The rich repertoire of cell death events is a crucial biological process that deserves extensive review at a formative time for the development of our knowledge concerning the state-of-the-art data on their cellular and molecular mechanisms of action and the ways in which they can be manipulated in and by protozoan parasites. We have attempted to provide a comprehensive overview to reveal intervention points that could be exploited to discover novel therapies, vaccine strategies and prophylactic intervention points for protozoan parasites, and to understand the parasitic disease progression and the infection consequence.
cell death, protozoan parasites, novel therapies, vaccine strategies and prophylactic intervention points
The facing of division (mitosis), specialization (differentiation) or cell death are three choices for any cell. To retain the functional equilibrium in the number of host cells, the balance between these processes is needed. The term cell death refers to several different phenomena, such as necrosis and programmed cell death (PCD). The former is disorganized way to die that provokes a strong inflammatory response, albeit the latter represents a tightly coordinated and regulated process with marked cellular pathways. Furthermore, the PCD is mainly classed into apoptosis and autophagy. The autophagy - apoptosis - necrosis continuum depends on parasite insult, response stringency and type, and cellular constituent and signaling pathway influences. Interestingly, autophagic (autophagosomes), apoptotic-like (membrane blebbing) phenotypes, and necrosis have been reported in Naphthoimidazoles-treated Trypanosoma cruzi [1]. It has been demonstrated that an intense cross-talking between autophagy, apoptosis, and necrosis in the protozoa in response to diverse drug stimuli that cover the same pathway of the previous processes [2]. Intriguingly, in the light of the fact that apoptosis and autophagy are interconnected, a significant analysis and discussion on the proteins with a dual role in autophagy and apoptosis were presented by Li et al. and Tait et al. [3,4]. Furthermore, several recent studies have reported the continuity of apoptosis and necrosis stemming from the diversion of the core apoptotic pathway by intracellular ATP and caspase depletion [5-7]. In line with these observations, current reports provide compelling evidence of the intricate interplay between apoptotic, necrotic and autophagic cell deaths [8,9]. The morphological features and the interconnection of these types are diagrammed below (Figure 1).
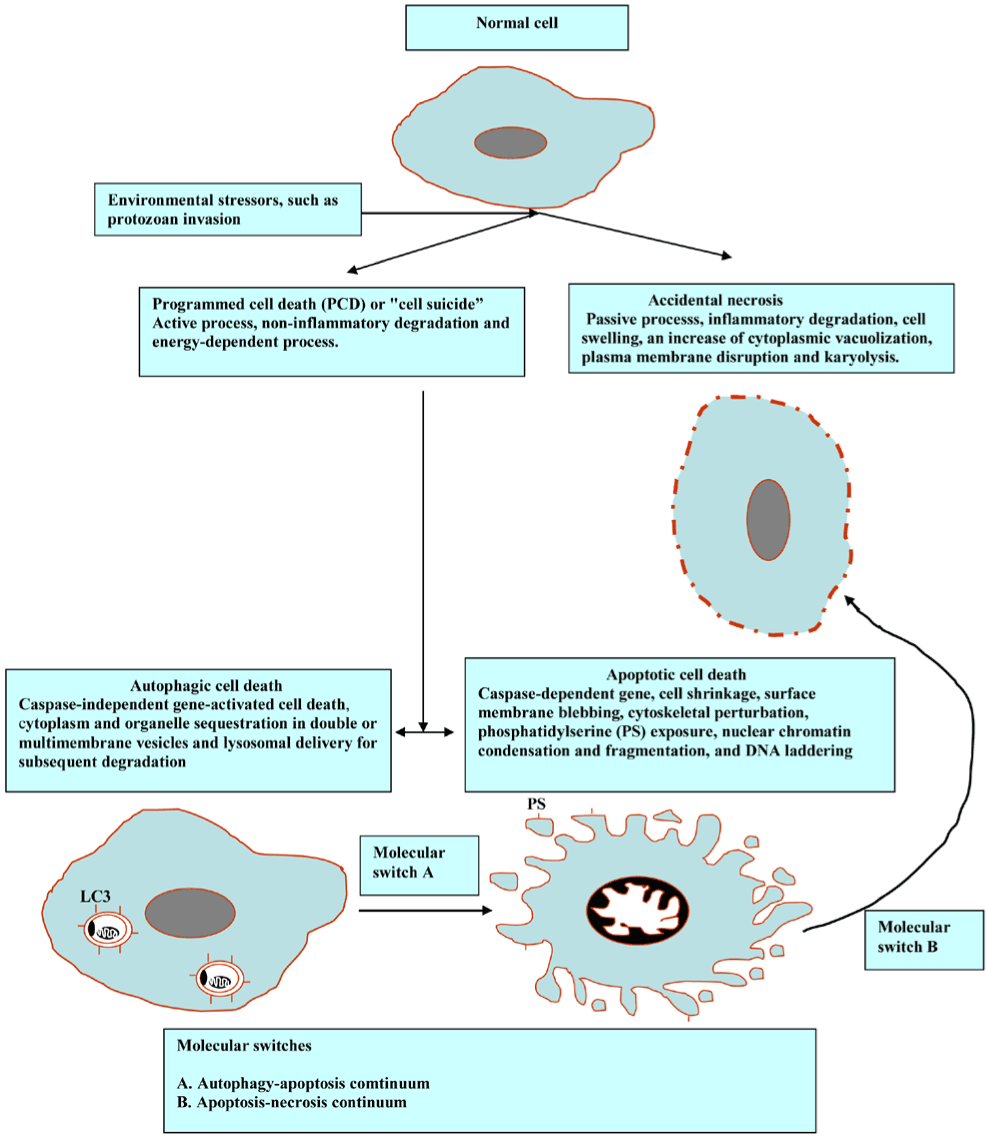
Figure 1. Morphological features and cross-talks among the main types of PCD.
Diverse pathologies, for example, cancer, autoimmunity, neurodegeneration and injury can result from too much or too little cell death. The diversified morphological feautres of cell death may be attributed, in part, to distinct mechanical/immunological, biochemical and molecular events. Programmed cell death (PCD) serves as an integral part of host-pathogen interactions since microorganisms, for example, protozoan can hijack the genetically controlled cell suicide programs of their host cells, triggering, delaying or even preventing it. Clearly, understanding how parasites and host cells deal with cell death might allow us to prophylactically and therapeutically manipulate them in various processes, including protozon parasites.
Apoptosis “self-killing”
The main features of apoptosis are the plasma membrane blebbing, nucleus fragmentation, release of cytochrome c from the mitochondria into the cytosol and the exposure of phosphatidyl serine on the cell surface (attack me/eat-me signal) to trigger phagocytosis and anti-inflammatory response (Figure 2).
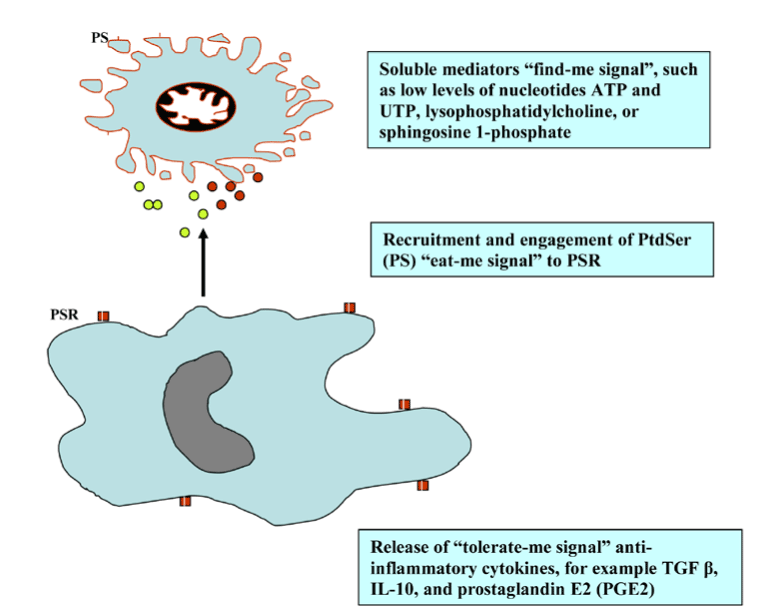
Figure 2. Find-, eat-, and tolerate- me signals.
Apoptosis is required for normal development, such as metamorphosis in tadpole and endometrial sloughing off during menstrual cycle and also; it is required to eliminate cells that represent a threat to the organism integrity and during immunocmpetency in the primary lymphoid organs. The outcome of balance between the absence of IL-2 mediated cell proliferation and the presence of internal or external death stimulators leads to apoptosis and the demise of the cells. Furthermore, apoptosis is essential to control the development of diseases like intracellular pathogen and cancer, whereas excessive PCD is an overriding element in neuro-degenerative diseases when deregulated in neurons (Figure 3).
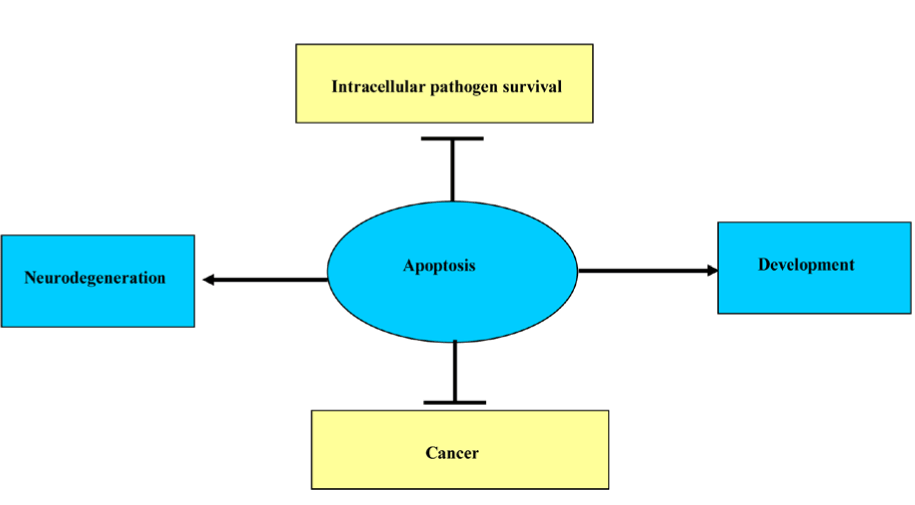
Figure 3. Different effects of apoptosis.
There are different types of general signals that initate apoptosis, including intracellular stress signals resulting in mitochondrial membrane permeabilization, extracellular ligands integrating to death receptors, such as stimulation of the Fas receptor or the TNF receptor (TNFR), and apoptotic stimuli, such as perforin and granzymes. There is a cross talk between the mitochondria pathways and death receptor mediated; for example, by caspase-8 cleavage of the protein Bid and cell death regulator Smac/DIABLO (second mitochondria derived activator of caspases / direct IAP Binding protein with low PI). During the apoptosis, the mitochondrial Smac/DIABLO inhibits the inhibitors of apoptosis proteins (IAP), thereby breaking the brake on apoptosis. For further information, we accredit the reader to the review articles [10,11]
The internal death stimulators “mitochondrial pathway”:
As in the case reactive oxygen species (ROS), in the literature, the term tends to be used to refer to molecular oxygen-derived reactive molecules and free radicals, including superoxide; hydrogen peroxide; hydroxyl radical; hydroxyl ion; and nitric oxide. Bak and Bax, which are Bcl-2 family members, play a crucial role in mitochondrial outer membrane permeabilisation (MOMP) [12]. Mitochondrial reactive oxygen species (mROS) has been demonstrated to play a role in diverse cellular processes, including differentiation, autophagy, metabolic adaptation, and immune cell activation in response to various stimuli; for example, immunoreceptor ligation, cytokine stimulation, and hypoxia [13]. mROS causes internal damage and mitochondrial cytochrome c (Cyto c) is released to the cytosol. The later binds to apoptotic protease activating factors-I (Apaf-1) to form apoptosome, which mobilizes the initiator caspases, such as procaspase 9 (inactive form) to caspase 9 (active form) resulting in the proteolytic activation of the executioner caspases, such as caspase 3. The final outcome is the degradation of the inhibitor of the caspase activating DNAse (I-CAD) by caspase 3, which in turn leads to nuclear DNA degradation by caspase activating DNase [14-18]. These contribute to the hallmark changes in apoptotic cells, including phosphatidyl serine (PS) residue externalization.
The external death stimulators “death receptor pathway”:
The extracellular ligands, such as Tumour necrosis factor (TNF-α) and Lymphotoxin (TNF-β), and Fas ligand (FasL) engage the intergral membrane proteins; TNF and Fas (CD95) receptors respectively. The adaptor proteins, Fas-associated death domain (FADD) and TNFR1-associated death domain (TRADD) engage caspases-8 and -10 to construct the death inducing signalling complex (DISC) at the active receptor [19]. Thereby, the downstream activation of caspase 3, which in turn accelerates DNA fragmentation through a cascade of reactions (Figure 4).
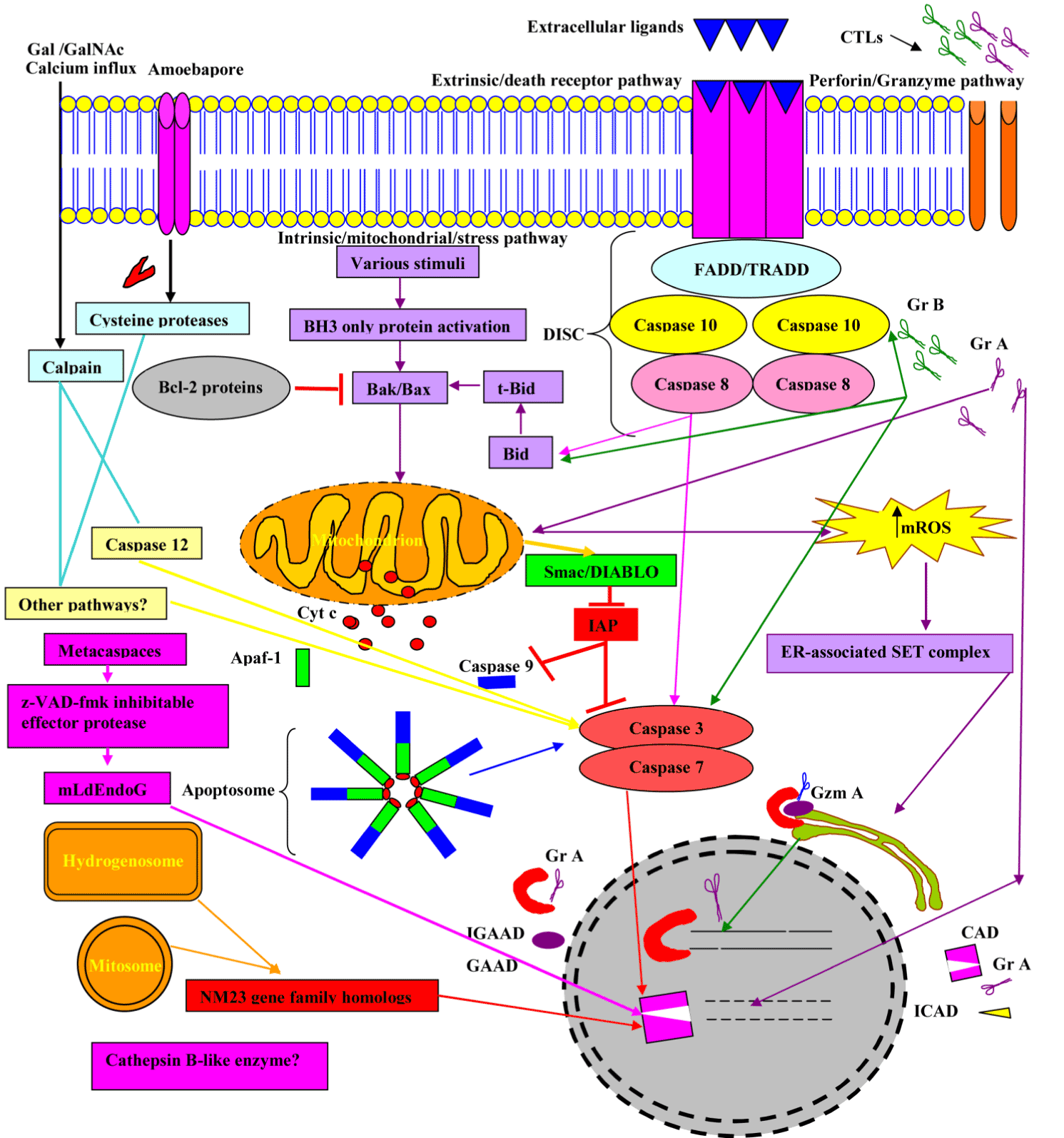
Figure 4. Overview of major molecular events involved in apoptosis.
Abbreviations: FasL, Fas Ligand; DISC, death inducing signalling complex; FADD, Fas-associated via death domain, TRADD, TNFR1-associated death domain; Cyto c, Cytochrome c; ROS, reactive oxygen species; Apaf-1, apoptotic protease activating factor-1; CAD, caspase activating DNAse, also known as DFF40; ICAD, inhibitor caspase activating DNAse; GAAD, granzyme A-activated DNase, for example NM23-H1; IGAAD, inhibitor caspase activating DNAse; GrA, Granzyme A; GrB, Granzyme B and mROS, mitochondrial reactive oxgen species
Organelles of mitochondrial origin
Hydrogenosomes and Mitosomes are double membrane-bounded organelles of mitochondrial origin that are found in a phylogenetically broad range of protists, such as diplomonad and trichomonad species. Hydrogenosomal component evolutionary analyses suggested that G. intestinalis mitosomes have evolved from hydrogenosomes, which predated parabasalid and diplomonad split split [20]. The hydrogenosomes produce molecular hydrogen, H 2 and ATP; however, mitosomes (also named crypton or relictual mitochondria) are not involved in ATP production (Figure 5).
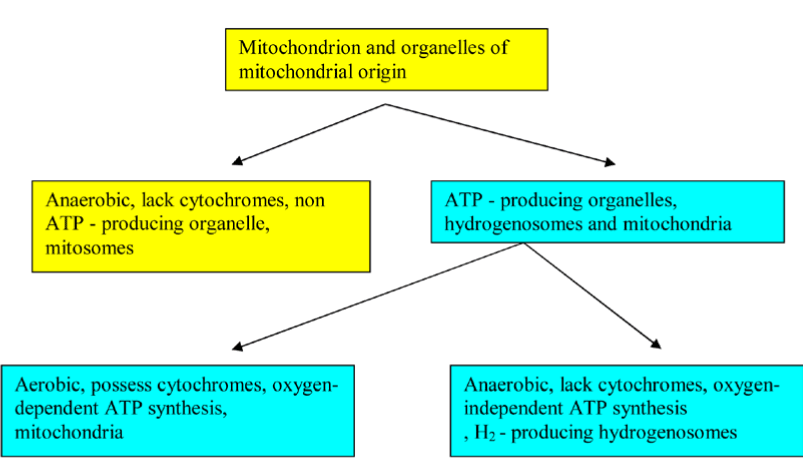
Figure 5. A Flowchart illustrates the functional classification of mitochondrion and organelles of mitochondrial origin.
Strikingly, E. histolytica mitosomes possess a compartmentalized sulfate activation pathway, which plays an important role in the parasite proliferation and in amebic stage conversion [21,22]. The distinction between mitochondria and organelles of mitochondrial origin no longer lies in the existence of a compartmentalized cell plan (i.e rudimentary/residual versus well-developed organelle), but rather in its complexity. Mitochondria of anaerobic eukaryotes, for example, hydrogenosomes and mitosomes are found in a wide variety of anaerobic/micro-aerophilic eukaryotes, such as Entamoeba histolytica, Giardia intestinalis, Trichomonas vaginalis, and Cryptosporidium parvum. They are oxygen-deprived or oxygen-poor environment dwellers. Such environmental constraints in the mammalian intestine and the genital organs drive the evolution of eukaryotes and their organelles [23]. Recent evidence suggests that T. vaginalis possesses osmotically inducible protein (OsmC) homologues in its hydrogenosomes that are sensitive to oxidative damage formed during host-parasite interactions [24]. Comparative bioinformatics examinations in Trichomonas vagina and Giardia intestinalis have shown the existence of the nuclease, NM23 gene family homologs, which has been implicated in DNA fragmentation. The in-depth transcriptomic signatures in Tetracycline (TET) treated-T. vaginalis showed the involvement of hydrogenosome in programmed cell death [25]. In response to beta-Lapachone treatment and starvation, Giardia lamblia presented some autophagic-like (myelinic figures in large vacuoles and LC-3 staining) and apoptotic-like (Cell shrinkage, chromatin condensation, membrane blebbing and vacuolization) programmed cell death characteristics suggesting the possible role of mitosomes, a relic organelle in these processes [26]. Bax, a member of the Bcl-2 gene family, induces permeabilization of the mitochondrial outer membrane resulting in release of cytochrome c. In the early-diverged Giardia lamblia, off-target membrane permeabilization of encystation-specific vesicles (ESVs) by Bax leads to cell death completely independently of mitochondrial relic, mitosome [27]. Functional and evolutionary studies of mitosomes, hydrogenosome, and further organelles of mitochondrial origin of metamonads and other parasitic protozoa are needed to evaluate the evolutionary role of these organelles in PCD and to reveal the full spectrum of different pathways that are involved in PCD. Pursuit of all approaches will allow dissection of the underlying PCD mechanisms; consequently, parasite-mediated refractory PCD can be circumvented by inducing other PCD pathways. Mitochondria and organelles of mitochondrial origin - specific therapy would provide an efficient strategy for treating parasite-mediated PCD-resistant.
The caspases have been known to be activated through the extrinsic, intrinsic pathway, and perforin/granzyme pathways. The immune cells responsible for cell-mediated cytotoxicity are collectively called cytotoxic lymphocytes (CLs), including natural killer (NK) cells and cytotoxic T lymphocytes (CTL). NK cells and CTLs can either kill their targets by ligation of death receptors or granule exocytosis (e.g. the proapoptotic proteins, including perforin and granzymes). GrA and GrB are important contributors to granule-mediated programmed cell death. The increased production of mitochondrial reactive oxygen species (mROS) in response to Granzyme A (GrA) mediates the translocation of the SET complex, namely HMG2, Ape1, and SET from the ER to the nucleus. GrA activates a DNase (NM23-H1) in the complex to nick DNA [28-31]. Readers who are interested in more detailed information about the family of Granzymes should refer to various exceptional reviews [32-34]. Apoptosis is morphologically and biochemically distinct from necrosis and plays a pivotal role during viral, bacterial and parasitic infections, tissue homeostasis, aging of multicellular organisms and tissue development [14]. Furthermore, apoptosis has been observed as a crucial defense mechanism during adaptive and innate immunity [15,35]. However, bacteria, parasites and viruses have implemented a variety of strategies with respect to apoptosis [15,17]. Clearly, protozoan parasites possess highly sophisticated systems for adapting to their host harsh environments that allow them to multiply and disseminate. Plasmodium species, including Plasmodium falciparum and Plasmodium berghei induce Fas-Fas ligand-mediated apoptosis in various host immune cells [36-39]. Moreover, apoptotic induction has been reported in Cryptosporidium parvum infected- biliary and intestinal epithelial cells [40-43], Leishmania donovani-infected splenic CD4 T cells, Leishmania braziliensis- infected CD4 and CD8 T cells [44], Plasmodium sporozoite-infected Kupffer cells apoptosis [45], to escape the host defense systems. One reason why Plasmodium berghei - infected hepatocytes become resistant to apoptosis is the activation of hepatocyte growth factor receptor (HGF), which binds receptor tyrosine kinase MET and results in hepatocyte proliferation, survival and contribute to transmission success [46,47]. However, some protozoan parasites are able to fine-tune the apoptotic signals in their host cells to establish a continual interaction, for example; Theileria parva intracellularly replicates within lymphocytes and disseminates, especially in the T cells throughout the host protecting T. parva -infected T cells from apoptosis [48,49]. Studies also suggest that Cryptosporidium parvum-infected human ileocecal adenocarcinoma tumor cell line (HCT-8) and intestinal epithelial cells have been manipulated in a way to inhibit apoptosis at the trophozoite stage and promote it at the sporozoite and merozoite stages [50,51]. Insights into the mechanisms of this biphasic modulation of apoptotic pathways may be a novel and potential target for curative interventions to alter the delicate balance between early prevention and late stimulation of apoptosis. Thereby, a better understanding of the molecular mechanisms that underlie apoptosis and other PCD may facilitate induction of T. parva -infected T cells to die and ultimately promote the discovery of unconventional therapeutic targets for theleriosis treatment. Programmed cell death in protozoan parasites seems to be similar in complexity to apoptosis in multicellular organisms but also has unique features may be related to their recurrent exposure to environmental stressors. A recent study by Rathore et al. involved the role of caspase-like VAD-FMK-binding proteases in TSN- (Tudor staphylococcal nuclease) homolog in P. falciparum and spliceosomal complex degradation causes apoptosis-like cell death of Plasmodium under cellular stress [52]. It is also worth noting that in contrast to viable parasites, which stimulate the CD4+T-cell proliferation, the apoptotic-like Leishmania induces anti-inflammatory mediator production by hijacking the host cells´ autophagy machinery, likely to silence host phagocytes contributing to the survival of the overall population [53]. A small scale study by Ni Nyoman and Lüder reached different conclusions, finding the possession of ancient apoptosis-like cell death machinery by T. gondii and its ability to be targeted by chemotherapeutic agents, such as clindamycin [54]. Strikingly, Plasmodium yoelii, rodent malaria parasite perturbs hepatocyte pro-death protein (p53) pathway, thereby decreasing its level in infected hepatocytes to foster survival [18]. It has also been shown that the disguised parasite-filled vesicles (merosomes) budding from Plasmodium-manipulative host hepatocytes (i.e. phosphatidylserine exposure/"eat me" signals) into the sinusoid lumen to ensure red blood cell infectious form transmission, responsible for triggering clinical malaria and host immunity circumvention [55]. In 2006, Carmen et al. published a paper in which they reported the hijacking of the initiation and execution phases of mitochondria-dependent PCD pathway by Toxoplasma gondii. Kaushansky et al. [47] were the first to report - in contrast to T. gondii-mediated resistant to mitochondria-initiated host cell apoptosis- that liver stage (LS) - Plasmodium infected hepatocytes upsurge the susceptibility of host hepatocytes to mitochondria - triggered apoptosis Kaushansky et al. [18].
Apoptosis that is elicited in T-lymphocytes and other leukocytes results in immune response abrogation to T. gondii. This parasite has evolved strategies to inhibit host cell apoptosis, including interference with the caspase cascade, upregulation of anti-apoptotic molecules by infected host cells (Bcl-2 protein family, e.g. Bfl-1 or Mcl-1) and IAPs, inhibition of the cytochrome c release from mitochondria, and a decreased activity of the poly(ADP-ribose) polymerase ( PARP). T. gondii-mediated apoptosis might be associated with the pathogenesis of neurodegeneration and neuropathology [56]. It has been demonstrated that apoptotic programmed cell death originated not only before multi- cellularity, but also prior to the divergence of unicellular lineages [57]. Evidence gained from the study of protozoan parasites, and free living protists are contributed to understand the key molecules (e.g.NM3 gene family homologs of free living prtotozoa, metacaspases and endonuclease G of Leishmania spp, Plasmodium spp, and Trypanosoma spp and cytochrome c of Plasmodium spp) of the initiation and execution of apoptosis-like programmed cell death. Various pro-apoptotic stimuli, such as chemotherapeutic agents elicit ancient apoptosis-like cell death in T. Gondii [54]. E. histolytica trophozoites themselves die by a process of PCD. Calcium influx and oxidative stress activate diverse non-caspase proteases (Ca2+-dependent proteases), such as calpain. Once activated, calpain substrates in the cytosol, nucleus and membrane are hydrolysed resulting in apoptosis [58]. Whereas it was shown that calpains activate the initiator (caspase 12), which subsequently activate downstream effector caspase, caspase 3 [59,60]. Amoebapore and cysteine proteases may serve to directly activate caspase 3. Relatedly, resveratrol treated - E. histolytica trophozoites leads to activated calpain, phosphatidylserine externalization and DNA fragmentation (apoptosis-like death) [61]. Interestingly, for malaria parasites of the genus Plasmodium that is transmitted through an infected mosquito bite, apoptosis of both the Plasmodium parasite (limiting the infection load to prevent vector death before sporozoite maturation) and cells in the mosquito vector (changing in resource partitioning via follicular epithelial cell death, thus fecundity reduction) work together to augment the probability of consecutive Plasmodium transmission. Unicellular eukaryotes possess several factors from both host and vector could participate to apoptosis-like death, including Anopheles NOS, mammalian NOS II, Ca2+ and NOS-independent factors, such as ROS and TGF-b1 [62,63]. Mitochondrial protease machinery in Plasmodium falciparum has been associated with apoptosis-like cell death [64]. The upstream regulators, caspase-like proteases, the metacaspases (MCA), are considered to exert the caspase function in protozoan parasites, plant and fungi. They are calcium-activated and arginine-specific peptidases. MCA releases the brakes on autophagy resulting in vacuolar programmed cell death in plants [65]. T. gondii metacaspase (TgMCA) contributes to Toxoplasma-apoptotic-like cell death [66]. The unicellular organism Leishmania donovani undergoes apoptosis-like cell death in response to external stress or exposure to anti-leishmanial agents, such as 3-O, 28-O-disuccinylbetulin (DiSB) resembled the PCD of higher eukaryotes. High level of metacaspase results in the translocation of endonuclease G (LdEndoG) from mitochondria to the nucleus for DNA degradation [67]. Surprisingly, the initiator proteins, for example, MCA-Arginine protease from both protozoan parasite species, such as Plasmodium falciparum and Leishmania major can elicit apoptotic cell death through a Z-VAD-FMK inhibitable effector protease [68].
Data from several sources have been reported that Trypanosoma cruzi cruzipain, a cysteine proteinase inhibition leads to mouse survival and parasite programmed cell death and additional peptidases, including Cathepsin B-like enzyme, two metacaspases and two autophagins in T. cruzi. A better utilization of cruzipain inhibitors will be helpful in developing intervention strategies against the parasites. Emerging study has revealed that autophagins and metacaspases of trypanosomiases play important roles in the regulation of the autophagy pathway and apoptotic cell death respectively [69]. The hallmark features of programmed cell death in P. falciparum and mammals have revealed fascinating analogies between them across both subkingdoms. The diagram below exemplifies programmed cell death- mediated by cys protease (s) in CQ-sensitive laboratory strain of P. falciparum, 3D7 (MRA-102, MR4, ATCC, Manassas, Va, USA) and caspase-3 in animals (Figure 6) [70].
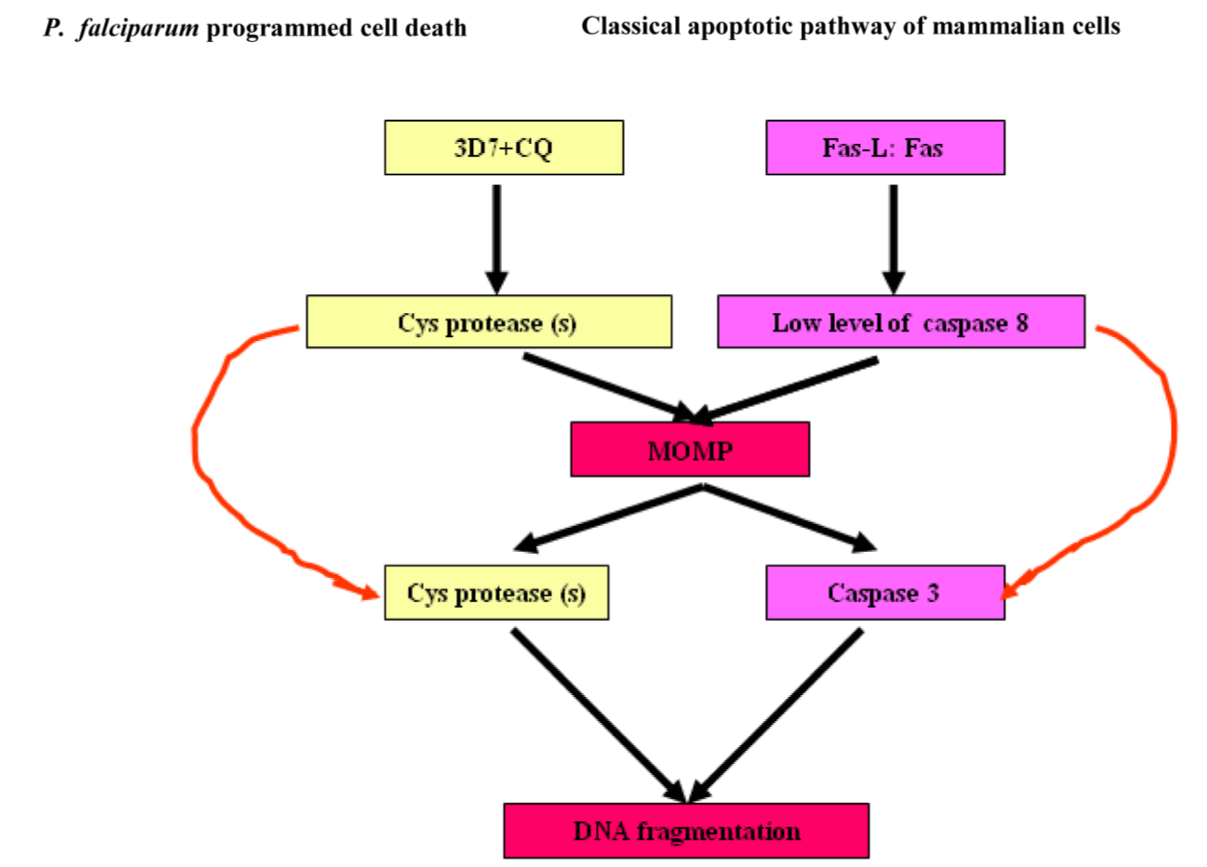
Figure 6. Analogies of program cell deaths between P. falciparum and mammalian apoptosis.
Abbreviations: CQ-sensitive laboratory strain of P. falciparum, 3D7 (MRA-102, MR4, ATCC, Manassas, Va, USA) and MOMP, mitochondrial outer membrane permeabilisation.
Adherence (Gal/GalNAc adherence lectin), contact-dependent cytolysis (induction of host cell’s apoptotic machinery), and phagocytosis are a multi-step process resulting in host tissue damage during Entamoeba histolytica infection. Gal/GalNac lectin, calcium fluxes, amoebapores, cysteine proteinases, caspase activation, phosphatidylserine exposure (PS) and morphological changes are involved in Entamoeba histolytica-triggered apoptosis (Figure 7) [71,72].
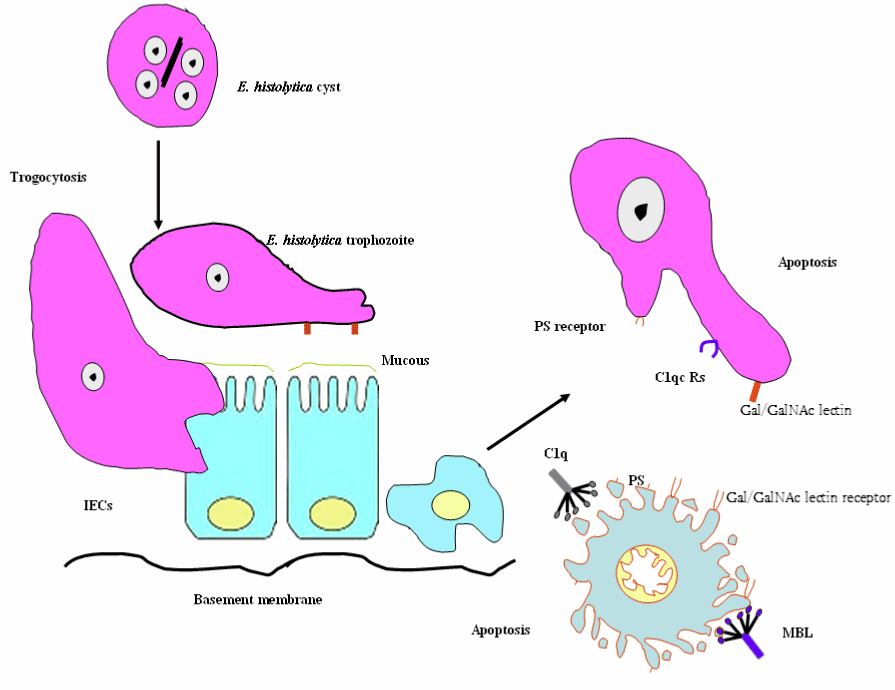
Figure 7. Schematic diagram of Entamoeba histolytica - elicited host cell apoptosis.
Abbreviations: MBL, mannose binding lectin; C1q, C1qRs; PS, phosphatidylserine exposure and IECs, Intestinal epithelial cells
Furthermore, opsonins, such as collectin family members (e.g., mannose binding lectin (MBL)) and the structurally related protein C1q bind to Entamoeba histolytica (Eh) C1q receptors (C1qRs) allowing and facilitating the uptake of apoptotic intestinal cells by E. histolytica [73,74]. In the literature, the term trogocytosis tends to be used to refer to the plasma membrane and membrane protein active [75]. In a follow-up review, it has been illustrated the role of trogocytosis in the innate immune signaling horizontal transfer, the presentation of antigens, including the possible transfer of the MHC between cells, and the spread of the bacteria within the host [76,77]. Furthermore, the parasite and host cells membrane exchange (i.e., trogocytosis) has been reported in Trypanosoma cruzi invasion. Interestingly, E. histolytica trophozoites bite off and internalize distinct pieces of living human cells, contributing to intracellular calcium increase and eventual cell death. Thus, amoebic trogocytosis is the mediator of tissue damage [78,79]. Signal transduction for amebic trogocytosis and phagocytosis are mediated by PI3K and C2-domain-containing protein kinase (C2PK) for actin polymerization; however Eh trogocytosis is prevalent in living cells. The transfer of materials from the Plasmodium falciparum- infected red blood cells (IRBC) to the brain endothelial cells (ECs) plasma membrane in a trogocytosis-like process, resulting in the transformation of EC into a target for the cytotoxic T cells and contributing to peri-vascular oedema and cerebral malaria (CM) [80]. The process of intercellular exchange, trogocytosis is an evolutionarily conserved process, benefits some pathogens by enhancing dissemination early during infection, and improves our understanding of how this process impacts CM. It will be important to decipher the molecular mechanisms underlying trogocytosis.
Autophagy is a highly controlled process, directing numerous intra-cellular cargo, such as misfolded or aggregated proteins, damaged organelles to the lysosome for degradation and subsequent recycling. Interestingly, under extreme environmental conditions, such as salinity increase, temperature shock, oxygen deprivation, and starvation, certain ciliated protozoa enter into an encystment-excystment cycle. Vegetative cell undergoes several drastic changes during encystment, including controlled autophagy of several intra-cellular organelles, resorption of oral and somatic cilia [81]. Autophagy is elicited as a cellular response to stressors, such as starvation, misfolded protein, endoplasmic reticulum (ER) stress, intracellular pathogens, and reactive oxygen species. In a review conducted by Kelekar it was shown those mediators of class I and class III PI3 kinase signaling pathways and trimeric G proteins mediate autophagosome formation regulation during the stress response. There are three main types of autophagy: chaperone-mediated autophagy (CMA), macro-autophagy, and micro-autophagy. CMA is a process where cargo proteins are translocated across the lysosomal membrane in a complex with a cytosolic chaperone protein, HSC70 [82]. The following section will mainly focus on macroautophagy process, which will subsequently be referred to as autophagy. Glick et al. describes five key stages of autophagy: (a) phagophore formation and nucleation controlled by Beclin-1/VPS34; (b) autophagy-related genes, Atg5–Atg12 conjugation and interaction with Atg16L at the phagophore; (c) LC3 processing and insertion into the extending phagophore membrane to act as a receptor to selectively interact with the cargo; (d) capture of random or selective cargo for degradation; and (e) autophagolysosome formation [83]. As shown in Figure 8 below, the mammalian target of rapamycin (mTOR) [84,85], the major negative regulator of autophagy through Unc-51-Like Kinase (ULK) complex, including ULK, Autophagy-related Protein 13 (Atg13) and FAK-family Interacting Protein of 200 kDa (FIP200) is regulated by nutrient status and stress. The ULK complex phosphorylates Activating Molecule in Beclin-1-Regulated Autophagy (AMBRA1), which activates the phosphatidylinositol-3-kinase (PI3K) complex (Vacuolar Protein Sorting 15 (VPS15), VPS34, Beclin-1 and AMBRA1). The phospotidylinositol 3-phosphate kinase (P13K) III mediates the phagophore nucleation. The autophagy process involves the coordinated actions of a variety of autophagy genes (ATG) that mediate the catabolic process of these stored energy sources. Ubiquitin-like ligases Atg7/Atg10 activate the conjugation of Atg12 onto Atg5, which complex in pairs with Atg16L. The latter complex elicits curvature into the growing phagophore membrane through recruitment of Atg4 - cleaved LC3B-II/Atg8, which is conjugated to phosphatidylethanolamine (PE) for initiating the formation and maturation of autophagic vesicles following its activation by Atg7/Atg3 (Figure 8) [86] for more detailed discussion of the molecular mechanisms of autophagy in unicellular eukaryotes).
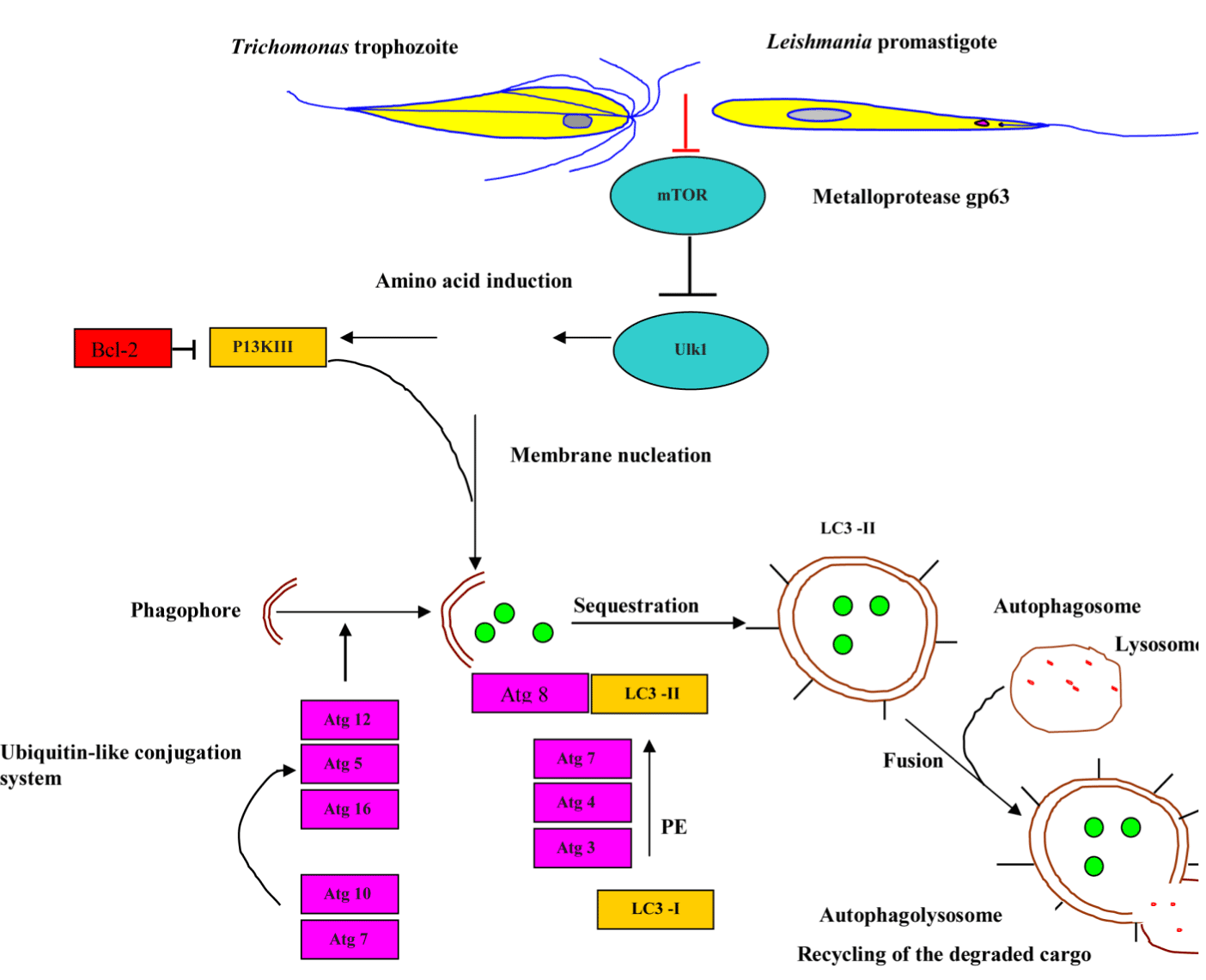
Figure 8. Schematic model of auto-phagosome formation and maturation.
Abbreviations: MOMP, mitochondrial outer membrane permeabilisation ; mTOR, mammalian target of rapamycin ; PI3KIII, phosphatidylinositol-3-kinase III ; Bcl-2, B-cell lymphoma 2 ; ULK 1, Unc-51-Like Kinase 1; ATG, autophagy-related gene ; PE, phosphatidylethanolamine ; LC3-I, microtubule-associated protein light chain 3-I and LC3-II, microtubule-associated protein light chain 3-II.
The bioinformatic evidence disclosed the existence of putative homologs of ATG genes, including TgATG1, TgATG3, TgATG7, TgATG8, TgATG18, TgATG20, and TgVPS34 and the absence of TgATG5 and TgATG12 [87, 88], which supports the notion of the evolutionarily conservation of autophagy. It has been demonstrated that L. major and Trichomonas vaginalis employed metalloprotease gp63 to disrupt the Akt/mTOR pathway [89,90], thereby enhancing parasite infectivity and survival. Not surprisingly, hijacking the Tor signaling cascade by certain protozoan parasites has thus far opened modern avenues for the development of new Tor-based therapies to combat protozoan infection. Besteiro et al. has reported that TgAtg3 plays a crucial role in maintaining mitochondrial integrity and developing T. gondii tachyzoites. Accordingly, manipulating autophagy could improve the control of T. gondii infection [87]. Accordingly, manipulating autophagy could improve the control of T. gondii infection. The interconnection between apoptosis and macrophagy has been observed in Tetrahymena programmed nuclear death during conjugation events in which only the parental macronucleus and micronuclei elimination is achieved by autophagy in collaboration with caspase-like enzymes and mitochondria [91]. Certain ciliated protozoa enter into the encystment-excystment cycle under unfavorable environmental conditions, such as salinity increase, oxygen deprivation, temperature shock and starvation. The dynamic rearrangement of oral, somatic cilia, infraciliatures and several cellular organelles to be sequestered within autophagosome and delivered to the lysosome where degradation occurs are critical for the systematic progression of autophagy [92,93]. It has been demonstrated that apoptotic-like Leishmania/amstigote hijacks the macrophages´ autophagy machinery by dampening T-cell-mediated parasite elimination [53]. As such, apoptotic-like cell death in a single-cell eukaryotic parasite could benefit the survival of the overall population and could be amenable to therapeutic intervention (Figure 9).
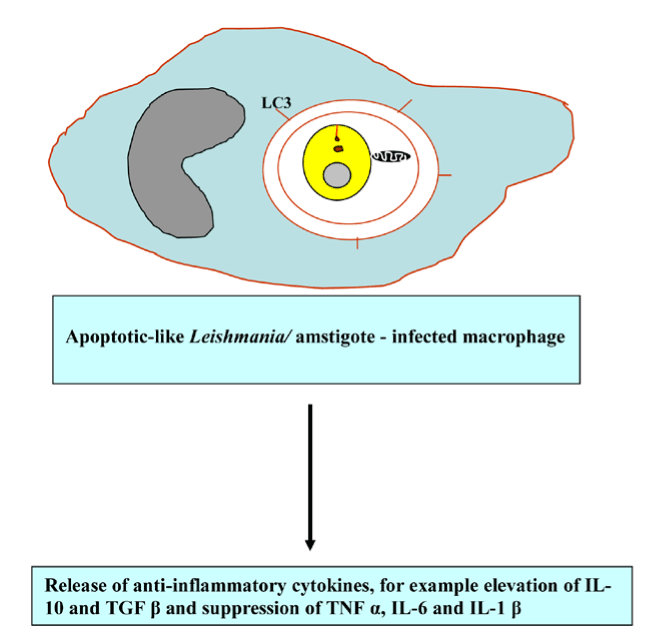
Figure 9. Apoptotic-like Leishmania elicits anti-inflammatory response.
Abbreviations: LC3, microtubule-associated protein light chain 3; IL-10, interleukin-10; TGF β, transforming growth factor beta; TNF α, tumor necrosis factor alpha and IL-6 interleukin-6 and IL-1 β, interleukin-1 β
It is the process of selective autophagic clearance of intracellular microorganisms and their targeting to the autophagic machinery for degradation. The colocalization of parasitophorous vacuoles and LC3, a marker of autophagy has been induced in T. gondii-infected mouse and human macrophage stimulated with CD40 molecule; therefore, phagophores capture T. gondii-residing parasitophorous vacuole and redirect them to the lysosomal degradation [94].
Harsh non-physiological insults, such as mechanical force, heat, or cold preferentially induce primary necrosis lacking the apoptic feature, while the failure of phagocytic clearance of apoptotic cells breaks down the integrity of the plasma membrane usually referred to as a secondary necrosis [95-97]. Except in primary necrosis, alteration of the intracellular ATP and caspase availability elicits secondary or post-apoptotic necrosis, which in turn represents the apoptosis-secondary necrosis continuum. Generally, apoptosis elicits anti-inflammatory response and diminishes the release of DAMP to actively down regulate immune responses [98]. However, apoptosis and necrosis interplay stimulates the pro-inflammatory response in the absence of removal of apoptotic cells by phagocystosis. The release of endogenous danger/damage-associated molecular patterns (DAMPs) or alarmins activates antigen-presenting cells, including macrophage and dendritic cells through TLR signalling and other mechanisms [99]. DAMPs activate and promote the maturation of DCs for subsequent migration to lymph nodes to initiate acquired immunity by processing and presenting antigens to antigen-specific T cells. A landmark study has implicated necrotic cells are sensed by the NLRP3 inflammasome resulting in the subsequent pro-inflammatory cytokine IL-1ß release (Figure 10) [100]. Early examples of research reported that caspases served to inactivate DAMPs ([101-103], thereby caspases could be viewed as cell death effectors and regulators of inflammation [103].
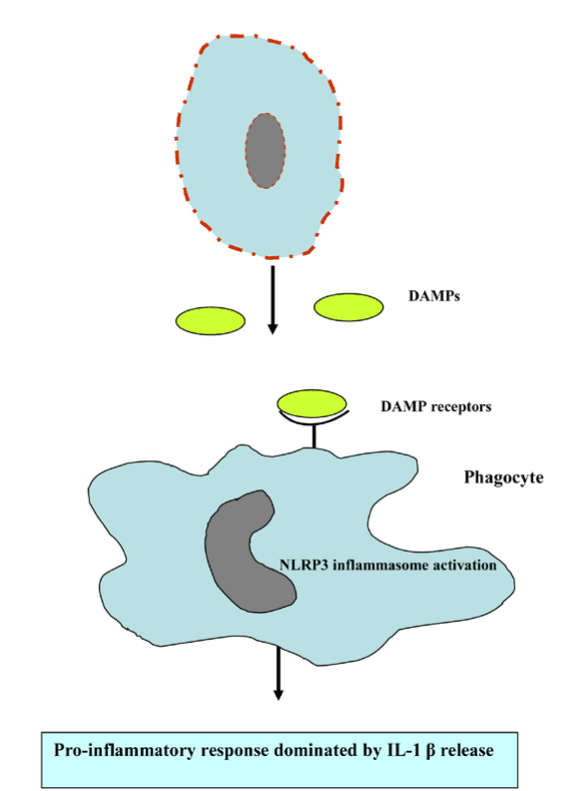
Figure 10. Necrotic cell death promotes proi-nflammatory response.
Abbreviations: DAMPs, danger/damage-associated molecular patterns and NLRP3, NOD-like receptors containing pyrin domain 3
It is an inflammatory form of caspase-independent, programmed necrotic cell death, which is mediated by the receptor-interacting protein kinase (RIP) 1-RIP3 complex, necrosome and is induced by death receptors, including tumor necrosis factor receptor (TNFR) 1, TNFR2, and Fas. The RIP1/RIP3 complex enhances the necroptosis; however, activation of caspase-8 drives the cleavage of necrosome and apotosis is formed [104]. Necroptosis and apoptosis both are essential for the elimination of damaged or protozoan parasitic - infected cells. The use of Necroptosis inhibitor necrostatin-1(Nec-1) as a tool has advanced scientific understanding of the physiological role of necroptosis. An emerging appreciation of the cross-talk relationships among different patterns of cell death and pathways will be imperative for understanding their roles in the inflammation process and protozoan parasitic infection. For slowly replicating intracellular pathogens, inhibition of apoptosis is necessary for life cycle completion (e.g., T. gondii); therefore, apoptosis stimulation displays pledge for the treatment of toxoplasmosis. With tremendous efforts devoted to researchers on our understanding of the molecular pathways of programmed cell death, it would be expected that approaches targeting cell survival and death signaling pathways will be developed for controlling the inflammatory diseases and protozoan parasites.
It is an inflammatory form of programmed lytic cell death, which is mediated by inflammasomes and caspase-1/11- mediated programmed cell death. Pyroptosis is characterized by distinct morphological and biochemical alterations to the cell, fluid influx, cell swelling and lysis, membrane pore formation, and DNA fragmentation without laddering. The inflammatory response is triggered by pyroptotic programmed cell death as a consequence of the release of cytosolic contents, for example ATP, high-mobility group box 1 (HMGB1) and IL-1α, lactate dehydrogenase (LDH), inflammatory cytokines IL-1β and IL-18 (Figure 11) [105,106].
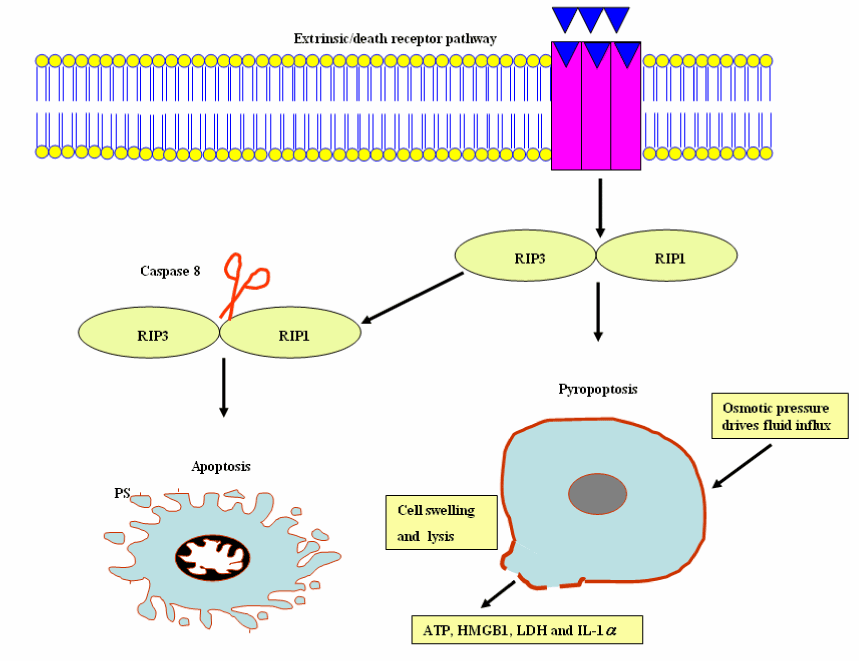
Figure 11. Pyroptosis and apoptosis cellular events.
Abbreviations: RIP 1, receptor-interacting protein kinase 1 and RIP 3, receptor-interacting protein kinase 3; HMGB1, high-mobility group box 1; LDH, lactate dehydrogenase; IL-1 α, interleukin-1 α
De Vasconcelos et al. have reported the inflammasomes, multi-protein platforms as polyvalent cell death platforms, including pyroptosis, pyronecrosis and apoptosis [107]. Programmed cell death is an important counterpart of inflammation and may be caused by inflammation. Multiple pathways of cell death induction have presumably evolved to alienate vacuolar or cytosolic protozoan parasitic evasion of cell death pathways.
The inflammasomes, an innate immune sensing cytosolic protein complexes, are composed of the effector caspases (caspase-1, caspase-11), an adapter molecule (such as ASC), and a sensor protein (such as NLRP1, NLRP3, NLRP12, NAIP1, NAIP2, NAIP5, or AIM2). Inflammasome assembly is initiated by cytosolic sensors (i.e. several nucleotide binding oligomerization domain-like receptor family members), including NLRP3, which recognizes pathogen- and danger-associated molecular patterns (PAMPs and DAMPs, respectively) and plays an instrumental role in inflammasome activation. The recruitment and activation of caspase-1 by inflammsomes promote IL-1β and IL-18 secretion and pyroptosis. In addition, recent research work shows the implication of inflammasomes in the induction of programmed cell death, such as apoptosis via recruit procaspase-8 [107]. Inflammasomes have now been described in details, and are reviewed elsewhere [108-111]. Inflammasomes counter parasite replication and clear infected cells through an inflammatory cell death program termed pyroptosis. As a countermeasure, various species of Leishmania have been reported to evolve virulence factors, such as surface metalloprotease GP63 to antagonize inflammasome pathways [112]. The parasite Gal (galactose)/GalNAc (N-acetylgalactosamine) lectin-mediated adherence, target cell calcium influx, putative cytotoxic effector proteins, such as amoebapores, proteases and other saposin-related proteins are responsible for parasite invasion and host cell death. As shown in Figure 12, the adherent parasite elicits calcium influx resulting in tyrosine dephosphorylation and caspase 3 activation [71]. Entamoeba histolytica -macrophage contact triggers the recruitment of α5β1 integrin, also called the fibronectin receptor, which is a member of type I transmembrane heterodimeric glycoprotein receptors to bind to cysteine protease on the parasite surface, termed EhCP5. An integrin receptor engagement by EhCP5 ligands induces the activation of the NLRP3 inflammasome, enabling the host to mobilize the highly pro-inflammatory response at the infection site [113]. Therefore, NLRP3 inflammasome activation by E. histolyitca is responsible for host-detrimental effects without restricting amoebiasis growth.
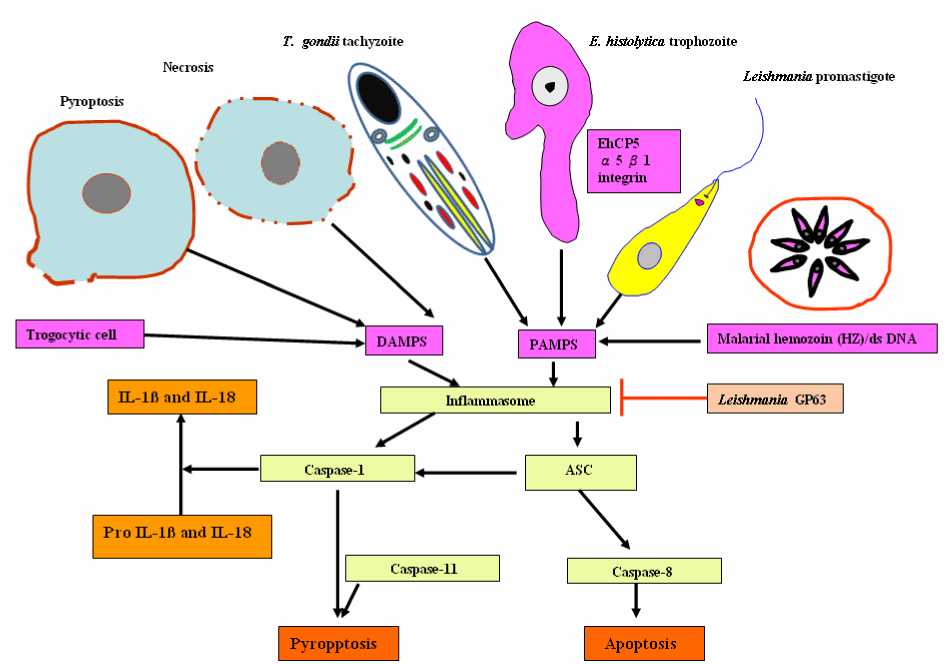
Figure 12. Inflammasome-mediated pyroptotic and apoptotic programmed cell death in response to a wide variety of protozoan parasitic stimuli and endogenous danger signals and its inhibition Leishmania gp63.
Abbreviations: DAMPs, danger/damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; EhCP5, cysteine protease on the parasite surface and ASC (apoptosis-associated speck-like protein containing C-terminal caspase recruitment domain [CARD])
Targeting α5β1 integrin may represent new potential therapeutic agents to fight the invasive trophozoite form of E. histolytica. Thus this micro-organism appears to drive apoptosis and necroptosis (and inflammation) as a mechanism to breach the intestinal epithelial cells and move to systemic sites. Relatedly, Toxoplasma gondii infection triggers the pattern recognition receptor, NLRP3 and NLRP1 inflammasome response that is protective to the host, controlling parasite replication, dissemination and persistence [114-116]. Malarial hemozoin and dsDNA can be sensed as a danger signal results in the activation of the NLRP3 and absent in melanoma 2, AIM 2 inflammasome complexes respectively, which triggers pyroptosis and IL-1β and IL-18 production [117,118]. However, inflammasome activation acts as a double-edged sword for various parasitic protozoan infections. NLRP3 inflammasome is activated in response to L. major infected susceptible BALB/c mice resulting in the production of IL-1β and IL-18. IL-18 elicits a Th2-biased adaptive immune response, characterized by IL-4 production by CD4 + T cells, which disseminates the disease and are potentially dangerous for the host. IL-18 neutralization may be a promising target for leishmaniasis [119]. Recently, L. major Seidman strain (LmSd) - infected C57BL/6 mice has resulted in a non-healing lesion. The most likely causes are the association of NLRP3 inflammasome-dependent IL-1β with localized neutrophil recruitment [120]. Considerably more work will need to be done to expand our current knowledge of inflammasome activation or regulation by protozoan parasites or their components to identify additional host molecules or parasitic protozoan components that activate and manipulate the NLRP3 inflammasome complex. Thereby, it will contribute to development of interventions to prevent tissue damage and host death in response to diverse parasitic protozoan infections.
PND is a type of autophagy in the ciliated protozoa, Tetrahymena thermophila. The hallmarks of the process include nuclear condensation, DNA fragmentation, lysosomal acidification, and final resorption. Diverse ciliated protozoa, including T. thermophila, develop a controlled parental somatic macronucleus degradation process during the sexual reproduction or conjugation, with differentiation of new macronuclei for the next generation [121]. The involvement of the same process in the disintegration of pre-zygotic micronuclei is exhibited in the diagram below (Figure 13). Moreover, colpodid ciliates show PND during encystment in response to diverse environmental stressors, for example, starvation. Recent evidence suggests that a novel mitochondrial nuclease-associated protein (TMN1), a major executor of the programmed nuclear death in T. thermophila participates in PND [122]. In a study which set out to determine central regulators of PND, Akematsu et al. [124] found that T. thermophila ortholog (TtVPS34) to yeast Vps34 and its human ortholog PIK3C3 is involved in the nuclear degradation events of PND within the autophagosome. TtVPS34 plays a critical role in autophagosome formation on the parental macronucleus and micronuclei together with two TtATG8s, sugars and phosphatidylserine (PS) exposure, allowing the lysosme to fuse with the macro-nuclear and micro-nuclear envelopes [123,124]. PND plays a key role in the events that occur during conjugation, which are fundamental to the vegetative cell genetic makeup. Thus, PND may best be viewed as a survival strategy for avoiding death.
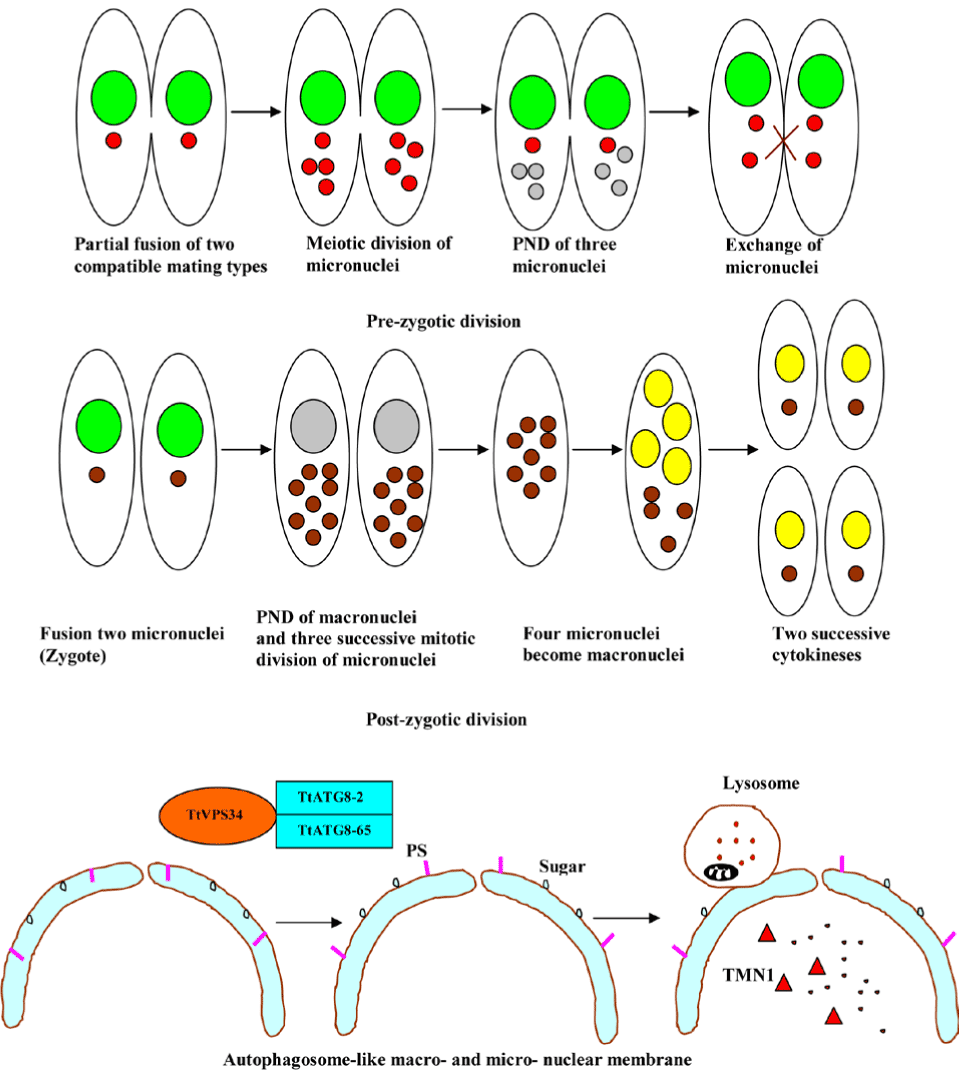
Figure 13. Programmed nuclear death in ciliated protozoa.
Abbreviations: PND; programmed nuclear death; TMN1, mitochondrial nuclease-associated protein; PS, phosphatidylserine; TtVPS34, T. thermophila ortholog (TtVPS34) to yeast Vps34;
TtATG8-2, T. thermophila autophagy gene 8-2 and TtATG8-65, T. thermophila autophagy gene 8-65
Taken together, the PND, which is genetically encoded process is worthy of study in its own right as an extreme example of PCD, which is useful as a source of understanding about the mechanisms involved in PCD of the unicellular protozoan parasites and can offer exciting new avenues of novel therapeutic strategies.
The molecular battle between the host and protozoan parasites has led to diverse measure/counter-measure responses in the context of cell death. The reader of this review may be perplexed by the paradoxical data, and the multitude of mechanisms involved in cell death and may ask the crucial question of what, as a concise blueprint, the plausible aproach actually is. A reasonable approach to exploit these cellular and molecular mechanisms of cell death could be to discover contemporary therapeutic and prophylactic intervention points for broad-spectrum host- oriented measures and parasite counter- measures, and to determine the infection outcome.
This research was supported by the National Key Basic Research Program (973 program) of China (Grant No. 2015CB150300). Additionally, the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that they have no competing interests.
- Menna-Barreto RF, Corrêa JR, Cascabulho CM, Fernandes MC, Pinto AV, et al. (2009) Naphthoimidazoles promote different death phenotypes in Trypanosoma cruzi. Parasitology 136: 499-510. [Crossref]
- Menna-Barreto RF, Salomão K, Dantas AP, Santa-Rita RM, Soares MJ, et al. (2009) Different cell death pathways induced by drugs in Trypanosoma cruzi: an ultrastructural study. Micron. 40: 157-168. [Crossref]
- Li M, Gao P, Zhang J (2016) Crosstalk between Autophagy and Apoptosis: Potential and Emerging Therapeutic Targets for Cardiac Diseases. Int J Mol Sci 17: pii: E332. [Crossref]
- Tait SW, Ichim G, Green DR3 (2014) Die another way--non-apoptotic mechanisms of cell death. J Cell Sci 127: 2135-2144. [Crossref]
- Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, et al. (1997) The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 15: 1573–1581.
- Zeiss CJ (2003) The apoptosis-necrosis continuum: insights from genetically altered mice. Vet Pathol 40: 481-495. [Crossref]
- Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495-516. [Crossref]
- Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N (2013) Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 1833: 3448-3459. [Crossref]
- Ouyang C, You J, Xie Z2 (2014) The interplay between autophagy and apoptosis in the diabetic heart. J Mol Cell Cardiol 71: 71-80. [Crossref]
- Hong X, Lei L, Glas R (2003) Tumors acquire inhibitor of apoptosis protein (IAP)-mediated apoptosis resistance through altered specificity of cytosolic proteolysis. J Exp Med 197: 1731-1743. [Crossref]
- Barras D, Widmann C (2011) Promises of apoptosis-inducing peptides in cancer therapeutics. Curr Pharm Biotechnol 12: 1153-1165. [Crossref]
- Dewson G, Kluck RM (2009) Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci 122: 2801-2808. [Crossref]
- Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48: 158-167. [Crossref]
- Vaux DL, Strasser A (1996) The molecular biology of apoptosis. Proc Natl Acad Sci U S A 93: 2239-2244. [Crossref]
- Liles WC (1997) Apoptosis -role in infection and inflammation. Curr Opin Infect Dis 10: 165-170.
- Barry M, McFadden G (1998) Apoptosis regulators from DNA viruses. Curr Opin Immunol 10: 422-430. [Crossref]
- Gao LY, Kwaik YA (2000) The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol 8: 306-313. [Crossref]
- Kaushansky A, Metzger PG, Douglass AN, Mikolajczak SA, Lakshmanan V, et al. (2013) Malaria parasite liver stages render host hepatocytes susceptible to mitochondria-initiated apoptosis. Cell Death Dis 4: e762. [Crossref]
- Peter ME, Krammer PH (2003) The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 10: 26-35. [Crossref]
- Jerlström-Hultqvist J, Einarsson E, Xu F, Hjort K, Ek B, et al. (2013) Hydrogenosomes in the diplomonad Spironucleus salmonicida. Nat Commun 4: 2493. [Crossref]
- Mi-ichi F, Makiuchi T, Furukawa A, Sato D, Nozaki T (2011) Sulfate activation in mitosomes plays an important role in the proliferation of Entamoeba histolytica. PLoS Negl Trop Dis 5: e1263. [Crossref]
- Santos HJ, Imai K, Hanadate Y, Fukasawa Y, Oda T, et al. (2016) Screening and discovery of lineage-specific mitosomal membrane proteins in Entamoeba histolytica. Mol Biochem Parasitol pii: S0166-6851(16): 30001-9. [Crossref]
- Makiuchi T, Nozaki T (2014) Highly divergent mitochondrion-related organelles in anaerobic parasitic protozoa. Biochimie 100: 3-17. [Crossref]
- Nývltová E, Smutná T, Tachezy J, Hrdý I (2016) OsmC and incomplete glycine decarboxylase complex mediate reductive detoxification of peroxides inhydrogenosomes of Trichomonas vaginalis. Mol Biochem Parasitol pii: 0166-6851(16)30006-8. [Crossref]
- Huang KY, Ku FM, Cheng WH, Lee CC, Huang PJ, et al. (2015) Novel insights into the molecular events linking to cell death induced by tetracycline in the amitochondriate protozoan Trichomonas vaginalis. Antimicrob Agents Chemother 59: 6891-903. [Crossref]
- Corrêa G, Vilela R, Menna-Barreto RF, Midlej V, Benchimol M (2009) Cell death induction in Giardia lamblia: effect of beta-lapachone and starvation. Parasitol Int 58: 424-437. [Crossref]
- Hehl AB, Regos A, Schraner E, Schneider A (2007) Bax function in the absence of mitochondria in the primitive protozoan Giardia lamblia. PLoS One 2: e488. [Crossref]
- Lowin B, Hahne M, Mattmann C, Tschopp J (1994) Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature 370: 650-652. [Crossref]
- Lieberman J, Fan Z (2003) Nuclear war: the granzyme A-bomb. Curr Opin Immunol 15: 553-559. [Crossref]
2021 Copyright OAT. All rights reserv
- Chowdhury D, Lieberman J (2008) Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol 26: 389-420. [Crossref]
- Ewen CL, Kane KP, Bleackley RC (2012) A quarter century of granzymes. Cell Death Differ 19: 28-35. [Crossref]
- Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, et al. (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell 23: 133-142. [Crossref]
- De Silva U, Choudhury S, Bailey SL, Harvey S, Perrino FW, et al. (2007) The crystal structure of TREX1 explains the 3' nucleotide specificity and reveals a polyproline II helix for proteinpartnering. J Biol Chem 282: 10537-10543. [Crossref]
- Yan N, Cherepanov P, Daigle JE, Engelman A, Lieberman J (2009) The SET complex acts as a barrier to autointegration of HIV-1. PLoS Pathog 5: e1000327. [Crossref]
- Williams GT (1994) Programmed cell death: a fundamental protective response to pathogens. Trends Microbiol 2: 463-464. [Crossref]
- Toure-Balde A, Sarthou JL, Aribot G, Michel P, Trape JF, et al. (1996) Plasmodium falciparum induces apoptosis in human mononuclear cells. Infect Immun 64: 744-750.
- Helmby H, Jönsson G, Troye-Blomberg M (2000) Cellular changes and apoptosis in the spleens and peripheral blood of mice infected with blood-stage Plasmodium chabaudi chabaudi AS. Infect Immun 68: 1485–1490. [Crossref]
- Kern P, Dietrich M, Hemmer C, Wellinghausen N (2000) Increased levels of soluble Fas ligand in serum in Plasmodium falciparum malaria. Infect Immun 68: 3061-3063. [Crossref]
- Matsumoto J, Kawai S, Terao K, Kirinoki M, Yasutomi Y, et al. (2000) Malaria infection induces rapid elevation of the soluble Fas ligand level in serum and subsequent T lymphocytopenia: possible factors responsible for the differences in susceptibility of two species of Macaca monkeys to Plasmodium coatneyi infection. Infect Immun 68: 1183-1188. [Crossref]
- Chen XM, Levine SA, Tietz P, Krueger E, McNiven MA, et al. (1998) Cryptosporidium parvum is cytopathic for human biliary epithelia via an apoptotic mechanism. Hepatology 28: 906–913. [Crossref]
- Chen XM, Gores GJ, Paya CV, LaRusso NF (1999) Cryptosporidium parvum induces apoptosis in biliary epithelia by a Fas/Fas ligand-dependent mechanism. Am J Physiol 277: 599–608. [Crossref]
- Ojcius DM, Perfettini JL, Bonnin A, Laurent F (1999) Caspase-dependent apoptosis during infection with Cryptosporidium parvum. Microbes Infect 1: 1163-1168. [Crossref]
- McCole DF, Eckmann L, Laurent F, Kagnoff MF (2000) Intestinal epithelial cell apoptosis following Cryptosporidium parvum infection. Infect Immun 68: 1710–1713. [Crossref]
- Bertho AL, Santiago MA, Da-Cruz AM, Coutinho SG (2000) Detection of early apoptosis and cell death in T CD4+and CD8+cells from lesions of patients with localized cutaneous leishmaniasis. Braz J Med Biol Res 33: 317-325.
- Klotz C, Frevert U (2008) Plasmodium yoelii sporozoites modulate cytokine profile and induce apoptosis in murine Kupffer cells. Int J Parasitol 38: 1639-1650. [Crossref]
- Carrolo M, Giordano S, Cabrita-Santos L, Corso S, Vigario AM, et al. (2003) Hepatocyte growth factor and its receptor are required for malaria infection. Nat Med 9: 1363-1369. [Crossref]
- Kaushansky A, Kappe SH (2011) The crucial role of hepatocyte growth factor receptor during liver-stage infection is not conserved among Plasmodium species. Nat Med 17: 1180-1. [Crossref]
- Heussler VT, Machado JJ, Fernandez PC, Botteron C, Chen CG, et al. (1999) The intracellular parasite Theileria parvaprotects infected T cells from apoptosis. Proc Natl Acad Sci USA 96: 7312–7317.
- Dobbelaere D, Baumgartner M (2009) Theileria. In: Schaible UH, A., editor. Intracellular Niches of Microbes - A Pathogens Guide through the Host Cell. Weinheim: Wiley-VCH 613-632.
- Mele R, GomezMorales MA, Tosini F, Pozio E (2004) Cryptosporidium parvum at different developmental stages modulates host cell apoptosis in vitro. Infect Immun 72: 6061-6067. [Crossref]
- Liu J, Deng M, Lancto CA, Abrahamsen MS, Rutherford MS, et al. (2009) Biphasic modulation of apoptotic pathways in Cryptosporidium parvum-infected human intestinal epithelial cells. Infect. Immun 77: 837-849.
- Rathore S, Datta G, Kaur I, Malhotra P, Mohmmed A (2015) Disruption of cellular homeostasis induces organelle stress and triggers apoptosis like cell-death pathways in malaria parasite. Cell Death Dis 6: e1803.
- Crauwels P, Bohn R, Thomas M, Gottwalt S, Jäckel F, et al. (2015) Apoptotic-like Leishmania exploit the host's autophagy machinery to reduce T-cell-mediated parasite elimination. Autophagy 2: 285-297. [Crossref]
- Ni Nyoman AD, Lüder CG (2013) Apoptosis-like cell death pathways in the unicellular parasite Toxoplasma gondii following treatment with apoptosis inducers and chemotherapeutic agents: a proof-of-concept study. Apoptosis 18: 664-680. [Crossref]
- Sturm A, Amino R, van de Sand C, Regen T, Retzlaff S, et al. (2006) Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science 313: 1287-1290. [Crossref]
- Dincel GC, Atmaca HT2 (2016) Role of oxidative stress in the pathophysiology of Toxoplasma gondii infection. Int J Immunopathol Pharmacol 29: 226-240. [Crossref]
- Taylor-Brown E, Hurd H (2013) The first suicides: legacy inherited by parasitic protozoans from prokaryote ancestors. Parasit Vectors 6: 108. [Crossref]
- Chan SL, Mattson MP (1999) Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res 58: 167-190. [Crossref]
- Váchová L, Palková Z (2007) Caspases in yeast apoptosis-like death: facts and artefacts. FEMS Yeast Res 7: 12-21. [Crossref]
- Kerbiriou M, Teng L, Benz N, Trouvé P, Férec C (2009) The calpain, caspase 1, caspase 3 cascade leading to apoptosis is altered in F508del-CFTR expressing cells. PLoS One 4: e8436. [Crossref]
- Pais-Morales J, Betanzos A, García-Rivera G, Chávez-Munguía B, Shibayama M, et al. (2016) Resveratrol Induces Apoptosis-Like Death and Prevents In Vitro and In Vivo Virulence of Entamoeba histolytica. PLoS One 11: e0146287. [Crossref]
- Kumar S, Christophides GK, Cantera R, Charles B, Han YS, et al. (2003) The role of reactive oxygen species on Plasmodium melanotic encapsulation in Anopheles gambiae. Proc Natl Acad Sci USA 100: 14139-14144. [Crossref]
- Gordeeva AV, Labas YA, Zvyagilskaya RA (2004) Apoptosis in unicellular organisms: mechanisms and evolution. Biochemistry (Mosc) 69: 1055-1066. [Crossref]
- Rathore S, Jain S, Sinha D, Gupta M, Asad M, et al. (2011) Disruption of a mitochondrial protease machinery in Plasmodium falciparum is an intrinsic signal for parasitecell death. Cell Death Dis 2: e231. [Crossref]
- Minina EA, Smertenko AP, Bozhkov PV1 (2014) Vacuolar cell death in plants: Metacaspase releases the brakes on autophagy. Autophagy 10: 928-929. [Crossref]
- Li M, Wang H, Liu J, Hao P, Ma L, et al. (2016) The Apoptotic Role of Metacaspase in Toxoplasma gondii. Front Microbiol 6: 1560. [Crossref]
- Chowdhury S, Mukherjee T, Chowdhury SR, Sengupta S, Mukhopadhyay S, et al. (2014) Disuccinyl betulin triggers metacaspase-dependent endonuclease G mediated cell death in unicellular protozoanparasite Leishmania donovani. Antimicrob Agents Chemother 58: 2186-201.
- Meslin B, Beavogui AH, Fasel N, Picot S (2011) Plasmodium falciparum Metacaspase PfMCA-1 Triggers a z-VAD-fmk Inhibitable Protease to Promote Cell Death. PLoS ONE 6: e23867. [Crossref]
- Alvarez VE, Niemirowicz GT, Cazzulo JJ (2013) Metacaspases, autophagins and metallocarboxypeptidases: potential new targets for chemotherapy of the trypanosomiases. Curr Med Chem 20: 3069-3077. [Crossref]
- Ch'ng JH, Kotturi SR, Chong AG, Lear MJ, Tan KS (2010) A programmed cell death pathway in the malaria parasite Plasmodium falciparum has general features of mammalian apoptosis but is mediated by clan CA cysteine proteases. Cell Death Dis 1: 26. [Crossref]
- Ralston KS, Petri WA (2011) The ways of a killer: how does Entamoeba histolytica elicit host cell death? Essays Biochem 51: 193-210. [Crossref]
- Begum S, Quach J, Chadee K (2015) Immune Evasion Mechanisms of Entamoeba histolytica: Progression to Disease. Front Microbiol . [Crossref]
- Teixeira JE, Huston CD (2008) Participation of the serine-rich Entamoeba histolytica protein in amebic phagocytosis of apoptotic host cells.Infect mmun 76: 959-66. [Crossref]
- Vaithilingam A, Teixeira JE, Miller PJ, Heron BT, Huston CD (2012) Entamoeba histolytica cell surface calreticulin binds human c1q and functions in amebic phagocytosis of host cells. Infect Immun 80: 2008-18. [Crossref]
- Joly E, Hudrisier D (2003) What is trogocytosis and what is its purpose? Nat Immunol 4: 815. [Crossref]
- Bourdonnay E, Henry T1 (2016) Catch me if you can. Elife 5. [Crossref]
- Steele S, Radlinski L, Taft-Benz S, Brunton J, Kawula TH (2016) Trogocytosis-associated cell to cell spread of intracellular bacterial pathogens. Elife 5: pii: e10625. [Crossref]
- Ralston KS (2015) Chew on this: amoebic trogocytosis and host cell killing by Entamoeba histolytica. Trends Parasitol 31: 442-452. [Crossref]
- Ralston KS (2015) Taking a bite: Amoebic trogocytosis in Entamoeba histolytica and beyond. Curr Opin Microbiol 28: 26-35. [Crossref]
- Jambou R, Combes V, Jambou M J, Weksler B B, Couraud PO, et al. (2010) Plasmodium falciparum adhesion on human brain microvascular endothelial cells involves transmigration-like cup formation and induces opening of intercellular junctions. PLoS Pathog 6: e1001021. [Crossref]
- González AM, Díaz S, Gallego A, Gutiérrez JC (2008) Programmed Nuclear Death and Other Apoptotic-Like Phenomena in Ciliated Protozoa. 1st edn., : Springer New York.
- Kelekar A (2005) Autophagy. Ann N Y Acad Sci 1066: 259-271. [Crossref]
- Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221: 3-12. [Crossref]
- De Virgilio C, Loewith R (2006) The TOR signalling network from yeast to man. Int J Biochem Cell Biol 38: 1476-1481. [Crossref]
- Shertz CA, Cardenas ME (2011) Exploiting and subverting Tor signaling in the pathogenesis of fungi, parasites, and viruses. PLoS Pathog 7: e1002269. [Crossref]
- Kiel JA (2010) Autophagy in unicellular eukaryotes. Philos Trans R Soc Lond B Biol Sci 365: 819-830. [Crossref]
- Besteiro S, Brooks CF, Striepen B, Dubremetz JF (2011) Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of Toxoplasma gondii tachyzoites. PLoS Pathog 7: e1002416. [Crossref]
- Ghosh D, Walton JL, Roepe PD, Sinai AP (2012) Autophagy is a cell death mechanism in Toxoplasma gondii. Cell Microbiol 14: 589-607. [Crossref]
- Matte C, Descoteaux A (2011) Disruption of the AKT/MTOR pathway by Leishmania major promastigotes. BMC Proc 5(Suppl 1): P44.
- Quan JH, Choi IW, Yang JB, Zhou W, Cha GH, et al. (2014) Trichomonas vaginalis metalloproteinase induces mTOR cleavage of SiHa cells.Korean J Parasitol 52: 595-603. [Crossref]
- Endoh H, Kobayashi T (2006) Death harmony played by nucleus and mitochondria: nuclear apoptosis during conjugation of Tetrahymena. Autophagy 2: 129-131. [Crossref]
- Martin-Gonzalez A, Benitez L, Gutierrez C (1992) Ultrastructural analisis of resting cysts and encystment in Colpoda inflata. 2. Encystment process and a review of ciliate resting cyst classification. Cytobios 72: 93-103.
- Gutierrez C, Callejas S, Bomiquel S Benítez L, Martín-González A (2001) Ciliate cryptobiosis: a microbial strategy against environmental starvation. Int Microbiol 4: 151-157. [Crossref]
- Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS (2006) CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest 116: 2366–2377. [Crossref]
- Majno G, Joris I (1995) Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146: 3-15. [Crossref]
- Walker NI, Harmon BV, Gobé GC, Kerr JF (1988) Patterns of cell death. Methods Achiev Exp Pathol 13: 18-54. [Crossref]
- Zong WX, Thompson CB (2006) Necrotic death as a cell fate. Genes Dev 20: 1-15. [Crossref]
- Green DR, Ferguson T, Zitvogel L, Kroemer G (2009) Immunogenic and tolerogenic cell death. Nat Rev Immunol 9: 353-363. [Crossref]
- Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, et al. (2013) Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 493: 547-551. [Crossref]
- Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, et al. (2009) Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A 48: 20388-20393. [Crossref]
- Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, et al. (2008) Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29: 21-32. [Crossref]
- Lüthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, et al. (2009) Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31: 84-98. [Crossref]
- Martin SJ, Henry CM, Cullen SP (2012) A perspective on mammalian caspases as positive and negative regulators of inflammation. Mol Cell 46: 387-397. [Crossref]
- Yang Y, Jiang G, Zhang P, Fan J (2015) Programmed cell death and its role in inflammation. Mil Med Res 2: 12. [Crossref]
- Miao EA, Rajan JV, Aderem A (2011) Caspase-1-induced pyroptotic cell death. Immunol Rev 243: 206-214. [Crossref]
- von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE (2013) Recognition of bacteria by inflammasomes. Annu Rev Immunol 31: 73-106. [Crossref]
- de Vasconcelos NM, Van Opdenbosch N, Lamkanfi M, et al. (2016) Inflammasomes as polyvalent cell death platforms. Cell Mol Life Sci 73: 2335-2347. [Crossref]
- Chen S, Sun B (2013) Negative regulation of NLRP3 inflammasome signaling. Protein Cell 4: 251-258. [Crossref]
- Chavarría-Smith J, Vance RE (2015) The NLRP1 inflammasomes. Immunol Rev 265: 22-34. [Crossref]
- Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677-687. [Crossref]
- Jo EK,, Kim JK,, Shin DM, et al. (2016) Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol 13: 148-159. [Crossref]
- Shio MT, Christian JG, Jung JY, Chang KP, Olivier M (2015) PKC/ROS-Mediated NLRP3 Inflammasome Activation Is Attenuated by Leishmania Zinc-Metalloprotease during Infection. PLoS Negl Trop Dis 9: e0003868. [Crossref]
- Mortimer L, Moreau F, Cornick S, Chadee K (2015) The NLRP3 Inflammasome Is a Pathogen Sensor for Invasive Entamoeba histolytica via Activation of a5ß1 Integrin at the Macrophage-Amebae Intercellular Junction. PLoS Pathog 11: e1004887. [Crossref]
- Ewald SE, Chavarria-Smith J, Boothroyd JC (2014) NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect Immun 82: 460-468. [Crossref]
- Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, et al. (2014) Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio 5: pii: e01117-13.
- Zamboni DS, Lima-Junior DS (2015) Inflammasomes in host response to protozoan parasites. Immunol Rev 265: 156-171. [Crossref]
- Olivier M, Van Den Ham K, Shio MT, Kassa FA, Fougeray S (2014) Malarial pigment hemozoin and the innate inflammatory response. Front Immunol 5: 25.
- Man SM, Karki R, Kanneganti TD (2016) AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol 46: 269-280. [Crossref]
- Gurung P, Karki R, Vogel P, Watanabe M, Bix M, et al. (2015) An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. J Clin Invest 125: 1329-1338. [Crossref]
- Charmoy M, Hurrell BP, Romano A, Lee SH, Ribeiro-Gomes F, et al. (2015) The Nlrp3 inflammasome, IL-1ß, and neutrophil recruitment are required for susceptibility to a non-healing strain of Leishmania major in C57BL/6 mice. Eur J Immunol. [Crossref]
- Davis MC, Ward JG, Herrick G, Allis CD (1992) Programmed nuclear death: apoptotic- like degradation of specific nuclei in conjugating Tetrahymena. Dev Biol 154: 419-432. [Crossref]
- Osada E, Akematsu T, Asano T, Endoh H (2014) A novel mitochondrial nuclease-associated protein: a major executor of the programmed nuclear death in Tetrahymena thermophila. Biol Cell 106: 97-109. [Crossref]
- Akematsu T, Pearlman RE, Endoh H (2010) Gigantic macroautophagy in programmed nuclear death of Tetrahymena thermophila. Autophagy 6: 901-911. [Crossref]
- Akematsu T, Fukuda Y, Attiq R, Pearlman RE (2013) Role of class III phosphatidylinositol 3-kinase during programmed nuclear death of Tetrahymena thermophila. Autophagy 10: 209-225. [Crossref]