Abstract
The pathogenesis of sporadic AD is believed to result from complex interactions between nutritional, environmental, epigenetic and genetic factors. Among those factors, elevated plasma cholesterol, independent of APOE genotypes, is strongly linked to the pathogenesis of sporadic AD, whereas chronic ingestion of caffeine is protective against sporadic AD. Others and we have shown that rabbits-fed cholesterol-enriched diet exhibit early AD-like pathological features including blood-brain barrier (BBB) leakage and endolysosome dysfunction, as well as, AD-like pathological hallmarks including synaptic disruption, increased intraneuronal and brain deposition of amyloid beta (Ab), and tau pathology. Further, we found that caffeine protects against the cholesterol-induced increases in BBB permeability. Here, we determined the extent to which caffeine affects cholesterol-enriched diet-induced increases in other pathological features of AD. We demonstrated that caffeine administration at low and moderate doses did not affect plasma levels of cholesterol, but did block significantly cholesterol-enriched diet-induced increases in brain levels of apoB and accumulation of apoB in neuronal endolysosomes. Furthermore, we demonstrated that caffeine administration at the two doses blocked cholesterol-enriched diet-induced abnormal accumulation of synaptophysin, Aβ, and phosphorylated tau in endolysosomes. Our findings suggest that caffeine exerts its protective effects against the development of sporadic AD-like pathological features, in part, by preventing the entrance of LDL cholesterol into brain parenchyma, the accumulation of LDL-cholesterol in neuronal endolysosomes, and structural and functional changes to endolysosomes.
Key words
sporadic Alzheimer’s disease, apoB, cholesterol, caffeine, blood-brain barrier, synpatophysin, amyloid beta, phosphorylated tau
Introduction
Alzheimer’s disease (AD), the most common neurodegenerative disorder of old age, is characterized clinically by a progressive decline in cognitive function, and pathologically by loss of synaptic integrity and neurons, amyloid plaques composed of amyloid beta (Aβ) protein, and neuronal tangles composed of hyperphosphorylated tau [1,2]. Although mutations in specific genes, including AβPP, PSEN1, and PSEN2, lead to rare early-onset familial forms of AD, the vast majority of AD cases are late in onset and sporadic in nature. The pathogenesis of sporadic AD is much more complex than that of familial AD and sporadic AD is believed to result from complex interactions between nutritional, environmental, epigenetic and genetic factors [3]. Of those factors, altered cholesterol homeostasis may have the strongest links to the pathogenesis of late-onset sporadic AD.
Evidence for the involvement of cholesterol dyshomeostasis in AD pathogenesis comes principally from the following findings. First, the ε4 allele of the APOE gene, which encodes the major cholesterol carrier protein in brain apolipoprotein E (apoE), is the strongest genetic risk factor yet identified for sporadic AD. Second, apoE4 is associated with elevated levels of plasma cholesterol [4,5]. Third, elevated levels of plasma cholesterol, independent of APOE genotypes, has been linked to the pathogenesis of sporadic AD [6-10], as supported by epidemiology studies [6,9] as well as experimental studies conducted in AβPP transgenic mice [11,12], in guinea pigs [13], in rabbits [14], and in rats [15].
Rabbits fed a high cholesterol diet are considered to be a good model for sporadic AD. We have shown that the cholesterol-fed rabbits develop pathological hallmarks of AD including disrupted synaptic integrity, brain deposition of Aβ, and tau pathology [7]. In addition, we have reported that cholesterol-fed rabbits exhibit very early pathological features found in people with AD including increased blood-brain barrier (BBB) leakage [7,16,17] and disturbed neuronal endolysosome structure and function [18]. Furthermore, using primary cultured neurons we demonstrated that LDL cholesterol, the major lipoprotein particle responsible for cholesterol transport in the blood, promoted AbPP internalization, enhanced BACE-1 activity, and increased amyloidogenic processing of AβPP in endolysosomes [8]. Collectively, our findings suggest that elevated levels of LDL cholesterol can enter brain parenchyma via a leaky BBB whereby the LDL cholesterol enters into endolysosomes via receptor-mediated endocytosis and disturbs the structure and function of neuronal endolysosomes thus promoting the development of AD-like pathology.
Human epidemiological and animal experimentation studies as well as studies conducted in cultured cell models have shown that caffeine protects against the onset and severity of AD [19-28]. Additionally, it has been shown that it can reverse behavioral and pathological features of AD [19,21,22,24,25,29-33]. Importantly, these protective effects of caffeine are observed using pharmacologically relevant doses [28,34]. Previously, using the cholesterol-fed rabbit model of sporadic AD, we reported that chronic ingestion of caffeine protects against cholesterol-induced increases in BBB leakage, oxidative stress, brain deposition of Aβ, and tau phosphorylation [16, 35,36]. The present studies were aimed to determine the effects of pharmacologically relevant very low and higher doses of caffeine on AD-like pathology using the cholesterol-fed rabbit model for sporadic AD with a specific focus on determining the extent to which endolysosomes are involved.
Material and methods
Animals and treatments: New Zealand White male rabbits (1.5–2 years of age and weighing 3 to 4 kg; Charles River Laboratories International, Wilmington, MA, USA) were assigned randomly to the following four groups (n=4 each); rabbits fed normal chow, rabbits fed a 2% cholesterol-enriched diet (Harlan Teklad Global Diets, Madison, WI, USA), rabbits fed a 2% cholesterol-enriched diet + 0.5 mg of caffeine/day, and rabbits fed a 2% cholesterol-enriched diet + 30 mg of caffeine/day. All rabbits were kept on their respective diet and caffeine treatment for 12 weeks after which time the rabbits were euthanized. Caffeine was prepared fresh daily and was administered in the drinking water. The very low (0.5 mg/day) and higher (30 mg/day) doses of caffeine were chosen based on findings that the basal metabolic rate in animals is proportional to the 3/4 power of their body mass; the metabolic rate for rabbit is about twice that of a human. Thus, rabbits ingesting 0.5 mg caffeine per day is roughly equivalent to a 70 Kg person ingesting 18 mg of caffeine; a dose well below that contained in a single serving of virtually all caffeinated beverages. At necropsy, animals were perfused with Dulbecco's phosphate-buffered saline at 37°C, and olfactory bulbs were dissected, frozen on a liquid nitrogen-cooled surface and stored at – 80°C until taken for experimentation; one olfactory bulb from each animal was taken for immunostaining and the other for immunoblotting. All animal procedures were carried out in accordance with the U.S. Public Health Service Policy on the Humane Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of North Dakota.
Plasma cholesterol levels: Total levels of plasma cholesterol in rabbits were measured in blood collected from an ear vein after overnight fasting immediately before euthanasia. The measurements were carried out using a Flex reagent cartridge and the Dimension Clinical Chemistry System (Dade Behring, Inc.).
Immunohistochemistry: Snap-frozen olfactory bulbs were embedded in Tissue-Tek Optimum Cutting Temperature (OCT) compound in cryomolds. Horizontal cryostat sections (thickness 14 m) were mounted on Superfrost Plus slides (Fisher). For immunofluorescent staining studies, sections were air-dried at room temperature for 40 minutes, fixed in ice-cold acetone for 10 minutes, air-dried for 30 minutes, and washed 3-times with PBS. Sections were then blocked with 5% donkey serum and incubated overnight at 4oC with primary antibodies targeting synaptophysin (1:1000, mouse monocolonal, Sigma), EEA1 (1:500, goat polycolonal, Santa Cruz), LAMP2 (1:500, goat polycolonal, Santa Cruz), apolipoprotein B (1:500, mouse monoclonal, Santa Cruz), Aβ/APP (6E10, 1:500, mouse monocolonal, Signet), and phospho-tau (AT8, 1:500, Pierce). After 3 washes with PBS, sections were incubated with corresponding fluorescence-conjugated secondary antibodies including Alexa 546-conjugated donkey anti-mouse antibodies (Invitrogen), Alexa 488-conjugated donkey anti-mouse antibodies (Invitrogen), and Alexa 546-conjugated donkey anti-goat antibodies (Invitrogen). Neurons were identified with fluorescence-based NeuroTrace Nissl stain (1:1000, Invitrogen). Sections were examined using an Olympus confocal microscope. Controls for specificity included staining olfactory bulb sections with primary antibodies without fluorescence-conjugated secondary antibodies (background controls), and staining olfactory bulb sections with only secondary antibodies – these controls eliminated auto-fluorescence in each channel and bleed-through (crossover) between channels. Fluorescence intensity was quantified using Image J software.
Immunoblotting: Olfactory bulbs were gently homogenized using a teflon homogenizer (Thomas) in 10 volumes of ice-cold suspension buffer (20 mM HEPES-KOH (pH 7.5), 250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1 mM PMSF, 2 mg/ml aprotinin, 10 mg/ml leupeptin, 5 mg/ml pepstatin, and 12.5 mg/ml of N-acetyl-Leu-Leu-Norleu-Al). Protein concentrations were determined with a Bradford protein assay (Bio-Rad). Proteins (10 μg) were separated by SDS-PAGE (12% gel), and following transfer to polyvinylidene difluoride membranes (Millipore) they were incubated overnight at 4°C with antibodies against synaptophysin (1:1000), N-terminal AβPP (1: 1000), C-terminal AbPP (1: 500, Sigma), apolipoprotein B (1:1000, Santa Cruz) or AT8 (1:1000, Pierce): β-actin (1: 10000, Abcam) was used as a gel loading control. The blots were developed with enhanced chemiluminescence, and bands were visualized and analyzed by LabWorks 4.5 software on a UVP Bioimaging System (Upland). Quantification of results was performed by densitometry and the results were analyzed as total integrated densitometric volume values (arbitrary units).
Statistical analysis: All data were expressed as means and SEMs. Statistical significance was analyzed with one-way ANOVA plus a Tukey post-hoc test. P < 0.05 was considered to be statistically significant.
Results
Caffeine prevented cholesterol-enriched diet-induced neuronal accumulation of apoB: We published previously that cholesterol-enriched diet disrupts the BBB [16]. Thus, elevated levels of plasma cholesterol may enter brain parenchyma and may promote neuronal uptake via receptor-mediated endocytosis. Indeed, we have shown that cholesterol-enriched diet increased neuronal endolysosome accumulation of LDL cholesterol derived from the systemic circulation as evidenced by findings that apoB, the exclusive apolipoprotein in LDL particles that is not normally found in brain [37], is accumulated in neuronal endolysosomes from rabbits fed a cholesterol-enriched diet. In addition, we found that the chronic injection of caffeine prevented cholesterol-enriched diet-induced disruption of the BBB [16]. Here, we determined the extent to which caffeine affected plasma levels of cholesterol and neuronal accumulation of cholesterol of peripheral origin using apoB as a marker. We found that the cholesterol-enriched diet markedly increased plasma cholesterol levels from 64 ± 7.4 to 658 ± 101.5 mg/dl, and that caffeine administration at the two different doses did not affect significantly plasma cholesterol levels in either control rabbits (84 ± 5.0 mg/dl for 0.5 mg/day and 88 ± 12.6 mg/dl for 30 mg/day) or cholesterol-fed rabbits (594 ± 124.6 mg/dl for 0.5 mg/day, and 743 ± 92.4 mg/dl for 30 mg/day). However, caffeine administration did affect cholesterol-enriched diet-induced brain accumulation of cholesterol of peripheral origin. Immunohistochemically, we demonstrated in olfactory bulb glomeruli that the cholesterol-enriched diet increased markedly neuronal accumulation of apoB, which was blocked by caffeine ingestion at both low (0.5 mg/day) and higher (30 mg/day) doses (Figure 1A). Furthermore, we demonstrated in rabbits fed a cholesterol-enriched diet that apoB was accumulated in EEA-1 positive endosomes and LAMP2 positive lysosomes (Figure 1B). Using immunoblotting methods, we quantified the protein levels of apoB in olfactory bulbs and found that the cholesterol-enriched diet increased significantly protein levels of apoB in olfactory bulbs and that this increase was blocked by caffeine ingestion at low (0.5 mg/day) and higher (30 mg/day) doses (Figure 1C). Consistent with our findings that caffeine protects against BBB leakage, these observations indicate that caffeine prevents accumulation in neurons and in brain of cholesterol derived from systemic circulation sources.
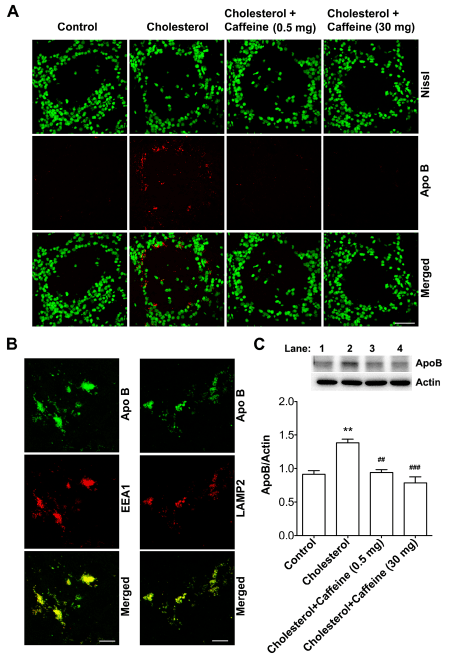
Figure 1. Caffeine prevented cholesterol-enriched diet-induced neuronal accumulation of apoB.
(A) Cholesterol-enriched diet markedly increased immunoreactivity of apoB (red) in neurons (Nissl, green) in olfactory bulb glomeruli, and such an effect was blocked by caffeine at two different doses (0.5 or 30 mg/day). Bar = 50 mm. (B) In olfactory bulbs from cholesterol-fed rabbits, immunoreactivity of apoB co-distributed with endosomes (EEA1) and lysosomes (LAMP2) in the glomerular layer. Bar = 20 mm. (C) Cholesterol-enriched diet increased significantly protein levels of apoB in olfactory bulbs, and such an effect was blocked by caffeine at two different doses (n=4, *p<0.05). Lane 1, Control; Lane 2, Cholesterol; Lane 3, Cholesterol + Caffeine 0.5 mg; Lane 4, Cholesterol + Caffeine 30mg.
Caffeine prevented cholesterol-enriched diet-induced synaptic loss: Synaptic loss is a pathological hallmark of AD. Here, we determined the extent to which caffeine affected synaptic integrity. Consistent with our previous findings, we demonstrated that cholesterol-enriched diet changed markedly the distribution of immunoreactive synaptophysin in glomeruli of olfactory bulb. In control rabbits, the distribution of synpatophysin immunoreactivity was fairly evenly distributed within individual glomeruli. In cholesterol-fed rabbits, the synpatophysin immunoreactivity was markedly decreased in the center of glomeruli. In addition, intense granule-shaped immunoreactivity was observed very close to the cell body of periglomerular cells in cholesterol-fed rabbits (Figure 2A). These distribution changes were almost completely blocked with low (0.5 mg/day) and higher (30 mg/day) doses of caffeine (Figure 2A). In olfactory bulbs from rabbits fed a cholesterol-enriched diet, granule-shaped synaptophysin immunoreactivity was co-distributed with EEA1 and with LAMP 2 (Figure 2B). Furthermore, caffeine at the two different doses prevented significantly cholesterol-enriched diet-induced decreases in protein levels of synpatophysin.
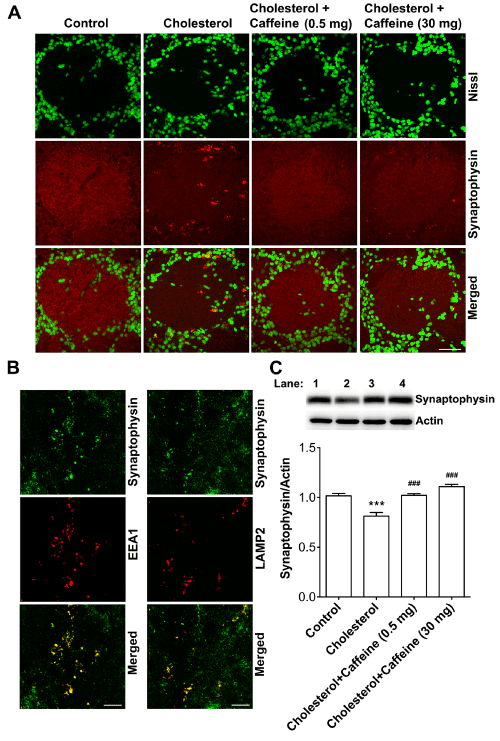
Figure 2. Caffeine prevented cholesterol-enriched diet-induced synaptic loss.
(A) Cholesterol-enriched diet changed markedly the distribution of synaptophysin in glomeruli of olfactory bulbs; the distribution pattern of synaptophysin immunoreactivity changed from an even distribution within individual glomeruli from control rabbits to decreased immunoreactivity in the center of glomeruli from cholesterol-fed rabbits, and intense granule-shaped immunoreactivity was observed very close to the cell body of periglomerular cells in cholesterol-fed rabbits, and such effects were blocked by caffeine at two different doses (0.5 or 30 mg/day). Bar = 50 mm. (B) In olfactory bulbs from cholesterol-fed rabbits, immunoreactivity of synaptophysin co-distributed with endosomes (EEA1) and lysosomes (LAMP2) in glomerular layer. Bar = 20 mm. (C) Caffeine at two different doses prevented significantly cholesterol-enriched diet-induced decreases in protein levels of synaptophysin (n=4, *p<0.05). Lane 1, Control; Lane 2, Cholesterol; Lane 3, Cholesterol + Caffeine 0.5 mg; Lane 4, Cholesterol + Caffeine 30mg.
Caffeine prevented cholesterol-enriched diet-induced brain deposition of Aβ: We reported previously that rabbits fed a cholesterol-enriched diet had increased brain deposition of Aβ, a pathological hallmark of AD [7, 38]. Consistent with those findings, we demonstrated here that the cholesterol diet increased markedly 6E10 positive immunopositive staining of Aβ/AβPP in olfactory bulbs (Figure 3A) and that these increases were blocked by ingestion of low (0.5 mg/day) and higher (30 mg/day) doses of caffeine (Figure 3A). Furthermore, intracellular Ab accumulated in EEA1 positive endosomes and LAMP2 positive lysosomes (Figure 3B). Using immunoblotting methods, we demonstrated that the cholesterol-enriched diet promoted amyloidogenic processing of AβPP (Figure 3C). Cholesterol-enriched diet did not affect significantly protein levels of full-length AβPP or C83 fragment, the product of the non-amyloidogenic β-secretase pathway. However, cholesterol-enriched diet increased significantly protein levels of C99 fragment, the cleavage product of AβPP catalyzed by β-site AβPP cleavage enzyme (BACE-1). Furthermore, the cholesterol-enriched diet increased significantly protein levels of BACE1, the rate-limiting enzyme in amyloidogenic processing of AβPP. Moreover, caffeine ingestion at low (0.5 mg/day) and higer (30 mg/day) doses blocked cholesterol-enriched diet-induced increases in protein levels of C93 fragment of AβPP and BACE-1 (Figure 3C).
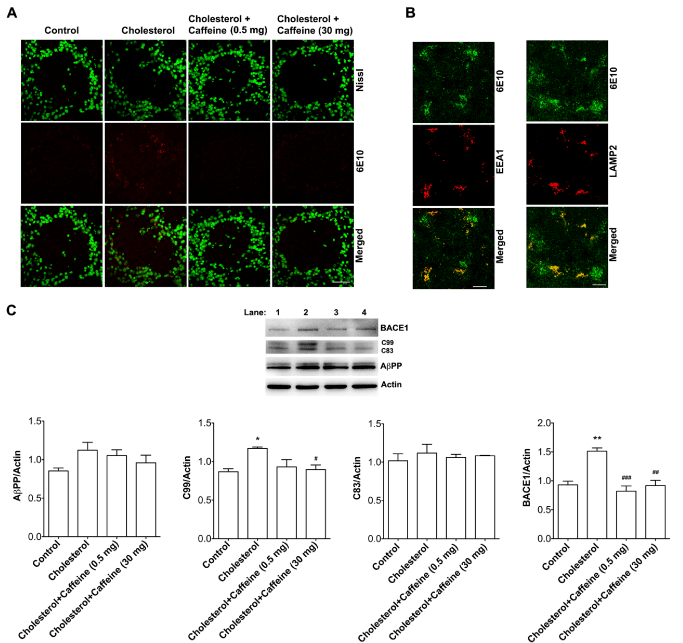
Figure 3. Caffeine prevented cholesterol-enriched diet-induced brain deposition of Aβ/APP.
(A) Cholesterol-enriched diet increased markedly 6E10 positive immunopositive staining of Ab/AbPP in olfactory bulbs, and such effects were blocked by caffeine at two different doses (0.5 or 30 mg/day). Bar = 50 mm. (B) In olfactory bulbs from cholesterol-fed rabbits, intracellular Ab/AbPP immunoreactivity co-distributed with EEA1 positive endosomes and LAMP2 positive lysosomes. Bar = 20 mm. (C) Cholesterol-enriched diet did not affect significantly protein levels of full-length AbPP or C83 fragment, but increased significantly protein levels of C99 fragment of AbPP and BACE1, and such effects were blocked by caffeine at two different doses (n=4, *p<0.05). Lane 1, Control; Lane 2, Cholesterol; Lane 3, Cholesterol + Caffeine 0.5 mg; Lane 4, Cholesterol + Caffeine 30mg.
Caffeine prevented cholesterol-enriched diet-induced tau phosphorylation
Neurofibrillary tangles composed of phosphorylated tau are another pathological hallmark of AD and we showed previously that cholesterol-enriched diet increased tau phosphorylation in neurons [7,38]. Here, we determined the extent to which caffeine affects tau phosphorylation in cholesterol-fed rabbits. Consistent with our previous findings, we demonsrated immunohistochemically that cholesterol-enriched diet increased neuronal deposition of phosphorylated tau (Figure 4A) and that these increases were blocked by ingestion of low (0.5 mg/day) or higer (30 mg/day) doses of caffeine (Figure 4A). In addition, we demonstrated in olfactory bulbs from cholesterol-fed rabbits increased accumulation of phosphorylated tau in EEA1 positive endosomes and LAMP2 positive lysosomes (Figure 4B). Furthermore, we found that caffeine ingestion at low (0.5 mg/day) and higer (30 mg/day) doses blocked cholesterol-enriched diet-induced increases in tau phosphorylation (Figure 4C).
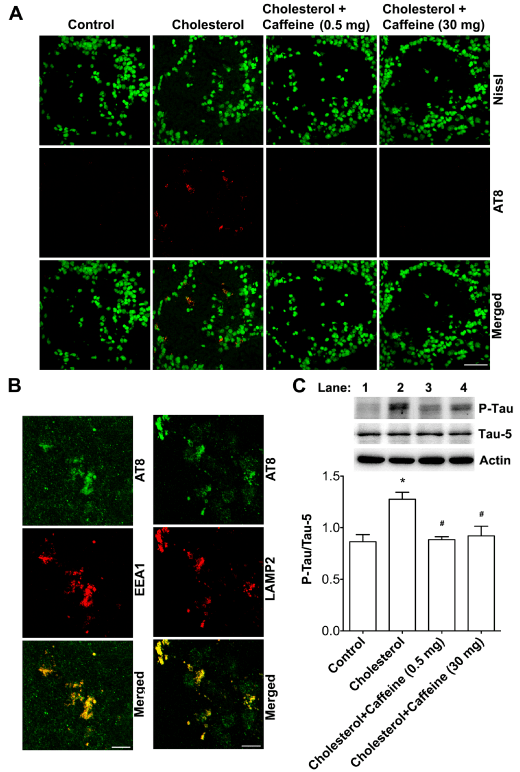
Figure 4. Caffeine prevented cholesterol-enriched diet-induced tau phosphorylation.
(A) Cholesterol-enriched diet increased markedly AT8 positive immunopositive staining of phosphorylated tau in olfactory bulbs and such effects were blocked by caffeine at two different doses (0.5 or 30 mg/day). Bar = 50 mm. (B) In olfactory bulbs from cholesterol-fed rabbits, immunoreactivity of AT8 co-distributed with EEA1 positive endosomes and LAMP2 positive lysosomes. Bar = 20 mm. (C) Cholesterol-enriched diet increased significantly protein levels of phosphorylated tau in olfactory bulbs, and such an effect was blocked by caffeine at two different doses (n=4, *p<0.05). Lane 1, Control; Lane 2, Cholesterol; Lane 3, Cholesterol + Caffeine 0.5 mg; Lane 4, Cholesterol + Caffeine 30mg.
Discussion
Consistent with findings from epidemiological and experimental studies that caffeine is protective against AD [29,34,39], we demonstrated here that chronic ingestion of caffeine at even very low doses exerts protective effects against the development of pathological hallmarks of AD, including synaptic loss, brain deposition of Aβ, and tau pathology, in a cholesterol-fed rabbit model of sporadic AD.
The pathogenesis of sporadic AD is believed to result from complex interactions between nutritional, environmental, epigenetic and genetic factors [3]. Among those factors, elevated levels of circulating LDL cholesterol, independent of APOE genotypes, is strongly linked to the pathogenesis of sporadic AD [6-10]. Of mechanistic importance, we have shown that elevated levels of LDL cholesterol, the essential lipoprotein transporting circulating cholesterol in blood, (1) induces blood-brain barrier (BBB) leakage [7,16] - an early pathological feature of sporadic AD that precedes brain deposition of Aβ [17], (2) disturbs neuronal endolysosome structure and function - another early pathological features of sporadic AD [18], and (3) promotes the development of pathological hallmarks of AD including disrupted synaptic integrity, brain deposition of Aβ, and tau pathology [7]. Furthermore, we have demonstrated in cultured neurons that treatment with LDL cholesterol increased cholesterol accumulation in endolysosomes, disturbed the structure and function of endolysosomes, promoted amyloidogenic processing of AβPP, increased levels of phosphorylated tau, and decreased levels of the presynaptic protein synaptophysin [8].
Oflactory dysfunction has been deemed to be the earliest symptom of AD [40,41]. Deficits in ordor identification have been shown to increase the likelihood of subsequent cognitive decline, especially in patients carring one or two copies of the β4 allele of ApoE [40,42,43], the major genetic risk factor of sporadic AD. Thus, altered cholesterol homeotasis could contribute to the development of olfactory dysfunction in sporadic AD. Pathologically, oxidative damage, the presence of Aβ plaque, and accumulation of phosphorylated tau as neurofilrillary tangles all have been documented in olfactory epithelium and olfactory bulbs of AD patients [44-51]. Previously we showed in olfactory bulb from cholesterol-fed rabbits that elevated levels of circulating cholesterol disrupted the blood-brain barrier [16], increased accumulations of cholesterol in endolysosomes of neurons in olfactory bulb originated from peripheral sources, and contributed to the development of AD-like pathology including synaptic loss, elevated Aβ production, and increased tau phosphorylation [7].
Consistent with our previous findings, here we demonstrated in olfactory bulbs that cholesterol-enriched diet increased brain levels of apoB containing LDL cholesterol and promoted the accumulation of apoB, synpatophysin, Aβ, and phosphorylated-tau in endolysosomes. Collectively, our findings suggest that elevated levels of LDL cholesterol, when it enters brain parenchyma via a leaky BBB, are internalized by neurons via receptor-mediated endocytosis. Because LDL cholesterol is not normally present in brain and is foreign to neurons, the accumulation of LDL cholesterol in neuronal endolysosomes may induce a “traffic jam” in intracellular cholesterol transport, thereby disturbing endolysosome structure and function. Such LDL cholesterol-induced endolysosome dysfunction could contribute directly to the development of pathological hallmarks of AD including brain deposition of Aβ, synaptic loss, and tau pathology [8]. In support, (1) endolysosomes are the major site, where Aβ is produced [52,53], and endolysosome dysfunction leads to brain deposition of Aβ [17]; (2) Endolysosomes are responsible for synaptic protein recycling [54-56], and endolysosome dysfunction leads to disruption of synaptic integrity in brain [57-61]; (3) Tau can be degraded by cathepsin D in autophagosomes-lysosomes [62-65] and increased levels of endolysosome cholesterol, as occurs in Niemann-Pick type C disease, leads to lysosome dysfunction and tau-pathology [66-71].
Substantial evidence from human epidemiological studies and from experimental studies conducted in animals and cultured cell models indicate that caffeine, when ingested chronically, can decrease Aβ levels, protect against the onset and severity of AD, and in some cases it can reverse behavioral and pathological features of AD [19,21,24,29]. Less clear, however, are the mechanisms by which caffeine exhibits these protective features. Previously, we have shown, in the cholesterol-fed rabbit model of sporadic AD, that chronic ingestion of caffeine protects against the disruption of the BBB [16]. As such caffeine may prevent the entrance of the foreign LDL cholesterol into brain parenchyma, thus protecting against subsequent LDL cholesterol-induced endolysosome dysfunction as well as the development of pathological hallmarks of AD. In support, we demonstrated that caffeine ingestion at two different doses did not affect plasma levels of cholesterol in either control rabbits or cholesterol-fed rabbits, but blocked cholesterol-enriched diet-induced elevation of brain levels of apoB, the key apolipoprotein of LDL particles. Importantly, we demonstrated that chronic ingestion of caffeine prevented cholesterol-enriched diet induced synaptic disruption, amyloidogenic processing of AβPP, and tau-pathology. In addition to its protective effects on the BBB, we have shown in neurons that caffeine inhibits LDL cholesterol internalization. As such caffeine could prevent abnormal accumulation of LDL cholesterol in neuronal endolysosomes, thus preventing LDL cholesterol-induced endolysosome dysfunction and subsequent development of AD-like pathology. Indeed, we demonstrated that caffeine blocked cholesterol-enriched diet-induced abnormal accumulation of synatophysin, Aβ, and phosphorylated tau in endolysosomes.
Together, our findings suggest that caffeine exerts its protective effects against the development of sporadic AD, in part, by preventing against entrance of LDL cholesterol into brain parenchyma and the accumulation of LDL-cholesterol in neuronal endolysosomes. Further elucidation of the involved mechanism may provide insight into the pathogenesis of sporadic AD and may lead to new effective therapeutic strategies against this devastating neurodegenerative disease.
Acknowledgment
Supported by P30GM103329, R01NS065957 and R21AG043338.
References
2021 Copyright OAT. All rights reserv
- Goate A, Hardy J (2012) Twenty years of Alzheimer's disease-causing mutations. J Neurochem 120 Suppl 1: 3-8. [Crossref]
- Holtzman DM, Morris JC, Goate AM (2011) Alzheimer's disease: the challenge of the second century. Sci Transl Med 3: 77sr1. [Crossref]
- Reitz C, Brayne C, Mayeux R (2011) Epidemiology of Alzheimer disease. Nat Rev Neurol 7: 137-152. [Crossref]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921-923. [Crossref]
- Marzolo MP, Bu G (2009) Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer's disease. Semin Cell Dev Biol 20: 191-200. [Crossref]
- Solomon A, Kivipelto M, Wolozin B, Zhou J, Whitmer RA (2009) Midlife serum cholesterol and increased risk of Alzheimer's and vascular dementia three decades later. Dement Geriatr Cogn Disord 28: 75-80. [Crossref]
- Chen X, Wagener JF, Morgan DH, Hui L, Ghribi O, et al. (2010) Endolysosome mechanisms associated with Alzheimer's disease-like pathology in rabbits ingesting cholesterol-enriched diet. J Alzheimers Dis 22: 1289-1303. [Crossref]
- Hui L, Chen X, Geiger JD (2012) Endolysosome involvement in LDL cholesterol-induced Alzheimer's disease-like pathology in primary cultured neurons. Life Sci 91: 1159-1168. [Crossref]
- Reed B, Villeneuve S2, Mack W3, DeCarli C, Chui HC3, et al. (2014) Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol 71: 195-200. [Crossref]
- Matsuzaki T, Sasaki K, Hata J, Hirakawa Y, Fujimi K, et al. (2011) Association of Alzheimer disease pathology with abnormal lipid metabolism: the Hisayama Study. Neurology 77: 1068-1075. [Crossref]
- Barbero-Camps E, Fernández A, Martínez L, Fernández-Checa JC, Colell A (2013) APP/PS1 mice overexpressing SREBP-2 exhibit combined Abeta accumulation and tau pathology underlying Alzheimer's disease. Hum Mol Genet 22: 3460-3476. [Crossref]
- Thirumangalakudi L, Prakasam A, Zhang R, Bimonte-Nelson H, Sambamurti K, et al. (2008) High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J Neurochem 106: 475-485. [Crossref]
- Sharman MJ, Moussavi Nik SH, Chen MM, Ong D, Wijaya L, et al. (2013) The Guinea Pig as a Model for Sporadic Alzheimer's Disease (AD): The Impact of Cholesterol Intake on Expression of AD-Related Genes. PLoS One 8: e66235. [Crossref]
- Sparks DL, Scheff SW, Hunsaker JC 3rd, Liu H, Landers T, et al. (1994) Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol 126: 88-94. [Crossref]
- Ullrich C, Pirchl M, Humpel C (2010) Hypercholesterolemia in rats impairs the cholinergic system and leads to memory deficits. Mol Cell Neurosci 45: 408-417. [Crossref]
- Chen X, Gawryluk JW, Wagener JF, Ghribi O, Geiger JD (2008) Caffeine blocks disruption of blood brain barrier in a rabbit model of Alzheimer's disease. J Neuroinflammation 5: 12. [Crossref]
- Ujiie M, Dickstein DL, Carlow DA, Jefferies WA (2003) Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 10: 463-470. [Crossref]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, et al. (2000) Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 157: 277-286. [Crossref]
- Ritchie K, Carrière I, de Mendonca A, Portet F, Dartigues JF, et al. (2007) The neuroprotective effects of caffeine: a prospective population study (the Three City Study). Neurology 69: 536-545. [Crossref]
- Santos C, Costa J, Santos J, Vaz-Carneiro A, Lunet N (2010) Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis 20 Suppl 1: S187-204. [Crossref]
- Santos C, Lunet N, Azevedo A, de Mendonça A, Ritchie K, et al. (2010) Caffeine intake is associated with a lower risk of cognitive decline: a cohort study from Portugal. J Alzheimers Dis 20 Suppl 1: S175-185. [Crossref]
- Gelber RP, Petrovitch H, Masaki KH, Ross GW, White LR (2011) Coffee intake in midlife and risk of dementia and its neuropathologic correlates. J Alzheimers Dis 23: 607-615. [Crossref]
- Eskelinen MH, Ngandu T, Tuomilehto J, Soininen H, Kivipelto M (2009) Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis 16: 85-91. [Crossref]
- Arendash GW, Mori T, Cao C, Mamcarz M, Runfeldt M, et al. (2009) Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer's disease mice. J Alzheimers Dis 17: 661-680. [Crossref]
- Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, et al. (2006) Caffeine protects Alzheimer's mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 142: 941-952. [Crossref]
- Laurent C, Eddarkaoui S, Derisbourg M, Leboucher A, Demeyer D, et al. (2014) Beneficial effects of caffeine in a transgenic model of Alzheimer's disease-like tau pathology. Neurobiol Aging 35: 2079-2090. [Crossref]
- Han K, Jia N, Li J, Yang L, Min LQ (2013) Chronic caffeine treatment reverses memory impairment and the expression of brain BNDF and TrkB in the PS1/APP double transgenic mouse model of Alzheimer's disease. Mol Med Rep 8: 737-740. [Crossref]
- Espinosa J, Rocha A, Nunes F, Costa MS, Schein V, et al. (2013) Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J Alzheimers Dis 34: 509-518. [Crossref]
- Cao C, Cirrito JR, Lin X, Wang L, Verges DK, et al. (2009) Caffeine suppresses amyloid-beta levels in plasma and brain of Alzheimer's disease transgenic mice. J Alzheimers Dis 17: 681-697. [Crossref]
- Arendash GW, Cao C (2010) Caffeine and coffee as therapeutics against Alzheimer's disease. J Alzheimers Dis 20 Suppl 1: S117-126. [Crossref]
- Arendash GW, Mori T, Cao C, Mamcarz M, Runfeldt M, et al. (2009) Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer's disease mice. J Alzheimers Dis 17: 661-680. [Crossref]
- Wostyn P, Van Dam D, Audenaert K, De Deyn PP (2011) Increased Cerebrospinal Fluid Production as a Possible Mechanism Underlying Caffeine's Protective Effect against Alzheimer's Disease. Int J Alzheimers Dis 2011: 617420. [Crossref]
- Eskelinen MH, Kivipelto M (2010) Caffeine as a protective factor in dementia and Alzheimer's disease. J Alzheimers Dis 20 Suppl 1: S167-174. [Crossref]
- Flaten V, Laurent C, Coelho JE, Sandau U2, Batalha VL, et al. (2014) From epidemiology to pathophysiology: what about caffeine in Alzheimer's disease? Biochem Soc Trans 42: 587-592. [Crossref]
- Chen X, Ghribi O, Geiger JD (2010) Caffeine protects against disruptions of the blood-brain barrier in animal models of Alzheimer's and Parkinson's diseases. J Alzheimers Dis 20 Suppl 1: S127-141. [Crossref]
- Prasanthi JR, Dasari B, Marwarha G, Larson T, Chen X, et al. (2010) Caffeine protects against oxidative stress and Alzheimer's disease-like pathology in rabbit hippocampus induced by cholesterol-enriched diet. Free Radic Biol Med 49: 1212-1220. [Crossref]
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH (1987) Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem 262: 14352-14360. [Crossref]
- Ghribi O, Larsen B, Schrag M, Herman MM (2006) High cholesterol content in neurons increases BACE, beta-amyloid, and phosphorylated tau levels in rabbit hippocampus. Exp Neurol 200: 460-467. [Crossref]
- Cao C, Loewenstein DA, Lin X, Zhang C, Wang L, et al. (2012) High Blood caffeine levels in MCI linked to lack of progression to dementia. J Alzheimers Dis 30: 559-572. [Crossref]
- Mesholam RI, Moberg PJ, Mahr RN, Doty RL (1998) Olfaction in neurodegenerative disease: a meta-analysis of olfactory functioning in Alzheimer's and Parkinson's diseases. Arch Neurol 55: 84-90. [Crossref]
- Peters JM, Hummel T, Kratzsch T, Lotsch J, Skarke C, et al. (2003) Olfactory function in mild cognitive impairment and Alzheimer's disease: an investigation using psychophysical and electrophysiological techniques. Am J Psychiatry 160: 1995-2002. [Crossref]
- Graves AB, Bowen JD, Rajaram L, McCormick WC, McCurry SM, et al. (1999) Impaired olfaction as a marker for cognitive decline: interaction with apolipoprotein E epsilon4 status. Neurology 53: 1480-1487. [Crossref]
- Gilbert PE, Murphy C (2004) The effect of the ApoE epsilon4 allele on recognition memory for olfactory and visual stimuli in patients with pathologically confirmed Alzheimer's disease, probable Alzheimer's disease, and healthy elderly controls. J Clin Exp Neuropsychol 26: 779-794. [Crossref]
- Perry G, Castellani RJ, Smith MA, Harris PL, Kubat Z, et al. (2003) Oxidative damage in the olfactory system in Alzheimer's disease. Acta Neuropathol 106: 552-556. [Crossref]
- Wesson DW, Wilson DA, Nixon RA (2010) Should olfactory dysfunction be used as a biomarker of Alzheimer's disease? Expert Rev Neurother 10: 633-635. [Crossref]
- Arnold SE, Lee EB, Moberg PJ, Stutzbach L, Kazi H, et al. (2010) Olfactory epithelium amyloid-beta and paired helical filament-tau pathology in Alzheimer disease. Ann Neurol 67: 462-469. [Crossref]
- Kovács T, Cairns NJ, Lantos PL (1999) beta-amyloid deposition and neurofibrillary tangle formation in the olfactory bulb in ageing and Alzheimer's disease. Neuropathol Appl Neurobiol 25: 481-491. [Crossref]
- Kovács T, Cairns NJ, Lantos PL (2001) Olfactory centres in Alzheimer's disease: olfactory bulb is involved in early Braak's stages. Neuroreport 12: 285-288. [Crossref]
- Thomann PA, Dos Santos V, Seidl U, Toro P, Essig M, et al. (2009) MRI-derived atrophy of the olfactory bulb and tract in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis 17: 213-221. [Crossref]
- Thomann PA, Dos Santos V, Toro P, Schönknecht P, Essig M, et al. (2009) Reduced olfactory bulb and tract volume in early Alzheimer's disease--a MRI study. Neurobiol Aging 30: 838-841. [Crossref]
- Fujishiro H, Tsuboi Y, Lin WL, Uchikado H, Dickson DW (2008) Co-localization of tau and alpha-synuclein in the olfactory bulb in Alzheimer's disease with amygdala Lewy bodies. Acta Neuropathol 116: 17-24. [Crossref]
- Rajendran L, Annaert W (2012) Membrane trafficking pathways in Alzheimer's disease. Traffic 13: 759-770. [Crossref]
- Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, et al. (2013) Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun 4: 2250. [Crossref]
- Murthy VN, Stevens CF (1998) Synaptic vesicles retain their identity through the endocytic cycle. Nature 392: 497-501. [Crossref]
- Kuromi H and Kidokoro Y (1998) Two distinct pools of synaptic vesicles in single presynaptic boutons in a temperature-sensitive Drosophila mutant, shibire. Neuron 20: 917-925. [Crossref]
- Blumstein J, Faundez V, Nakatsu F, Saito T, Ohno H, et al. (2001) The neuronal form of adaptor protein-3 is required for synaptic vesicle formation from endosomes. J Neurosci 21: 8034-8042. [Crossref]
- Callahan LM, Vaules WA, Coleman PD (1999) Quantitative decrease in synaptophysin message expression and increase in cathepsin D message expression in Alzheimer disease neurons containing neurofibrillary tangles. J Neuropathol Exp Neurol 58: 275-287. [Crossref]
- Bahr BA, Bendiske J (2002) The neuropathogenic contributions of lysosomal dysfunction. J Neurochem 83: 481-489. [Crossref]
- Bendiske J and Bahr BA (2003) Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis--an approach for slowing Alzheimer disease? J Neuropathol Exp Neurol,. 62: 451-463. [Crossref]
- Bendiske J, Caba E, Brown QB, Bahr BA (2002) Intracellular deposition, microtubule destabilization, and transport failure: an "early" pathogenic cascade leading to synaptic decline. J Neuropathol Exp Neurol 61: 640-650. [Crossref]
- Kanju PM, Parameshwaran K, Vaithianathan T, Sims CM, Huggins K, et al. (2007) Lysosomal dysfunction produces distinct alterations in synaptic alpha-amino-3-hydroxy-5-methylisoxazolepropionic acid and N-methyl-D-aspartate receptor currents in hippocampus. J Neuropathol Exp Neurol 66: 779-788. [Crossref]
- Oyama F, Murakami N, Ihara Y (1998) Chloroquine myopathy suggests that tau is degraded in lysosomes: implication for the formation of paired helical filaments in Alzheimer's disease. Neurosci Res 31: 1-8. [Crossref]
- Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, et al. (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18: 4153-4170. [Crossref]
- Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, et al. (2008) Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci 27: 1119-1130. [Crossref]
- Kenessey A, Nacharaju P, Ko LW, Yen SH (1997) Degradation of tau by lysosomal enzyme cathepsin D: implication for Alzheimer neurofibrillary degeneration. J Neurochem 69: 2026-2038. [Crossref]
- Distl R, Treiber-Held S, Albert F, Meske V, Harzer K, et al. (2003) Cholesterol storage and tau pathology in Niemann-Pick type C disease in the brain. J Pathol 200: 104-111. [Crossref]
- Bi X, Liao G (2007) Autophagic-lysosomal dysfunction and neurodegeneration in Niemann-Pick Type C mice: lipid starvation or indigestion? Autophagy 3: 646-648. [Crossref]
- Liao G, Yao Y, Liu J, Yu Z, Cheung S, et al. (2007) Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am J Pathol 171: 962-975. [Crossref]
- Vance JE (2006) Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett 580: 5518-5524. [Crossref]
- Bu B, Li J, Davies P, Vincent I (2002) Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci 22: 6515-6525. [Crossref]
- Sawamura N, Gong JS, Garver WS, Heidenreich RA, Ninomiya H, et al. (2001) Site-specific phosphorylation of tau accompanied by activation of mitogen-activated protein kinase (MAPK) in brains of Niemann-Pick type C mice. J Biol Chem 276: 10314-10319. [Crossref]