 30 single nucleotide polymorphism (SNPs), common alleles containing a single nucleotide polymorphism (SNPs) in FGFR2 (rs2981582), TNRC9 (rs3803662), and MAP3K1 (rs889312) were associated with increased breast cancer risk in the general population [66]. To further investigate if these loci are also linked with breast cancer risk in BRCA1 and BRCA2 mutation carriers, genotyping was performed to assess the SNPs identified. The minor alleles of SNP rs2981582 and rs889312 were each associated with increased breast cancer risk in BRCA2 mutation carriers, but not in BRCA1 carriers. The SNP rs3803662 was associated with increased breast cancer risk in both BRCA1 and BRCA2 mutation carriers [67]. In 2009 a pericentromeric SNP on chromosome 1p11.2 was identified in a large linkage disequilibrium block neighboring NOTCH2 and FCGR1B genes [68]. A large-scale genotyping study where 29807 SNPs were identified and further genotyped revealed 41 new loci associated with increased breast cancer risk [69]. Taken together, these studies reveal that advanced sequencing studies will likely continue to identify new loci that confer the risk of breast cancer. With the decreasing cost of genomic technologies and the ability to detect genetic variation in patients at high accuracy and reduced cost, clinical decision making may be fundamentally altered by these technologies in the near future.
Inherited BRCA1 mutant breast cancer usually presents a basal-like transcriptomic signature which is defined by the high expression of basal layer genes, and frequently results in triple-negative breast cancers – approximately 80% of BRCA1 mutation cases [70,71]. Histological characterization of germline BRCA1 mutant tumors has been well defined, featuring a high histological grade, atypical medullary features, high proliferation indices, invasive borders and lymphocytic infiltrates. BRCA2 mutation carriers present tumors with a higher risk of contralateral breast cancer and estrogen-receptor positivity in most cases [72,73].
A multivariate study, including 223 breast cancer patients carrying BRCA mutations and 446 controls with sporadic breast cancer matched for age and year of diagnosis, showed no difference in terms of specific breast cancer survival between BRCA1 mutation carriers and sporadic cases, or between overall survival for BRCA2 mutation carriers and sporadic controls [72]. A second study using a cohort of 491 patients (86 BRCA-mutants and 391 non-mutants) suggested that BRCA1 mutation carriers had higher nuclear grade tumors than the other two groups of patients, and that BRCA2 mutated patients were older at the time they were diagnosed with breast cancer, in comparison with BRCA1 mutants and non-mutants [74].
Two recent studies demonstrated different results with respect to the role of BRCA mutations on breast cancer prognosis. In the first study, which utilized a database containing the mutation status of 105,220 breast cancer patients with 3.4% BRCA-carriers, BRCA1 mutation carriers displayed a worse overall survival than patients with a non-mutated BRCA1 allele. The same study also suggested that BRCA2 mutation carriers have worse disease-specific survival than patients with a non-mutated BRCA1 allele, but they present a similar overall survival. In the same year, Templeton et al. evaluated a total of 16 studies comprising data from 10,180 patients concluding that BRCA mutations were not associated with worse overall survival [75]. Taken together, the results suggest that BRCA mutation may be inadequate as an independent outcome predictor [76,77].
A clinical diagnosis of hereditary breast and ovarian cancer occurs when one or more of the following features are present in a family: i) early onset breast cancer (less than 50 years of age) including both invasive and ductal carcinoma in situ breast cancers; ii) two breast primary or breast and other related cancer (ovarian, fallopian tube or primary peritoneal) in a single individual, or two or more breast primary or other related cancer in close relatives (first- to third-degree) from the same side of family; iii) populations at risk (Ashkenazi Jewish); iv) member of a family with a known BRCA1; v) any male breast cancer; vi) ovarian, fallopian tube or primary peritoneal cancer at any 15]. BRCA mutations are diagnosed using molecular genetic testing to assess potential genomic rearrangements in the BRCA1 or BRCA2 genes [15]. The NCCN recently updated their guidelines for genetic/familial high-risk assessment and provide recommendations for genetic testing, counseling, and risk assessment [78].
Primary prevention strategies to reduce breast cancer risk in individuals who carry BRCA1 or BRCA2 mutations include prophylactic mastectomy, surveillance, and chemoprevention [79]. A recent study of 1504 patients with germline BRCA1 or BRCA2 mutations showed a reduced risk of 50% for developing contralateral breast cancer when taking tamoxifen as adjuvant [80]. Currently, for postmenopausal women, the decision to treat with tamoxifen therapy depends on the stage of the disease, risk of recurrence, age or personal choice. Additionally, ASCO guidelines recommend a switch to an aromatase inhibitor at some point during the anti-estrogen therapy. In the case of premenopausal women, tamoxifen therapy for 10 years may decrease the risk of breast cancer recurrence [81].
Surgery
Several studies have demonstrated differences between breast cancers with and without BRCA1 or BRCA2 mutations. For instance, women who carry BRCA mutations are more likely to develop a secondary cancer – either in the same breast (ipsilateral) or in the opposite breast (contralateral). For these women, a bilateral mastectomy is recommended, since studies have suggested that women who are BRCA1/2 mutation carriers and receive a bilateral mastectomy are less likely to die from breast cancer than women who were treated with unilateral mastectomy [82,83].
Chemotherapy
Taxanes: Taxanes are microtubule stabilizing chemotherapy agents that block cell proliferation, leading to apoptosis. The most common taxanes used for breast cancer treatment are docetaxel and paclitaxel, which were approved for medical use in 1993 and 1995. BRCA1 mutation carriers in the subgroup of hormone-negative cancers showed less sensitivity to taxane chemotherapy than non-BRCA1 mutation carriers hormone-negative patients. Conversely, in the subgroup of hormone-positive cancers, both hereditary and sporadic cases show similar sensitivities to taxane therapy [84] . An approach for neoadjuvant chemotherapy used a combination of anthracycline-taxane, and 46% of the BRCA1 mutation carriers showed pathological complete response (pCR), while the sporadic breast cancer patients showed 22% pCR [85]. However, a recent meta-analysis study suggested that a taxane-based therapy is potentially a better option than the anthracycline-taxane regimen for advanced breast cancer cases, since both produce similar clinical outcomes, and taxane is less toxic [86].
Platinum agents
Platinum agents bind directly to DNA, forming DNA/platinum adducts that results in inter-strand DNA crosslinks and subsequent o double strand breaks. A study showed that neoadjuvant chemotherapy promotes enhanced response to platinum agents and a reduced response to taxanes in hereditary BRCA1-associated breast cancer. Although this work used a small cohort of patients, the pCR for cisplatin was 83%, while women treated with doxorubicin and docetaxel presented 8% of pCR. Interestingly, combinatorial therapy involving doxorubicin and cyclophosphamide, and in certain cases fluorouracil, showed a pCR of only 22% [87]. Another work focusing neoadjuvant cisplatin therapy showed that decreased BRCA1 expression may help to identify subsets of triple negative cancers that are cisplatin-sensitive [88]. Further evidence was provided with a follow-on clinical trial using cisplatin that showed that BRCA1 mutation carriers are highly sensitive to this chemotherapeutic agent [89]. A systematic review and meta-data analysis of all published studies employing platinum agents in addition to standard neoadjuvant chemotherapy in triple-negative cancer was conducted, and it showed that the pCR increases significantly by including cisplatin or carboplatin in triple negative breast cancer, rather than any other neoadjuvant chemotherapy [90]. In contrast, a recent study reported a BRCA1 reversion mutation in a recently diagnosed triple negative breast cancer patient, that developed over 18 weeks of platinum-based neoadjuvant therapy, resulting in poor response, early relapse and death [91].
PARP inhibitors
Poly(ADP-ribose) polymerases (PARPs) are important enzymes in DNA damage repair mechanisms. In general, PARP activation is promoted by DNA damage, particularly through PARP-1 to PARP-3, initiators of the DNA damage response. PARP synthesizes a polymer (ADP-ribose polymer) that attracts the assembly of DNA repair complexes at sites of damage [92]. PARP inhibitors block the repair of DNA damage, resulting in chromosomal instability, cell cycle arrest and subsequent apoptosis, leading to the persistence of DNA lesions normally repaired by homologous recombination. PARP inhibitors attack tumors defective in the BRCA1 or BRCA2 genes by a concept termed ‘synthetic lethality’. PARP inhibitors cause an increase in DNA single-strand breaks (SSBs), which are converted during replication to irreparable toxic DNA double-strand breaks (DSBs) in BRCA1/2 defective cells. Clinical trials have shown that PARP inhibitors are beneficial in the treatment of patients that are carriers of germline BRCA mutations. Moreover, PARP inhibitors are also likely to be useful for non-BRCA mutations carriers [93].
Several clinical trials are focused on the use of PARPs inhibitors, in the adjuvant, neoadjuvant and metastatic settings for the treatment of ovarian, BRCA-mutated breast cancer and other cancers [92]. Although there is excitement around this new class of drugs, Iniparib by Sanofi-aventis, the most advanced PARP inhibitor in clinical trials in 2011, failed to prolong survival in phase III in triple-negative breast cancer. The failure was related with a resistance event suggested in a study from 2013, where they showed clinical observations of PARPs blocking drugs resistance correlating with the emergence of a secondary BRCA2 mutation. This mutation will likely restore the wild-type protein function, compromising the synthetic lethality approach [94].
A follow-on study showed that Iniparib and its metabolites do not inhibit PARP in intact cells [95], suggesting PARP inhibitors should be given additional consideration in clinical studies. Current clinical trials are testing the potential of seventeen new PARPs inhibitors in early and advanced breast cancer, such as Olaparib (Phase III in germline BRCA mutated breast cancer), Veliparib (Phase III in neoadjuvant setting standard or in combination with carboplatin in triple-negative breast cancer), Niraparib (Phases II/III in combination therapy in germline BRCA mutated breast cancer), Talazoparib (Phases II/III for different settings in germline BRCA mutated breast cancer), and Rucaparib (Phase II in germline BRCA mutated solid breast cancer) [92]. Olaparib received FDA approval in 2014, and Rucaparib was approved in December 2016 [96]. Long term exposure and strategies to expand PARP therapies beyond breast and ovarian cancer are being intensively investigated [92].
It is clear that BRCA mutation status can provide valuable insight in terms of prevention and treatment options. With appropriate management and surveillance, BRCA mutation carriers have options to prevent or detect cancer at earlier stages, when there is a greater chance for successful treatment. The decreased cost of genome-sequencing and advances in bioinformatics will likely change the landscape for tailored treatment strategies not only for BRCA mutations carriers but also for patients with unique genetic mutations that have not been previously considered. The ultimate goal is to identify aberrations that make each individual’s cancer more vulnerable to particular drugs — and to match individual patients with available therapies or clinical trials that will most benefit them.
Our work has been supported by R00-CA181352 (DMG).
- Allred DC (2010) Ductal carcinoma in situ: terminology, classification, and natural history. J Natl Cancer Inst Monogr 2010: 134-138. [Crossref]
- Reis-Filho JS, Pusztai L (2011) Gene expression profiling in breast cancer: Classification, prognostication, and prediction. Lancet 378: 1812-1823. [Crossref]
- Kittaneh M, Montero AJ, Glück S (2013) Molecular profiling for breast cancer: a comprehensive review. Biomark Cancer 5: 61-70. [Crossref]
- Ghoussaini M, Pharoah PD, Easton DF (2013) Inherited genetic susceptibility to breast cancer: the beginning of the end or the end of the beginning? Am J Pathol 183: 1038-1051. [Crossref]
- Penrose H (1954) The Annals of Human Genetics has an archive of material originally published in print format by the Annals of Eugenics (1925-1954). This material is available in specialised libraries and archives. We believe there is a clear academic interest in makin.
- Lane-Claypon JE (1926) A Further Report on Cancer of the Breast, With Special Reference To Its Associated Antecedent Conditions. London, Reports on Public Health and Medical Subjects No. 32 Ministry of Health, His Majesty’s Stationery Office: 18,57,84,111-112.
- Pharoah PD, Day NE, Duffy S, Easton DF, Ponder BA (1997) Family history and the risk of breast cancer: a systematic review and meta-analysis. Int J Cancer 71: 800-809. [Crossref]
- Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, et al. (1990) Linkage of early-onset familial breast cancer to chromosome 17q21. Science 250: 1684-1689. [Crossref]
- Easton DF, Bishop DT, Ford D, Crockford GP (1993) Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet 52: 678-701. [Crossref]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, et al. (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266: 66-71. [Crossref]
- Stratton MR, Ford D, Neuhasen S, Seal S, Wooster R, et al. (1994) Familial male breast cancer is not linked to the BRCA1 locus on chromosome 17q. Nat Genet 7: 103-107. [Crossref]
- Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, et al. (1994) Localization of a Breast Cancer Susceptibility Gene , BRCA2 , to Chromosome 13q12-13. Science 265: 2088-2090. [Crossref]
- Albertsen H, Plaetke R, Ballard L, Fujimoto E, Connolly J, et al. (1994) Genetic mapping of the BRCA1 region on chromosome 17q21. Am J Hum Genet 54: 516-525. [Crossref]
- Shattuck-Eidens D, Mcclure M, Simard J, Labrie F, Narod S, et al. (1995) A Collaborative Survey of 80 Mutations in the BRCA1 Breast and Ovarian Cancer Susceptibility Gene Implications for Presymptomatic Testing and Screening. JAMA 273: 535-541. [Crossref]
- Petrucelli N, Daly MB, Feldman GL (2010) Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet Med 12: 245-259. [Crossref]
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE (1994) Risks of cancer in BRCA1 mutation carriers. Lancet 343: 692-695. [Crossref]
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, et al. (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378: 789-792. [Crossref]
- Breast Cancer Linkage Consortium (1999) Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 91: 1310-1316. [Crossref]
- Antoniou A, Pharoah PDP, Narod S, Risch HA, Eyfjord JE, et al. (2003) Average Risks of Breast and Ovarian Cancer Associated with BRCA1 or BRCA2 Mutations Detected in Case Series Unselected for Family History: A Combined Analysis of 22 Studies. Am J Hum Genet 72: 1117-1130. [Crossref]
- Chen S, Parmigiani G (2007) Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol 25: 1329-1333. [Crossref]
- Metcalfe KA, Lynch HT, Ghadirian P, Tung N, Olivotto IA, et al. (2005) The risk of ovarian cancer after breast cancer in BRCA1 and BRCA2 carriers. Gynecol Oncol 96: 222-226. [Crossref]
- Kast K, Rhiem K, Wappenschmidt B, Hahnen E, Hauke J, et al. (2016) Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J Med Genet 53: 465-471. [Crossref]
- Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, et al. (2006) Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 295: 1379-1388. [Crossref]
- Mazoyer S (2005) Genomic rearrangements in the BRCA1 and BRCA2 genes. Hum Mutat 25: 415-422. [Crossref]
- Ewald IP, Ribeiro PL, Palmero EI, Cossio SL,2021 Copyright OAT. All rights reservearrangements in BRCA1 and BRCA2: A literature review. Genet Mol Biol 32: 437-446. [Crossref]
- Sluiter MD, Van Rensburg EJ (2011) Large genomic rearrangements of the BRCA1 and BRCA2 genes: Review of the literature and report of a novel BRCA1 mutation. Breast Cancer Res Treat 125: 325-349. [Crossref]
- Roy R, Chun J, Powell SN (2012) BRCA1 and BRCA2: important differences with common interests. Nat Rev Cancer: 372-372.
- Roy R, Chun J, Powell SN (2015) BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 12: 68-78. [Crossref]
- Starita LM, Parvin JD (2003) The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Curr Opin Cell Biol 15: 345-350. [Crossref]
- Larson JS, Tonkinson JL, Lai MT (1997) A BRCA1 mutant alters G2-M cell cycle control in human mammary epithelial cells. Cancer Res 57: 3351-3355. [Crossref]
- Yamane K, Katayama E, Tsuruo T (2000) The BRCT regions of tumor suppressor BRCA1 and of XRCC1 show DNA end binding activity with a multimerizing feature. Biochem Biophys Res Commun 279: 678-684. [Crossref]
- Shamoo Y (2003) Structural insights into BRCA2 function. Curr Opin Struct Biol 13: 206-211. [Crossref]
- Nasmyth K, Peters JM, Uhlmann F (2000) Splitting the chromosome: cutting the ties that bind sister chromatids. Science 288: 1379-1385. [Crossref]
- Welcsh PL, King MC (2001) BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet 10: 705-713. [Crossref]
- Scully R, Livingston DM (2000) In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 408: 429-432. [Crossref]
- Prakash R, Zhang Y, Feng W, Jasin M (2015) Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7: a016600. [Crossref]
- Lord CJ, Ashworth A (2016) BRCAness revisited. Nat Rev Cancer 16: 110-120. [Crossref]
- Welcsh PL, Owens KN, King MC (2000) Insights into the functions of BRCA1 and BRCA2. Trends Genet 16: 69-74. [Crossref]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, et al. (1997) Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88: 265-275. [Crossref]
- Zhong Q, Chen CF, Li S, Chen Y, Wang CC, et al. (1999) Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 285: 747-750. [Crossref]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, et al. (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14: 927-939. [Crossref]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, et al. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10: 886-895. [Crossref]
- Zhang H, Somasundaram K, Peng Y, Tian H, Zhang H, et al. (1998) BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 16: 1713-1721.
- Aprelikova ON, Fang BS, Meissner EG, Cotter S, Campbell M, et al. (1999) BRCA1-associated growth arrest is RB-dependent. Proc Natl Acad Sci U S A 96: 11866-11871. [Crossref]
- Wang Q, Zhang H, Kajino K, Greene MI (1998) BRCA1 binds c-Myc and inhibits its transcriptional and transforming activity in cells. Oncogene 17: 1939-1948. [Crossref]
- Ouchi T, Lee SW, Ouchi M, Aaronson S a, Horvath CM (2000) Collaboration of signal transducer and activator of transcription 1 (STAT1) and BRCA1 in differential regulation of IFN-gamma target genes. Proc Natl Acad Sci U S A 97: 5208-5213. [Crossref]
- Wang H, Shao N, Ding QM, Cui J, Reddy ES, et al. (1997) BRCA1 proteins are transported to the nucleus in the absence of serum and splice variants BRCA1a, BRCA1b are tyrosine phosphoproteins that associate with E2F, cyclins and cyclin dependent kinases. Oncogene 15: 143-157. [Crossref]
- Scully R, Anderson SF, Chao DM, Wei W, Ye L, et al. (1997) BRCA1 is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci U S A 94: 5605-5610. [Crossref]
- Fan S, Wang J, Yuan R, Ma Y, Meng Q, et al. (1999) BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science 284: 1354-1356. [Crossref]
- Yeh S, Hu YC, Rahman M, et al. (2000) Increase of androgen-induced cell death and androgen receptor transactivation by BRCA1 in prostate cancer cells. Proc Natl Acad Sci U S A 97: 11256-11261. [Crossref]
- Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, et al. (2000) BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell 102: 257-265. [Crossref]
- Yarden RI, Brody LC (1999) BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci U S A 96: 4983-4988. [Crossref]
- Zhang H, Wang Q, Kajino K, Greene MI (2000) VCP, a weak ATPase involved in multiple cellular events, interacts physically with BRCA1 in the nucleus of living cells. DNA Cell Biol 19: 253-263. [Crossref]
- Rahman N, Seal S, Thompson D, Kelly P, Renwick A, et al. (2007) PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet 39: 165-167. [Crossref]
- Atipairin A, Canyuk B, Ratanaphan A (2011) The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by the platinum-based anticancer drugs. Breast Cancer Res Treat 126: 203-209. [Crossref]
- Boardman LA, Thibodeau SN, Schaid DJ, Lindor NM, McDonnell SK, et al. (1998) Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med 128: 896-899. [Crossref]
- Lynch ED, Ostermeyer EA, Lee MK, Arena JF, Ji H, et al. (1997) Inherited mutations in PTEN that are associated with breast cancer, cowden disease, and juvenile polyposis. Am J Hum Genet 61: 1254-1260. [Crossref]
- Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, et al. (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250: 1233-1238. [Crossref]
- Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, et al. (2006) ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 38: 873-875. [Crossref]
- Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, et al. (2002) Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 31: 55-59. [Crossref]
- Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, et al. (2014) Breast-cancer risk in families with mutations in PALB2. N Engl J Med 371: 497-506. [Crossref]
- Kolinjivadi AM, Sannino V, de Antoni A, Técher H, Baldi G, et al. (2017) Moonlighting at replication forks: a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. [Crossref]
- Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, et al. (2017) A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer. [Crossref]
- Couch FJ, Kuchenbaecker KB, Michailidou K, Mendoza-Fandino GA, Nord S, et al. (2016) Identification of four novel susceptibility loci for oestrogen receptor negative breast cancer. Nat Commun 7: 11375. [Crossref]
- Tomczak K, Czerwinska P, Wiznerowicz M (2015) The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) 19: A68-77. [Crossref]
- Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, et al. (2007) Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447: 1087-1093. [Crossref]
- Antoniou AC, Spurdle AB, Sinilnikova OM, et al. (2008) Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCA1 and BRCA2 mutation carriers. Am J Hum Genet 82: 937-948. [Crossref]
- Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, et al. (2009) A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat Genet 41: 579-584. [Crossref]
- Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, et al. (2013) Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 45: 353-361, 361e1-2. [Crossref]
- Turner NC, Reis-Filho JS (2006) Basal-like breast cancer and the BRCA1 phenotype. Oncogene 25: 5846-5853. [Crossref]
- Bayraktar S, Glück S (2012) Systemic therapy options in BRCA mutation-associated breast cancer. Breast Cancer Res Treat 135: 355-366. [Crossref]
- Brekelmans CTM, Tilanus-Linthorst MMA, Seynaeve C, et al. (2007) Tumour characteristics, survival and prognostic factors of hereditary breast cancer from BRCA2-, BRCA1- and non-BRCA1/2 families as compared to sporadic breast cancer cases. Eur J Cancer 43: 867-876. [Crossref]
- Goodwin PJ, Phillips KA, West DW (2007) Prognosis of breast cancer in carriers of BRCA1 and BRCA2 mutations. N Engl J Med 357: 1555. [Crossref]
- Atchley DP, Albarracin CT, Lopez A, Valero V, Amos CI, et al. (2008) Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J Clin Oncol 26: 4282-4288. [Crossref]
- Templeton AJ, Gonzalez LD, Vera-Badillo FE, Tibau A, Goldstein R, et al. (2016) Interaction between Hormonal Receptor Status, Age and Survival in Patients with BRCA1/2 Germline Mutations: A Systematic Review and Meta-Regression. PLoS One 11: e0154789. [Crossref]
- Reddy KB (2011) Triple-negative breast cancers: an updated review on treatment options. Curr Oncol 18: e173-179. [Crossref]
- Trainer AH, Lewis CR, Tucker K, Meiser B, Friedlander M, et al. (2010) The role of BRCA mutation testing in determining breast cancer therapy. Nat Rev Clin Oncol 7: 708-717. [Crossref]
- Daly MB, Pilarski R, Axilbund JE, Berry M, Buys SS, et al. (2016) Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2015. J Natl Compr Canc Netw 14: 153-162. [Crossref]
- Winship I, Southey MC (2016) Gene panel testing for hereditary breast cancer. Med J Aust 204: 188-190. [Crossref]
- Gronwald J, Robidoux A, Kim-Sing C, Tung N, Lynch HT, et al. (2014) Duration of tamoxifen use and the risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat 146: 421-427. [Crossref]
- Smith GL (2014) The Long and Short of Tamoxifen Therapy: A Review of the ATLAS Trial. J Adv Pract Oncol 5: 57-60. [Crossref]
- Rebbeck TR, Friebel T, Lynch HT, Neuhausen SL, van 't Veer L, et al. (2004) Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: The PROSE study group. J Clin Oncol 22: 1055-1062. [Crossref]
- Metcalfe K, Gershman S, Ghadirian P, Lynch HT, Snyder C, et al. (2014) Contralateral mastectomy and survival after breast cancer in carriers of BRCA1 and BRCA2 mutations: retrospective analysis. BMJ 348: g226. [Crossref]
- Kriege M, Jager A, Hooning MJ, Huijskens E, Blom J, et al. (2012) The efficacy of taxane chemotherapy for metastatic breast cancer in BRCA1 and BRCA2 mutation carriers. Cancer 118: 899-907. [Crossref]
- Arun B, Bayraktar S, Liu DD, Gutierrez Barrera AM, Atchley D, et al. (2011) Response to neoadjuvant systemic therapy for breast cancer in BRCA mutation carriers and noncarriers: A single-institution experience. J Clin Oncol 29: 3739-3746. [Crossref]
- Zheng R, Han S, Duan C, Chen K, You Z, et al. (2015) Role of taxane and anthracycline combination regimens in the management of advanced breast cancer: a meta-analysis of randomized trials. Medicine (Baltimore) 94: e803. [Crossref]
- Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, et al. (2010) Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol 28: 375-379. [Crossref]
- Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, et al. (2010) Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol 28: 1145-1153. [Crossref]
- Turner NC, Tutt ANJ (2012) Platinum chemotherapy for BRCA1-related breast cancer: do we need more evidence? Breast Cancer Res 14: 115. [Crossref]
- Petrelli F, Coinu A, Borgonovo K, Cabiddu M, Ghilardi M, et al. (2014) The value of platinum agents as neoadjuvant chemotherapy in triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res Treat 144: 223-232. [Crossref]
- Afghahi A, Timms KM, Vinayak S, Jensen KC, Kurian AW, et al. (2017) Tumor BRCA1 Reversion Mutation Arising During Neoadjuvant Platinum-Based Chemotherapy in Triple-Negative Breast Cancer Is Associated with Therapy Resistance. Clin Cancer Res. [Crossref]
- Livraghi L, Garber JE (2015) PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med 13: 188. [Crossref]
- Lord CJ, Tutt AN, Ashworth A (2015) Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med 66: 455-470. [Crossref]
- Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, et al. (2013) Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol 229: 422-429. [Crossref]
- Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH (2012) Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clin Cancer Res 18: 1655-1662. [Crossref]
- Savage P (2017) Approvals in 2016: cost-benefit challenges of new anticancer agents. Nat Rev Clin Oncol 14: 133-134. [Crossref]