Although biliary canal cysts were first described around the 1720s, the aetiology, physiopathology, natural course, and treatment options of the disease remain controversial. These cysts are becoming more common and can now be more easily diagnosed thanks to recent developments in imaging methods. Nevertheless, if left undiagnosed, the risk of progressive complications such as spontaneous perforation, cholelithiasis, choledocholithiasis, cholangitis, secondary biliary cirrhosis, portal hypertension, and development of malignancies should be considered. In this review, we discuss the epidemiology, classification, physiopathology, carcinogenesis, and clinical course of biliary cysts.
choledochal cyst, todani classification, choledochocel
Choledochal cysts (CCs) are rare medical conditions, which are congenital cystic dilatations of any portion of the bile ducts, most often occurring in the main portion of the common bile duct. Although choledochal cysts are considered a disorder of childhood and infancy, the ages in reported cases range from newly born to 80 years old; however 60% of such cysts are diagnosed in patients less than 10 years old [1-6].
Choledochal cysts (CCs) are extremely rare with an incidence of about 1/100–150,000 in Western societies. The disease affects 1 in 13,500 live births in the USA and 1 in 15,000 live births in Australia. It is seen more frequently in Asians; two out of three cases are of Japanese origin despite the reported incidence of 1/1,000. There is significant female gender predominance (F/M: 3–4/1). The cause of this female and Asian origin predominance is unknown [6].
Alonso-Lej defined three types of biliary dilatations in 1959; this classification system has since been widely accepted. Todani expanded this classification in 1977 and divided the CCs into five subgroups. Todani re-modified the classification to include pancreatic junctional abnormalities, and the resulting system became the final and most commonly used classification method [6] (Table 1) (Figure 1). According to the Todani classification, CCs are classified as follows:
Table 1. Todani classification of the bile duct cysts
|
Type IA Cystic dilatation of the extrahepatic bile ducts
|
|
Type IB Extrahepatic distal focal - segmental biliary dilatation
|
|
Type IC Extrahepatic fusiform biliary dilatation
|
|
Type II Extrahepatic biliary diverticula
|
|
Type III Intraduodenal portion of the common bile duct dilatation ( Choledococel)
|
|
Type IVA Multiple cystic dilatation of the intrahepatic and extrahepatic bile duct
|
|
Type IVB Multiple cystic dilatation of the only extrahepatic bile duct
|
|
Type V Cystic dilatation of the intrahepatic bile ducts ( Caroli's disease)
|
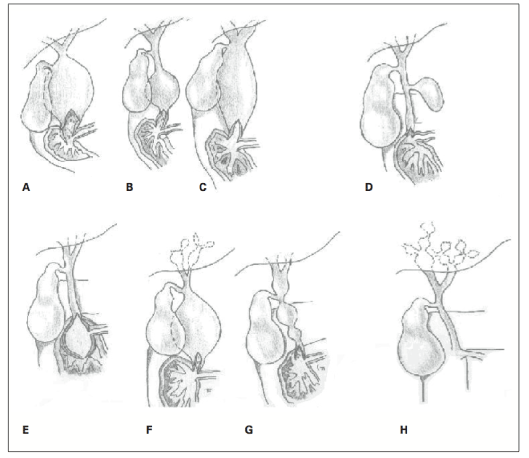
Figure 1. Todani classification of the bile duct cysts.
A: Type IA B: Type IB C: Type IC D: Type II E: Type III F: Type IVA G: Type IVB H: Tip V
Type IA Choledochal Cysts: Characterised by cystic dilatation of the extrahepatic biliary tract; the intrahepatic biliary tract is preserved. Gallbladder opens into a dilated hepatic duct.
Type IB Choledochal Cysts: Characterised by focal-segmental dilatation in the extrahepatic biliary tree. Cyst may be located anywhere within the biliary tract, but is typically distal. The cystic duct of the gallbladder opens into a biliary duct of normal diameter which is proximal to the cyst. The intrahepatic biliary tree is preserved.
Type IC Choledochal Cysts: Characterised by a regular and fusiform dilatation extending from the pancreaticobiliary junction into the intrahepatic biliary tract.
Type II Choledochal Cysts: Characterised by a diverticulum originating from the extrahepatic biliary tract that is generally connected to the tract via a narrow peduncle.
Type III Choledochal Cysts: Characterised by a dilatation limited to the duodenal wall in the distal part; named as a choledochal cell since it resembles a ureterocele morphologically and aetiologically. The outer wall of the cyst contains almost exclusively duodenal mucosa while the inner wall may include duodenal or biliary epithelium. This lesion has been divided into five subgroups by some authors according to its associations with the choledococele, ampulla Vateri and pancreatic channel; this classification has gained much support [7].
Type IVA Choledochal Cysts: Characterised by multiple intra-extra hepatic dilatations. The intrahepatic dilatation may be cystic, fusiform or irregular. In addition, Tadoni reported that these cysts could be classified as cystic-cystic, cystic-fusiform or fusiform-fusiform according to the shape of the intrahepatic and extrahepatic dilatations [8].
Type IVB Choledochal Cysts: Characterised by multiple dilatations including only the extrahepatic biliary tracts. The morphology of this type of CC can be described as “beads” or as “a bunch of grapes” [9].
Type V Choledochal Cysts: These cysts, also known as Caroli disease, are characterised by multiple intrahepatic saccular or cystic dilatations. Caroli disease describes isolated biliary dilatations, while Caroli syndrome describes biliary dilatations along with congenital hepatic fibrosis [10]. Some authors have described Caroli disease in addition to extrahepatic CC; however, they were unable to differentiate this entity from Type IVA CC. On the other hand, some authors have reported that differentiation can be based on an extrahepatic diffuse fusiform dilatation diameter < 3 cm, in addition to intrahepatic saccular dilatation [11,12].
Furthermore, a subgroup known as the “coarse form” refers to cases that present with abdominal pain and obstructive jaundice and that include pancreatobiliary junction abnormalities but no dilatation of the biliary tract. Patients in this group have the same clinical findings as with CCs; histological inflammation and malignancy potential are believed to represent another facet of the disease [13,14]. Other than these, combined types including Type I and II CCs have also been described [15].
Incidences have been reported as 50–80% for Type I, 2% for Type II, 1.4–4.5% for Type III, 15–35% for Type IV and 20% for Type V.
Although the exact aetiology of CC is unknown, many theories have been proposed in the pathophysiology of the condition. The most widely accepted hypothesis is Babbitt's theory, which states that the long common channel develops due to a pancreatobiliary junction abnormality. According to this theory, the long common channel allows mixing of the pancreatic secretions and bile for longer than usual, activating pancreatic enzymes. The activated enzymes then cause inflammation and destruction in the wall of the biliary tract, causing dilatation [16]. Also, high pressure inside the pancreatic channel causes progressive dilatation of the weak wall of the cyst [17].
The amylase level in the cystic common bile duct has also been found to be higher in this patient population than in control groups in some studies [18,19]. Furthermore, high amylase levels have been associated with early clinical findings and the degree of dysplasia. According to this theory, symptoms are seen at an earlier age, when the severity of the pancreaticobiliary reflux and the amylase level are higher and the disease is more asymptomatic, and becomes complicated at an older age when the severity of reflux and the amylase level are lower.
Since amylase can be used to determine the severity of the pancreaticobiliary reflux, trypsinogen and phospholipase-A2 levels have also been investigated as disease markers and were found to be increased in patients with CC [19-21]. Interestingly, trypsinogen was found to be activated by trypsin, in 61% of cases in the biliary tract and in 65% of cases in the gallbladder [19]. Enterokinase is necessary for this activation and normally it cannot be produced in mucosa other than the duodenal wall. Enterokinase is secreted by the dysplastic biliary epithelium, which is secondary to the pancreatic reflux and is activated by trypsinogen and lecithin-lisolecithin activation through phospholipase-A2, inflammation and destruction in the wall of the biliary tract has been proposed. This theory is further supported by animal experiments in which a pancreaticobiliary junction abnormality is produced surgically, leading to a biliary tract dilatation [21,22].
The presence of a pancreaticobiliary reflux has been confirmed in patients with a CC following administration of secretin, which increases pancreatic secretions in those patients. Secretin causes dilatation of the biliary tract and gallbladder. In the control group, on the other hand, the duodenum alone was shown to be filled, confirming the presence of a pancreaticobiliary reflux. This also supports the hypothesis that the presence of a pancreaticobiliary reflux is responsible for the pathogenesis of the “coarse form” of the disease [23,24].
Only 50–80% of CCs demonstrate an association with a pancreaticobiliary junction abnormality. In addition, the presence of antenatally diagnosed CCs despite a lack of immature pancreatic secretions also suggests that this theory is not fully adequate [25,26]. Also, when evaluating the long common channel theory, it is unclear which length defines “long”, since common channels of 10–45 mm have been demonstrated. Therefore, it has been suggested by some authors that a junction at a level other than the duodenal wall should be accepted as long, since this might allow mixing of pancreatic secretions with bile, leading to reflux [21].
Another theory is related to the congenital origin of CCs. Excess growth of the immature epithelium in the biliary tract during the development phase or an absence at any phase has been suggested to cause biliary tract dilatation [27,28]. A study that evaluated neonatal cystic CCs found that the amounts of neurons and ganglia were decreased in these cases [29]. Based on this finding, cystic dilatations were suggested to develop secondary to a dilatation at the distal part of the biliary tract, similar to Hirschprung disease, rather than fusiform ones that are acquired due to abnormal reflux. Another study found that elastin fibrils in the biliary tract are absent before the first year of life; cystic dilatations were proposed to develop before an individual is 1 year old, while fusiform dilatations were suggested to develop after 1 year of life secondary to increased pressure in the biliary tract [29,30].
Another theory proposes that dilatations seen in adults develop due to obstructions at the distal biliary tract secondary to various abnormalities (Oddi sphincter dysfunction, scar tissue and gallstones) and that a long narrow stenosis results in a cystic dilatation while a short wide stenosis results in a fusiform dilatation [8,29]. According to this theory, both distal and hilar-intrahepatic stenoses are necessary for the development of Type IV A cysts.
In general, those theories are meant to explain Type I and Type IV CCs. Type II cysts, on the other hand, are diverticular cysts that demonstrate minimal inflammation and carginogenic potential histologically. Therefore, it is unclear whether these cysts develop secondary to causes explained in the theories stated above or whether they are true biliary duplications [31].
Type II cysts (choledococeles) have been proposed to develop secondary to a pressure increase in the distal intramural biliary tract due to an ampullary obstruction or sphincter dysfunction. Some authors, on the other hand, suggest that choledococeles may actually be duodenal or biliary duplication cysts, since they can contain duodenal or biliary inner epithelium [32,33].
The premalignant nature of CC has been widely recognised; not only is the development of malignancy more frequent, the age of development of malignancy is also earlier than in the normal population [34]. Malignancy is a result of chronic inflammation, which leads to dysplasia and may also develop secondary to recurrent cholangitis and pancreatic reflux [35-37]. The risk of malignancy associated with a CC has been reported to be 10 to 15% in the overall population; however, this rate increases with increasing age [21,38]. The risk of malignancy is 23% at the age of 20 to 30 years, while it can increase up to 75% at the age of 70 to 80 years [35,39]. Malignancies include adenocarcinomas in 73 to 84% of cases, anaplastic carcinomas in 10%, undifferentiated carcinomas in 5 to 7%, squamous cell carcinomas in 5%, and other types of carcinoma in 1.5% [40,41]. These malignancies affect the extrahepatic biliary tract in 50 to 62% of cases, the gallbladder in 38 to 46%, the intrahepatic biliary tract in 2.5%, and the liver and pancreas in 0.7% [35]. The presence of a pancreaticobiliary junction abnormality carries a risk of malignancy in 16 to 55% of cases, regardless of whether a biliary dilatation is present [35,42,43]. The malignancy risk in the coarse form without biliary tract dilatation is 12 to 39%. While malignancies usually develop inside the cyst, in the coarse form they develop in the gallbladder. Therefore, some authors have suggested that tumours are most common in areas of highest exposure to biliary irritation (inside the cyst in patients with CC, or in the gallbladder if there is no cyst] [36-38]. The risk of malignancy is 7–15% and 2.5% in Caroli disease and in a choledococele, respectively [44-47].
Although the symptoms of biliary cysts can be seen at any age, they manifest before the age of 10 years in 80% of cases. Although the triad of abdominal pain, jaundice and an intraabdominal palpable mass is known as the classical clinical presentation, it is rare for a patient to present with all three signs (∼20%); however, two out of three of those symptoms are present in 8% of cases [48,49]. In the neonatal period, patients often present with abdominal pain and mechanical jaundice (< 12 months), while older patients present with abdominal pain, nausea and vomiting, and jaundice [50-52].
Clinical findings of CC develop secondary to ascending cholangitis and pancreatitis, which occur due to biliary stasis, development of gallstones, inflammation and development of secondary inflammation [49-66]. Pancreatitis develops due to obstruction of the pancreatic channel secondary to gallstones and protein-rich secretions from the dysplastic epithelium [52,61]. The cause of recurrent cholangitis in patients with intrahepatic involvement (Types IVA and V) is bacterial colonisation secondary to gallstones and obstruction due to intrahepatic biliary stasis [44,67]. The clinical presentation in those patients may advance to portal hypertension and biliary cirrhosis. Portal hypertension may develop without cirrhosis due to mechanical pressure of the cyst on the portal vein [69-73].
The initial symptoms may be abdominal pain and signs of peritonitis due to cyst rupture in 1-2% of cases [74]. Cyst rupture is thought to be due to the fact that the cyst wall, which becomes more fragile secondary to a distal obstruction in the biliary tract or increased intraabdominal pressure, cannot withstand the tension [75]. Perforation is often seen at the junction of the cystic duct and the main hepatic duct, which has the weakest blood supply in the biliary tract [74-75]. In cases of perforation, although the clinical findings are extremely aggressive, radiographic diagnosis is challenging because dilatations in the biliary tract disappear.
Patients with a choledococele (Type III) are usually asymptomatic. Rarely, findings of gastric outlet obstruction or duodenal obstruction may be seen in these patients due to cystic obstruction of the duodenal lumen [76-82].
Choledochal cysts are associated with many different developmental anomalies, which have given to rise some additional etiological theories. Such associations include colonic atresia, duodenal atresia, imperforate anus, pancreatic arteriovenous malformation, multiseptate gallbladder, OMENS plus syndrome, ventricular septal defect, aortic hypoplasia, pancreatic divisum, pancreatic aplasia, focal nodular hyperplasia, congenital absence of the portal vein, heterotopic pancreatic tissue and familial adenomatous polyposis [83-87].
- Lee HK, Park SJ, Yi BH, Lee AL, Moon JH, et al. (2009) Imaging features of adult choledochal cysts: a picto-rial review. Korean J Radiol 10: 71-80. [Crossref]
- Wiseman K, Buczkowski AK, Chung SW, Francoeur J, Schaeffer D, et al. (2005) Epidemiology, presentation, diagnosis, and outcomes of choledochal cysts in adults in an urban environment. Am J Surg 189: 527-531. [Crossref]
- Flanigan PD (1975) Biliary cysts. Ann Surg 182: 635-643. [Crossref]
- Powell CS, Sawyers JL, Reynolds VH (1981) Management of adult choledochal cysts. Ann Surg 193: 666-676. [Crossref]
- Crittenden SL, McKinley MJ (1985) Choledochal cyst--clinical features and classification. Am J Gastroenterol 80: 643-647. [Crossref]
- Singham J, Yoshida EM, Scudamore CH (2009) Choledochal cysts: part 1 of 3: classification and pathogenesis. Can J Surg 52: 434-440. [Crossref]
- Sarris GE, Tsang D (1989) Choledochocele: case report, literature review, and a proposed classification. Surgery 105: 408-414. [Crossref]
- Todani T, Watanabe Y, Toki A, Morotomi Y (2003) Classification of congenital biliary cystic disease: special reference to type Ic and IVA cysts with primary ductal stricture. J Hepatobiliary Pancreat 10: 340-344. [Crossref]
- Levy AD, Rohrmann CA Jr (2003) Biliary cystic disease. Curr Probl Diagn Radiol 32: 233-263. [Crossref]
- Keane F, Hadzic N, Wilkinson ML, Qureshi S, Reid C, et al. (1997) Neonatal presentation of Caroli's disease. Arch Dis Child Fetal Neonatal Ed 77: F145-146. [Crossref]
- Levy AD, Rohrmann CA Jr, Murakata LA, Lonergan GJ (2002) Caroli's disease: radiologic spectrum with pathologic correlation. AJR Am J Roentgenol 179: 1053-1057. [Crossref]
- Lilly JR, Stellin GP, Karrer FM (1985) Forme fruste choledochal cyst. J Pediatr Surg 20: 449-451. [Crossref]
- Sarin YK, Sengar M, Puri AS (2005) Forme fruste choledochal cyst. Indian Pediatr 42: 1153-1155. [Crossref]
- Thomas S, Sen S, Zachariah N, Chacko J, Thomas G (2002) Choledochal cyst sans cyst--experience with six "forme fruste" cases. Pediatr Surg Int 18: 247-251. [Crossref]
- Kaneyama K, Yamataka A, Kobayashi H, Lane GJ, Miyano T (2005) Mixed type I and II choledochal cyst: a new clinical subtype? Pediatr Surg Int 21: 911-913. [Crossref]
- Babbitt DP (1969) [Congenital choledochal cyst: new etiological concept based on anomalous relationships of the common bile duct and pancreatic bulb]. Ann Radiol (Paris) 12: 231-240. [Crossref]
- Han SJ, Hwang EH, Chung KS, Kim MJ, Kim H (1997) Acquired choledochal cyst from anomalous pancreatobiliary duct union. J Pediatr Surg 32: 1735-1738. [Crossref]
- Sugiyama M, Haradome H, Takahara T, Izumisato Y, Abe N, et al. (2004) Biliopancreatic reflux via anomalous pancreaticobiliary junction. Surgery 135: 457-459. [Crossref]
- Todani T, Narusue M, Watanabe Y, Tabuchi K, Okajima K (1978) Management of congenital choledochal cyst with intrahepatic involvement. Ann Surg 187: 272-280. [Crossref]
- Ochiai K, Kaneko K, Kitagawa M, Ando H, Hayakawa T (2004) Activated pancreatic enzyme and pancreatic stone protein (PSP/reg) in bile of patients with pancreaticobiliary maljunction/choledochal cysts. Dig Dis Sci 49: 1953-1956. [Crossref]
- Okada A, Hasegawa T, Oguchi Y, Nakamura T (2002) Recent advances in pathophysiology and surgical treatment of congenital dilatation of the bile duct. J Hepatobiliary Pancreat Surg 9: 342-351. [Crossref]
- Mizuno M1, Kato T, Koyama K (1996) An analysis of mutagens in the contents of the biliary tract in pancreaticobiliary maljunction. Surg Today 26: 597-602. [Crossref]
- Oguchi Y, Okada A, Nakamura T, Okumura K, Miyata M, et al. (1988) Histopathologic studies of congenital dilatation of the bile duct as related to an anomalous junction of the pancreaticobiliary ductal system: clinical and experimental studies. Surgery 103: 168-173. [Crossref]
2021 Copyright OAT. All rights reserv
- Matos C, Nicaise N, Deviere J, Cassart M, Metens T, et al. (1998) Choledochal cysts: comparison of findings at MR cholangiopancreatog-raphy and endoscopic retrograde cholangiopancreatography in eight patients. Radiology 209: 443-448. [Crossref]
- Imazu M, Iwai N, Tokiwa K, Shimotake T, Kimura O, et al. (2001) Factors of biliary carcinogenesis in choledochal cysts. Eur J Pediatr Surg 11: 24-27. [crossref]
- Hosoki T, Hasuike Y, Michita T, et al. (2004) Visualization of pancreaticobiliary reflux in anomalous pancreaticobiliary junction by secretin stimulated dynamic magnetic resonance cholangiopancreatography. Acta Radiol 45: 375-382. [Crossref]
- Cheng SP, Yang TL, Jeng KS, Liu CL, Lee JJ, et al. (2004) Choledochal cyst in adults: aetiological considerations to intrahepatic involvement. ANZ J Surg 74: 964-967. [Crossref]
- Yotsuyanagi S (1936) Contribution to aetiology and pathology of idiopathic cystic dilatation of the common bile duct with report of three cases. Gann 30: 601-752.
- Davenport M, Basu R (2005) Under pressure: choledochal malformation manometry. J Pediatr Surg 40: 331-335. [Crossref]
- Ohkawa H, Sawaguchi S, Yamazaki Y, Ishikawa A, Kikuchi M (1982) Experimental analysis of the ill effect of anomalous pancreaticobiliary ductal union. J Pediatr Surg 17: 7-13. [Crossref]
- Jindal RM, Harris N, McDaniel HM, et al. (1996) Presentation of choledochal cysts without intrabiliary communica-tion on endoscopic retrograde cholangiopancreatography. Liver Transpl Surg 2: 468-471. [Crossref]
- Kagiyama S, Okazaki K, Yamamoto Y, Yamamoto Y (1987) Anatomic variants of choledochocele and manometric measurements of pressure in the cele and the orifice zone. Am J Gastroenterol 82: 641-649. [Crossref]
- Venu RP, Geenen JE, Hogan WJ, Dodds WJ, Wilson SW, et al. (1984) Role of endoscopic retrograde cholangiopancreatography in the diagnosis and treatment of choledochocele. Gastroenterology 87: 1144-1149. [Crossref]
- Tsuchiya R, Harada N, Ito T, Furukawa M, Yoshihiro I (1977) Malignant tumors in choledochal cysts. Ann Surg 186: 22-28. [Crossref]
- Todani T, Watanabe Y, Fujii M (1985) Carcinoma arising from the bile duct in choledochal cyst and anomalous arrangement of the pancreatobiliary ductal union. Biliary Tract Pancreas 6: 525-535.
- Miyano G, Yamataka A, Shimotakahara A (2005) Cholecystectomy alone is inadequate for treatment of form fruste choledochal cyst: evidence from a rare but important case. Pediatr Surg Int 21: 61-63. [Crossref]
- Franko J, Nussbaum ML, Morris JB (2006) Choledochal cyst cholangiocarcinoma arising from adenoma: case report and a review of the literature. Curr Surg 63: 281-284. [Crossref]
- Bismuth H, Krissat J (1999) Choledochal cystic malignancies. Ann Oncol 10: 94-98. [Crossref]
- Benjamin IS (2003) Biliary cystic disease: the risk of cancer. J Hepatobiliary Pancreat Surg 10: 335-339. [Crossref]
- Todani T, Watanabe Y, Tokai A, et al. (1987) Carcinoma related to choledochalcysts with internal drainage operations. Surg Gynecol Obstet 164: 61-64. [Crossref]
- Fieber SS, Nance FC (1997) Choledochal cyst and neoplasm: a comprehensive review of 106 cases and presentation of two original cases. Am Surg 63: 982-987. [Crossref]
- Okamura K, Hayakawa H, Kuze M (2000) Triple carcinomas of the biliary tract associated with congenital choledochal dilatation and pancreaticobiliary maljunction. J Gastroenterol 35: 465-471. [Crossref]
- Miyano G, Yamataka A, Shimotakahara A, Kobayashi H, Lane GJ, et al. (2005) Cholecystectomy alone is inadequate for treating forme fruste choledochal cyst: evidence from a rare but important case report. Pediatr Surg Int 21: 61-63. [Crossref]
- Tanaka T (1995) Pathogenesis of choledochal cyst. Am J Gastroenterol 90: 685. [Crossref]
- Fulcher AS, Turner MA, Sanyal AJ (2001) Case 38: Caroli disease and renal tubular ectasia. Radiology 220: 720-723. [Crossref]
- Totkas S, Hohenberger P (2000) Cholangiocellular carcinoma associated with segmental Caroli's disease. Eur J Surg Oncol 26: 520-521. [Crossref]
- Ohtsuka T, Inoue K, Ohuchida J, Nabae T, Takahata S, et al. (2001) Carcinoma arising in choledochocele. Endoscopy 33: 614-619. [Crossref]
- Shi LB, Peng SY, Meng XK, Peng CH, Liu YB, et al. (2001) Diagnosis and treatment of congenital choledochal cyst: 20 years' experience in China. World J Gastroenterol 7: 732-734. [Crossref]
- Visser BC, Suh I, Way LW, Kang SM (2004) Congenital choledochal cysts in adults. Arch Surg 139: 855-860. [Crossref]
- O'Neill JA Jr, Templeton JM Jr, Schnaufer L, Bishop HC, Ziegler MM, et al. (1987) Recent experience with choledochal cyst. Ann Surg 205: 533-540. [Crossref]
- Le L, Pham AV, Dessanti A (2006) Congenital dilatation of extrahepatic bile ducts in children. Experience in the central hospital of Hue, Vietnam. Eur J Pediatr Surg 16: 24-27. [Crossref]
- Lee HC, Yeung CY, Fang SB, Jiang CB, Sheu JC, et al. (2006) Biliary cysts in children--long-term follow-up in Taiwan. J Formos Med Assoc 105: 118-124. [Crossref]
- Rattan KN, Magu S, Ratan S, Chaudhary A, Seth A (2005) Choledochal cyst in children: 15-year experience. Indian J Gastroenterol 24: 178. [Crossref]
- Chaudhary A, Dhar P, Sachdev A, Kumar N, Vij JC, et al. (1996) Choledochal cysts--differences in children and adults. Br J Surg 83: 186-188. [Crossref]
- Lipsett PA, Pitt HA, Colombani PM, Boitnott JK, Cameron JL (1994) Choledochal cyst disease. A changing pattern of presentation. Ann Surg 220: 644-652. [Crossref]
- Stain SC1, Guthrie CR, Yellin AE, Donovan AJ (1995) Choledochal cyst in the adult. Ann Surg 222: 128-133. [Crossref]
- Sela-Herman S, Scharschmidt BF (1996) Choledochal cyst, a disease for all ages. Lancet 347: 779. [Crossref]
- Büyükyavuz I, Ekinci S, Ciftçi AO, Karnak I, Senocak ME, et al. (2003) A retrospective study of choledochal cyst: clinical presentation, diagnosis and treatment. Turk J Pediatr 45: 321-325. [Crossref]
- Söreide K, Körner H, Havnen J, Söreide JA (2004) Bile duct cysts in adults. Br J Surg 91: 1538-1548. [Crossref]
- Rha SY, Stovroff MC, Glick PL, Allen JE, Ricketts RR (1996) Choledochal cysts: a ten year experience. Am Surg 62: 30-34. [Crossref]
- Hewitt PM, Krige JE, Bornman PC, Terblanche J (1995) Choledochal cysts in adults. Br J Surg 82: 382-385. [Crossref]
- de Vries JS, de Vries S, Aronson DC, Bosman DK, Rauws EA, et al. (2002) Choledochal cysts: age of presentation, symptoms, and late complications related to Todani's classification. J Pediatr Surg 37: 1568-1573. [Crossref]
- Postema RR, Hazebroek FW (1999) Choledochal cysts in children: a review of 28 years of treatment in a Dutch children's hospital. Eur J Surg 165: 1159-1161. [Crossref]
- Kabra V, Agarwal M, Adukai TK, Dixit VK, Agrawal AK, et al. (2001) Choledochal cyst: a changing pattern of presentation. ANZ J Surg 71: 159-161. [Crossref]
- Horaguchi J, Fujita N, Kobayashi G, Noda Y, Ito K, et al. (2005) Clinical study of choledochocele: is it a risk factor for biliary malignancies? J Gastroenterol 40: 396-401. [Crossref]
- Pereira LH, Bustorff-Silva JM, Sbraggia-Neto L, Bittencourt DG, Hessel G (2000) [Choledochal cyst: a 10-year experience] J Pediatr (Rio J) 76: 143-148. [Crossref]
- Karim AS (2004) Caroli's disease. Indian Pediatr 41: 848-850. [Crossref]
- Gupta AK, Gupta A, Bhardwaj VK, Chansoria M (2006) Caroli's disease. Indian J Pediatr 73: 233-235. [Crossref]
- Aguilera V, Rayón M, Pérez-Aguilar F, Berenguer J (2004) Caroli's syndrome and imaging: report of a case. Rev Esp Enferm Dig 96: 74-76. [Crossref]
- Harjai MM, Bal RK (2000) Caroli syndrome. Pediatr Surg Int 16: 431-432. [Crossref]
- Metcalfe MS, Wemyss-Holden SA, Maddern GJ (2003) Management dilemmas with choledochal cysts. Arch Surg 138: 333-339. [Crossref]
- Rao KL, Chowdhary SK, Kumar D (2003) Choledochal cyst associated with portal hypertension. Pediatr Surg Int 19: 729-732. [Crossref]
- Kiresi DA, Karabacakoglu A, Dilsiz A, Karakose S (2005) Spontaneous rupture of choledochal cyst presenting in childhood. Turk J Pediatr 47: 283-286. [Crossref]
- Moss RL, Musemeche CA (1997) Successful management of ruptured choledochal cyst by primary cyst excision and biliary reconstruction. J Pediatr Surg 32: 1490-1491. [Crossref]
- Arda IS, Tuzun M, Aliefendioglu D, Hicsonmez A (2005) Spontaneous rupture of extrahepatic choledochal cyst: two pediatric cases and literature review. Eur J Pediatr Surg 15: 361-363. [Crossref]
- Ramos A, Castell J, Pinto I (1990) Intestinal intussusception as a presenting feature of choledochocele. Gastrointest Radiol 15: 211-214. [Crossref]
- Yamaoka K, Tazawa J, Koizumi K, Asahina Y, Tajiri K, et al. (1994) Choledochocele with obstructive jaundice: a case report and a review of the Japanese literature. J Gastroenterol 29: 661-664. [Crossref]
- Zheng X, Gu W, Xia H, Huang X, Liang B, et al. (2013) Surgical treatment of type IV-A choledochal cyst in a single institution: children vs. adults. J Pediatr Surg 48: 2061-2066. [Crossref]
- Rayamajhi A, Singh R, Prasad R, Basnet NB (2006) An unusual case of Type IV(A) choledochal cyst with subaortic ventricular septal defect. Pediatr Int 48: 187-189. [Crossref]
- Iwasaki Y, Shimoda M, Furihata T, Rokkaku K, Sakuma A, et al. (2002) Biliary papillomatosis arising in a congenital choledochal cyst: report of a case. Surg Today 32: 1019-1022. [Crossref]
- Sgro M, Rosetti S, Barozzino T, et al. (2004) Caroli’s disease: prenatal diagnosis, postnatal outcome and genetic analysis. Ultrasound Obstet Gynecol 23: 73-76. [Crossref]
- Wheeler W (1940) An unusual case of obstruction of the common bile duct (choledochocele?). Br J Surg 27: 446-448.
- Ninan VT, Nampoory MR, Johny KV, Gupta RK, Schmidt I, et al. (2002) Caroli's disease of the liver in a renal transplant recipient. Nephrol Dial Transplant 17: 1113-1115. [Crossref]
- Al-Wafi A, Morris-Stiff G, Lari A (1998) Colonic atresia secondary to a choledochal cyst. Pediatr Surg Int 13: 422-423. [Crossref]
- Oyachi N, Ohhama Y, Take H, Fukuzato Y, Murakami T, et al. (2006) Aplasia of the dorsal pancreas and choledochal cyst. Pediatr Surg Int 22: 557-559. [Crossref]
- Komuro H, Takahashi MI, Matoba K, Hori T, Hirai M, et al. (2006) Rare association of severe hypoplasia of the abdominal aorta with imperforate anus, colonic atresia, and choledochal cyst. Pediatr Surg Int 22: 289-292. [Crossref]
- Dalvi AN, Pramesh CS, Prasanna GS, Rege SA, Khare R, et al. (1999) Incomplete pancreas divisum with anomalous choledochopancreatic duct junction with choledochal cyst. Arch Surg 134: 1150-1152. [Crossref]