Aging-associated neurodegenerative diseases, such as Alzheimer’s disease, represent a challenge for modern medicine and neuroscience. It is considered that any disease is related to degeneration of specific neuronal populations. However, it is not clear what factors initiate this specific degeneration and at which stage therapeutic interventions may still reverse neurodegenerative process and rescue functional capacities of neurons. Here we review data showing that the early stages of Alzheimer’s disease are rather associated with a loss of cholinergic neuron phenotype in the forebrain than with neuronal death in this brain region. This is manifested in specific disappearance of choline acetyltransferase (ChAT), the key enzyme of acetylcholine synthesis, in these neurons. We hypothesize that this loss of cholinergic phenotype may be related to impairments in ChAT protein synthesis rather than in ChAT gene expression. Other potential mechanisms resulting in the loss of cholinergic phenotype are discussed.
aging, Alzheimer’s disease, cholinergic neurons, choline acetyltransferase, protein synthesis
Aging is associated with cognitive decline, memory deterioration, motor impairments, and homeostasis destabilization. Most of these manifestations are directly related to brain aging accompanied by decrease in the structural complexity of neurons and volumes of the hippocampus and neocortex. On the other hand, neurodegenerative processes gradually develop and underlie progression of severe cerebral diseases such as Alzheimer’s disease (AD), Parkinson disease, Huntington disease or amyotrophic lateral sclerosis only in the minority of the population. Though sometimes associated with mutations in specific genes, these brain pathologies cannot be entirely explained by mutations. Each disease starts with neurodegeneration in specific brain region(s), but the causes of disease initiation and the vulnerability of specific cell groups for definite disease still remain obscure.
Indeed, discussing and exploring specific factors inducing definite neurodegenerative disease is a hot topic in neurobiology and translational medicine. Most researchers suggest that the main trigger of most neurodegenerative pathologies, including sporadic AD, is brain aging per se [1-5]. K. Herrup hypothesized on the relationship between AD pathogenesis and aging processes [3]. According to his hypothesis, age-related natural decrease in functional capacities of brain cells is followed by a moderate cognitive decline under the ideal conditions (Figure 1). Three main factors are explicit for transformation of normal aging to AD. First, so called “initiating damage”, such as vascular impairments, microstrokes, brain traumatic lesions, should affect the brain. We also suppose that chronic stress inducing depression may be considered as such triggering event. All these factors promote activation of neuroinflammation, the second factor. On the cellular and molecular levels, it is expressed as activation of microglia and alterations in the cytokine system, respectively. Cytokines are actively produced and secreted by microglial cells and circulate in the nervous tissue. These alterations are followed by changes in the state of neurons and modifications of their functional properties (the third factor) associated with synaptic dysfunction and neuronal cell death. The manifestation of AD in the form of dementia is the result of successive events associated with the above three factors (Figure).
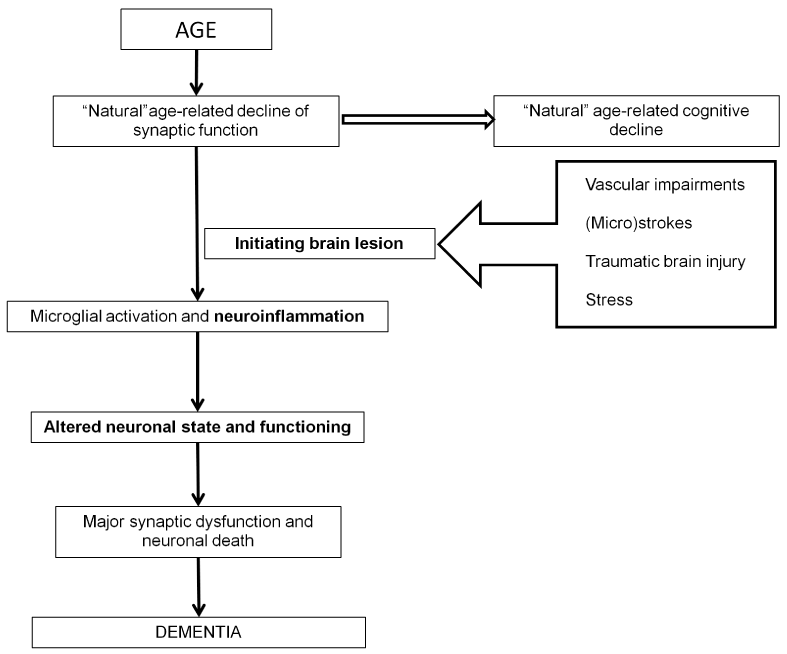
Figure 1. Schematic drawing of Herrup’s hypothesis on age-related pathogenesis of Alzheimer’s disease dementia with minor modifications.
K. Herrup [3] has stressed an important role of traumatic lesion and a unique role of neuroinflammation processes, which dramatically modify chemistry of neuronal microenvironment. Amyloid-β peptide (Aβ) is one of the triggers of neuroinflammation in the brain, while neuroinflammation in its turn may promote Aβ production [6]. AD manifestation is a result of significant alterations in functional capacities of all types of brain cells, including neurons, microglia, and astrocytes. The author of this hypothesis only pointed out that these changes in functional capacities lead to degeneration of a selective population of neurons in each specific neurodegenerative disease, however, did not consider region-specific mechanisms of selective neuronal death.
In 1982, a cholinergic hypothesis of geriatric memory impairments has been suggested [7]. Based on a large body of clinical studies on AD, the authors suggested that age-related memory impairments are associated with the degeneration of cholinergic neurons. Acetylcholine (ACh) is a neurotransmitter synthesized in cholinergic neurons in the reaction catalyzed by the enzyme choline acetyltransferase (ChAT). In AD patients, activity of ChAT is significantly decreased; while in individuals with “normal” cognitive aging decreases in ChAT activity were observed less frequently [8]. Similar patterns were found in aged rodents. The densities of muscarinic and nicotinic ACh receptors were relatively stable in aging while the binding of ligands with nicotinic receptors was significantly lower in AD patients [9].
Noteworthy, the number of cholinergic neurons in the basal nucleus of Meynert in aged individuals without cognitive impairments or in patients with mild cognitive impairments (MCI) was similar to that, observed in age-matched controls [10]. The early stages of AD pathogenesis are associated with a 15% decrease in the number of cholinergic neurons; however, the effect described in this study was statistically insignificant. Furthermore, in patients with MCI or at mild stage of AD ChAT activity is even increased in the hippocampus and frontal cortex demonstrating a compensatory capability of the cholinergic system [11]. At the early stage of AD, alterations in cholinergic cells are not limited to changes in expression and/or activity of ChAT, vesicular ACh transporter (VAChT) or ACh receptors only. Additionally, decreases in the ACh release and high-affinity choline uptake are observed as well as the attenuated response of neurons to neurotrophin signals, primarily to nerve growth factor (NGF) [8]. This is caused by decreased expression of TrkA, a high-affinity NGF receptor, in the neocortex and basal nuclei of AD patients [12,13]. Furthermore, the impaired NGF processing, i.e. formation of the mature NGF peptide from its precursor, and decreased expression of TrkA were found in MCI patients [14]. In the basal nuclei of patients with MCI or early AD, the number of neurons expressing the low-affinity p75NTR receptor was decreased by 38 and 43%, respectively [15]. However, the assessment of expression profile of proteins in a single cholinergic neuron of the basal nuclei demonstrated no changes in p75NTR expression and lower expression of TrkA in MCI or AD patients [16].
NGF/TrkA signaling is important for proper development and functioning of the cholinergic system [17]. NGF affects formation of the dendritic tree and modulates the activities of ChAT and acetylcholinesterase (AChE) in basal forebrain neurons [18]; therefore, cholinergic neurons critically depend on target-derived NGF. Interacting with TrkA receptor NGF activates a promoter of the cholinergic locus and thus, regulates expression of several genes essential for cholinergic transmission, including ChAT and VAChT as well as the gene encoding choline transporter. The effects of NGF on transcriptional activity may be mediated by phosphatidylinositol-3’-kinase-related phosphorylation of Akt kinase [19]. Furthermore, recent data from septal cholinergic-enriched cultures demonstrate that NGF withdrawal is followed by downregulation in the expression levels of several distinct presynaptic proteins involved in vesicles trafficking and neurotransmitter release, such as synapsin I, SNAP25 and α-synuclein, at early time-points [20]. These alterations are not associated with global changes in protein synthesis. Though the relationship between NGF, trk A and loss of ChAT activity is not completely established, alterations in NGF/TrkA signaling probably underlie the selective degeneration of the cholinergic basal forebrain neurons occurring in vivo in AD [21-23]. Therapeutic NGF gene intervention leads to a significant reduction of cognitive deficit along with an improvement of cholinergic hypofunction found in phase I clinical trial in humans affected from mild AD [24,25].
Studies on the molecular mechanisms underlying the time course of the above mentioned cellular alterations development in patients are extremely complicated. Therefore, various animal models were used to decipher the mechanism of cholinergic degeneration. It is well known that neuronal deafferentation results in retrograde degeneration. Disruption of the septo-hippocampal pathway by an experimental transection of the fimbria-fornix induces dysfunction of cholinergic neurons of the medial septal nucleus. Axotomy of cholinergic neurons results in a decrease in the number of neurons expressing cholinergic markers in the medial septum. In several studies, the reduction of cholinergic markers has been misinterpreted as analogous to the neurodegeneration of cholinergic cells [26,27]. However, a slow reduction of cholinergic cells labeled by ChAT and p75NTR demonstrated after fimbria-fornix transection was accompanied neither by neurodegenerative changes nor by a decrease in total number of neurons in the medial septum [27,28]. Although the remaining cells appeared healthy, they were unable to respond to delayed NGF infusion depending on the time of the start of the treatment. Hagg et al. [27] reported that the recovery of ChAT immunoreactivity was less pronounced if NGF was infused 14 or 21 days after the transection as compared to 7 days. The authors suggested that at the delayed time points, cholinergic neurons underwent to irreversible damage. These data were supported by a recent study [28]; however, the authors concluded that until 14 days after axotomy cholinergic neurons did not demonstrate activated caspase-3, p53 or Fluorojade C, markers of cell death. These results demonstrate that in axotomized cholinergic neurons down-regulation of NGF receptors occurs, precluding the possibility of a response to NGF. In other words, neurons lose their cholinergic phenotype without dying.
Another example of axotomy of the basal forebrain cholinergic neurons is olfactory bulbectomy (OBE) in rodents. The olfactory bulbs (OB) receive extensive cholinergic inputs from the basal forebrain via the nucleus of the horizontal limb of the diagonal band of Broca (HDB) innervating primarily glomerular and granule cell layers of the bulb [29]. In mice, OBE results in a decrease in the number of ChAT labeled neurons in the medial septal nucleus [30], intensity of ChAT immunostaining in the cerebral cortex, hippocampus, and amygdala [31] as well as ChAT content in the hippocampus [32]. Bobkova et al. [33] also reported that OBE in mice resulted in a decreased density of ChAT labeled cells in the HDB (55% of control), basal magnocellular preoptic nucleus (58.9%), and the caudate nucleus-putamen complex (68.2%). No significant changes in the vertical limb of the diagonal band of Broca and globus pallidus were observed. Recent data from this group demonstrate the involvement of mitochondrial dysfunction and oxidative stress in brain damage in an OBE model [34].
We have previously reported that in OBE mice, the number of ChAT-positive cells decreased by 25 and 48% in the medial septum and HDB, respectively [35] and the content of ChAT protein in the hippocampus was lower by 56% as compared to the respective control [36]. Furthermore, lower level of ChAT in the hippocampus was associated with an important trend to a decreased content of NGF by 23% in this brain region. These data are to some extent similar with the results of Antunes, et al. [37]. These authors demonstrated that OBE resulted in two-fold decreases in the contents of brain-derived neurotrophic factor (BDNF) and NGF in the hippocampus, associated with increased AChE activity. Lower expression of NGF mRNA has been also found in the hippocampus of OBE rats [38]. However, Hellweg et al. [39] demonstrated significantly increased BDNF, but normal NGF protein levels in hippocampus and frontal cortex of OBE mice as compared to sham-operated animals. This discrepancy may be due to different time points after surgery used to study neurotrophin levels, specifically 16 days in [39] versus more than 30 days in [36] and [37]. A principle failure of all these studies is the absence of a comprehensive study for all relevant indices in the same experiment, including NGF protein and mRNA, p75NTR and TrkA proteins and mRNAs, and ChAT and VAChT proteins and mRNAs. Based on the data from axotomy experiments it has been suggested that the physiological role of NGF in the adult septal cholinergic system is to support phenotypic differentiation but not neuronal survival, and this raises questions about the relationship between transcriptional regulation of the cholinergic phenotype by retrograde-derived trophic signaling and the transcriptional changes evident when retrograde transport is impaired due to neuropathological conditions [28].
Neurotrophic support, specifically NGF level and ratio between proNGF and mature NGF, substantially influence expression of ChAT in neurons. However, some cytokines produced in the brain may also significantly affect physiological properties of cholinergic neurons in the medial septum. Interleukin (IL)-2 is a potent stimulator of ACh release from septohippocampal neurons [40]. IL-2 gene knockout mice exhibited a marked reduction of ChAT-positive medial septum/diagonal band of Broca cell bodies as compared to wild-type littermates [41]. This loss of ChAT-positive neurons was selective for medial septum, since the cholinergic phenotype of wild-type and IL-2 knockout mice did not differ in a number of ChAT-positive neurons in the striatum, and GABAergic neurons in the medial septum/diagonal band of Broca did not differ between wild-type and IL-2 knockout mice [41]. However, total number of cells in the medial septum remained unchanged and the NGF content was even higher as compared to the control mice [42]. Thus, in spite of the previously cited hypothesis [28], the high content of NGF is not sufficient to maintain the cholinergic phenotype.
Loss of cholinergic phenotype is observed in another model of chronic neurodegeneration, induced by intracerebroventricular administration of Aβ(25-35). It is well known that injection of Aβ(25-35) to rats or mice results in impairments of learning and memory (see review [43]). This model allows reproducing some symptoms of early AD and studying the mechanisms of neurodegeneration. It has been shown that significantly lower number of ChAT labeled neurons in the medial septum is observed as early as 12 days after Aβ(25-35) administration into the rat cerebral ventricles [44]. The number of ChAT-positive neurons progressively decreased within a month after the treatment, whereas the total number of medial septal neurons remained relatively stable. Interestingly, the decrease in the number of cells with immunohistochemically detected ChAT expression was preceded by lower expression of ChAT and VAChT mRNAs in the medial septal area. Decreased expression of the high affinity NGF receptor TrkA mRNA accompanied these alterations of ChAT and VAChT expression. Later on, the expression of TrkA mRNA increased probably indicating a compensatory brain response. The time course of the compensatory increases in the contents of ChAT and VAChT mRNAs was associated with the appearance of a deficit in immunohistochemical staining for ChAT in the medial septum. On the other hand, expression of TrkA mRNA in the hippocampus increased only 28 days after Aβ(25-35) administration [45]. Importantly, decreased content of ChAT protein detected using immunohistochemistry was associated with high levels of ChAT mRNA transcripts in the brain. This may indirectly indicate impaired translation processes in cholinergic neurons of the basal forebrain in chronic neurodegeneration.
The importance of the NGF system was also evident in experiments with lentiviral vector-mediated transduction of hippocampal neurons. Administration of Aβ(25-35) into the cerebral ventricles or incubation of hippocampal slices with the peptide impaired long-term potentiation (LTP), a form of long-term synaptic plasticity, both in vivo and in slices [46,47]. However, preliminary transduction of neurons of the dentate gyrus with a vector containing the human NGF gene under the control of a neuron-specific CaMKII promoter allowed to prevent the LTP decline induced by Aβ(25-35). This protective effect was specific for chronically elevated NGF but not BDNF in the hippocampus [47].
Intrahippocampal injection of fibrillar Aβ induced direct lesions of the hippocampus, specifically CA1 field [48,49]. However, it also resulted in decreases in the medial septal ChAT and glutamate immunoreactive neurons as compared to controls. In contrast, the number of GAD67 inmunoreactive neurons was not significantly reduced [48]. Only scarce apoptotic cells were detected in the medial septal region of Aβ(1-40) treated animals but not in controls. These results show that limited Aβ-induced hippocampal lesions lead to an overall damage of vulnerable septal neuronal populations, as supposed by the authors, most likely by Aβ interaction with septo-hippocampal axon terminals. It has been shown that in the medial septum the expression of several genes related to oxidative stress was lower 24 h after injection of fibrillar Aβ(1-40) into the hippocampus, including superoxide dismutase-1 (SOD1), 8-oxoguanine DNA glycosylase, and monoamine oxidase A; however, expression of SOD1 was significantly increased 1 month after the treatment [50]. In the lateral septum, which does not contain cholinergic neurons, and in the hippocampus all these genes were overexpressed. These data suggest that cholinergic neurons of the basal forebrain may be specifically vulnerable to oxidative stress. Interestingly, this loss of cholinergic phenotype and expression of oxidative stress vulnerability are associated with an increased level of NGF observed in hippocampus after administration of Aβ(1-40) or Aβ(25-35) into this structure [51].
Cholinergic degeneration has been extensively studied in several genetic models of AD. Transgenic mice with overexpression of the human amyloid precursor protein (APP) gene develop plaques associated with activated microglia and astrocytes and other biomarkers of inflammation [52,53]. Only a few mouse models of AD have been shown to develop an actual neuron loss in the hippocampus evaluated by stereological quantification [54,55]. In aged mice expressing APP carrying Swedish mutation (APPswe or APP23), a modest decrease in cortical cholinergic enzyme activity as compared with age-matched wild-type mice has been reported [56,57]. In the brains of APP23 mice, the levels of α7 nAChR increased progressively over time most pronounced in areas of gliosis, reaching a 3- to 4-fold increase at 9 months of age. Interestingly, a decrease in α7 nAChR expression was observed in these mice at 12 months of age [58]. Total cholinergic fiber length was more severely affected, with 29 and 35% decreases in the neocortex of aged APP23 mice compared with age-matched wild-type mice and young transgenic mice, respectively. However, there was no loss of cholinergic basal forebrain neurons in these aged APP23 mice [56]. The presence of the APPswe gene did not augment the vulnerability of forebrain cholinergic neurons to the chronic neuroinflammation [59].
Cholinergic dysfunction has been studied in another AD mouse model APP_SweDI, overexpressing APP with the Swedish K670N/M671L, Dutch E693Q, and Iowa D694N mutations. A significant decrease in cholinergic neurons in the transgenic mouse model in comparison with the wild-type mice was revealed, identified by immunohistochemistry against ChAT and p75NTR as well as by in situ hybridization. Moreover, a significant decrease in cortical cholinergic fiber density was found in the transgenic mice as compared to the wild-type. In the cerebral cortex of APP_SweDI mice, swollen cholinergic varicosities were seen in the vicinity of Ab plaques [60]. In human APP transgenic mice expressing mutated Swedish K670M/N671L and London V717I human APP751, a loss of ChAT immunoreactivity in the basal nucleus correlated with the enhanced level of pro-NGF in the hippocampus [61]. The number of TrkA and p75NTR positive cells in the basal nucleus remained unchanged. Treatment of these mice with cerebrolysin, a drug normalizing pro-NGF/NGF ratio, reduced ChAT deficit, and no cell death was found in mice of this strain. Additionally, reductions in hippocampal ChAT protein levels associated with degeneration of cholinergic neurons, were analyzed in 5×FAD mice, overexpressing mutant human APP(695) with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) familial AD (FAD) mutations along with human PS1 harboring two FAD mutations, M146L and L286V [62]. These mice had lower hippocampal ChAT protein levels and ChAT-immunoreactive neurons in the medial septum and the vertical limb of the diagonal band (Ch1/2) providing cholinergic innervations to the hippocampus. However, death of cholinergic neurons has not been demonstrated in these models either.
In APP/PS1KI mice, expressing both APP, carrying Swedish and London mutations, and presenilin 1 (PS1) with the M233T and L235P FAD mutations, expression of the APP transgene was found in ChAT-positive neurons of motor nuclei accompanied by robust intracellular Aβ accumulation, whereas no APP expressing neurons and, thus, no intracellular Aβ accumulation were found in either the forebrain or pons complexes, or in the caudate putamen [63, 64]. Stereological cell count revealed a loss of ChAT-positive neurons in APP/PS1KI mice only in the motor nuclei Mo5 and 7N accumulating intracellular Aβ. Thus, intracellular Aβ accumulation is supposed being an early pathological alteration contributing to cell death in AD [63]. A specific capability of Aβ oligomers to attack cholinergic neurons and inhibit ChAT in them has been also demonstrated in cell cultures [65].
Though the reasons of loss of a cholinergic phenotype remain unclear, there are several possible factors which may induce this loss. The above data demonstrate that impaired protein synthesis may be one of the reasons. Early stages of AD are associated with endoplasmic reticulum stress [66]. Transient inhibition of protein synthesis due to suppression of translation is an adaptive response of the cell to endoplasmic reticulum stress [67]. Neurons are especially sensitive to this inhibition because normal cerebral functions including memory formation need continuous protein synthesis. Therefore, chronic inhibition of translation is followed by neuronal death [68]. Inhibition of protein synthesis due to ribosome dysfunction is one of the earliest features of metabolic alterations in the AD brain [69]. These alterations are caused by formation of heavy polyribosomes, total decrease in rRNA and tRNA, and a significant increase in the content of oxidized RNA, which can be observed in the brain of MCI and early AD patients [69,70]. rRNA is one of most abundant molecules in almost all types of cells and the molecule generally vulnerable to oxidative stress-induced damage in human and animal brain. 5S rRNA, the smallest molecule among all four forms of rRNAs, is responsible for formation of a ribosomal complex and its stability [71]. In AD, oxidative stress significantly damages 5S rRNA and, thus, specifically impairs functions of ribosomes. In addition to rRNA, up to 50% of mRNA is subjected to oxidative damage in AD brain [72] and ribosomes purified from Alzheimer hippocampus contained significantly higher levels of RNase-sensitive iron(II) and redox activity than control [73]. In cell cultures, oxidation of mRNA not only prevents normal protein synthesis but also serves as the first step triggering cell death [74].
It has been shown that administration of Aβ(25-35) into the lateral ventricles of rat brain induces oxidative and nitrosative stress [75,76] probably promoting RNA oxidation. Administration of Aβ into the rat brain leads to formation of heavy polyribosomes in hippocampal neurons followed by impaired protein synthesis [77]. Using proteome analysis Virok et al. [78] demonstrated that Aβ oligomers may potentially interact with 24 proteins involved in initiation of protein translation and elongation of polypeptides. These authors revealed that Aβ oligomers interact with ribosomes and inhibit translation on a concentration-dependent manner. Indeed, protein levels of translation initiation factors eIF2α, eIF3η and eIF5, and elongation factor eEF2, are altered in the CA1 region in AD [79]. Additionally, increased phosphorylation of the mammalian (mechanistic) target of rapamycin (mTOR), a protein kinase phosphorylating a wide spectrum of intracellular proteins, may also promote inhibition of protein synthesis in AD [80]. It is possible that mTOR phosphorylates the elongation factor 1A (EF1A), a crucial translation factor mediating peptide elongation by promoting GTP-dependent binding of aminoacyl tRNA to the ribosome, and eIF2α and thus, inhibits protein synthesis.
In AD and other taupathies, the pathological tau protein associates with ribosomes leading to impaired RNA translation. This may result in a decrease in synthesis of the synaptic protein PSD-95 [81]. In addition to this direct effect of tau on protein translation, it may indirectly inhibit ribosome functions due to chronic suppression of endoplasmic reticulum-associated degradation, which in its turn activates the unfolded protein response and subsequently the protein kinase RNA-like endoplasmic reticulum kinase (PERK) pathway [82]. The prolonged activation of the PERK pathway results in a reduction in RNA translation through phosphorylation of the initiation factor eIF2α [83]. However, elevated phosphorylation of eIF2α paradoxically causes translational activation of a subset of mRNAs such as the β-secretase enzyme, β-site APP-cleaving enzyme 1 (BACE1) and cAMP response element binding protein (CREB) repressor, activating transcription factor 4 (ATF4). In a 5×FAD mouse model, increased phospho-eIF2α level is associated with cholinergic dysfunction but not cell loss [62,84]. Unfortunately, it is not clear to which extent the data from AD brain autopsy and transgenic models could be correlated with selective lesion or loss of cholinergic neurons. Furthermore, silencing of ChAT expression using RNA interference was not followed by significant cell death in cultured cells or the rat brain [85]. In contrast to alterations evident for expression of NGF receptors [16], the changes in protein synthesis were demonstrated at the level of some cerebral structures such as cortex or hippocampus, but not clearly shown to be specific for cholinergic neurons.
It is not clear yet whether we can translate data from experimental studies into clinic and suggest that the early stages of AD are associated with loss of cholinergic phenotype rather than death of cholinergic neurons. Future studies are needed to answer the question whether inhibition of protein synthesis in forebrain cholinergic neurons is responsible for a loss of their phenotype at the early stages of neurodegeneration and to which extent. We believe that general efforts to tackle these problems will help to better understand neurobiology of AD and develop new strategies for early pharmacological interventions.
This study was supported by the Russian Foundation for Basic Research, grant #16-04-01054a.
- Bishop NA, Lu T, Yankner BA (2010) Neural mechanisms of ageing and cognitive decline. Nature 464: 529-535. [Crossref]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB (2014) Alzheimer's Disease Neuroimaging Initiative What is normal in normal aging? Effects of aging, amyloid and Alzheimer's disease on the cerebral cortex and the hippocampus. Prog Neurobiol 117: 20-40. [Crossref]
- Herrup K (2010) Reimagining Alzheimer's disease--an age-based hypothesis. J Neurosci 30: 16755-16762. [Crossref]
- Hunter S, Arendt T, Brayne C (2013) The senescence hypothesis of disease progression in Alzheimer disease: an integrated matrix of disease pathways for FAD and SAD. Mol Neurobiol 48: 556-570. [Crossref]
- Mattson MP (2004) Pathways towards and away from Alzheimer's disease. Nature 430: 631-639. [Crossref]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, et al. (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol 14: 388-405. [Crossref]
- Bartus RT, Dean RL, Beer B, Lippa AS (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217: 408-414. [Crossref]
- Schliebs R, Arendt T (2011) The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221: 555-563. [Crossref]
- Lagarde J, Sarazin M, Chauviré V, Stankoff B, Kas A, et al. (2017) Cholinergic Changes in Aging and Alzheimer Disease: An [18F]-F-A-85380 Exploratory PET Study. Alzheimer Dis Assoc Disord 31: 8-12. [Crossref]
- Gilmor ML, Erickson JD, Varoqui H, Hersh LB, Bennett DA, et al. (1999) Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer's disease. J Comp Neurol 411: 693-704. [Crossref]
- DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, et al. (2002) Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol 51: 145-155. [Crossref]
- Boissière F, Hunot S, Faucheux B, Hersh LB, Agid Y, et al. (1997) Trk neurotrophin receptors in cholinergic neurons of patients with Alzheimer's disease. Dement Geriatr Cogn Disord 8: 1-8. [Crossref]
- Mufson EJ, Li JM, Sobreviela T, Kordower JH (1996) Decreased trkA gene expression within basal forebrain neurons in Alzheimer's disease. Neuroreport 8: 25-29. [Crossref]
- Peng S, Wuu J, Mufson EJ, Fahnestock M (2004) Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol 63: 641-649. [Crossref]
- Mufson EJ, Ma SY, Dills J, Cochran EJ, Leurgans S, et al. (2002) Loss of basal forebrain P75(NTR) immunoreactivity in subjects with mild cognitive impairment and Alzheimer's disease. J Comp Neurol 443: 136-153. [Crossref]
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ (2006) Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J Neurochem 97: 475-487. [Crossref]
- Yuen EC, Howe CL, Li Y, Holtzman DM, Mobley WC (1996) Nerve growth factor and the neurotrophic factor hypothesis. Brain Dev 18: 362-368. [Crossref]
- Isaev NK, Stelmashook EV, Genrikhs EE (2017) Role of Nerve Growth Factor in Plasticity of Forebrain Cholinergic Neurons. Biochemistry (Mosc) 82: 291-300. [Crossref]
- Madziar B., Shah S., Brock M., Burke R., Lopez-Coviella I., et al. (2008) Nerve growth factor regulates the expression of the cholinergic locus and the high-affinity choline transporter via the Akt/PKB signaling pathway. J Neurochem 107: 1284-1293. [Crossref]
- Latina V, Caioli S, Zona C, Ciotti MT, Amadoro G, et al. (2017) Impaired NGF/TrkA Signaling Causes Early AD-Linked Presynaptic Dysfunction in Cholinergic Primary Neurons. Front Cell Neurosci 11: 68. [Crossref]
- Counts SE, Mufson EJ (2005) The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J Neuropathol Exp Neurol 64: 263-272. [Crossref]
- Mufson EJ, Counts SE, Perez SE, Ginsberg SD (2008) Cholinergic system during the progression of Alzheimer's disease: therapeutic implications. Expert Rev Neurother 8: 1703-1718. [Crossref]
- Niewiadomska G, Mietelska-Porowska A, Mazurkiewicz M (2011) The cholinergic system, nerve growth factor and the cytoskeleton. Behav Brain Res 221: 515-526. [Crossref]
- Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, et al. (2005) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 11: 551-555. [Crossref]
- Tuszynski MH, Yang JH, Barba D, U HS, Bakay RA, et al. (2015) Nerve Growth Factor Gene Therapy: Activation of Neuronal Responses in Alzheimer Disease. JAMA Neurol 72: 1139-1147. [Crossref]
- Gage FH, Armstrong DM, Williams LR, Varon S (1988) Morphological response of axotomized septal neurons to nerve growth factor. J Comp Neurol 269: 147-155. [Crossref]
- Hagg T, Fass-Holmes B, Vahlsing HL, Manthorpe M, Conner JM (1989) Nerve growth factor (NGF) reverses axotomy-induced decreases in choline acetyltransferase, NGF receptor and size of medial septum cholinergic neurons. Brain Res 505: 29-38.
- Lazo OM, Mauna JC, Pissani CA, Inestrosa NC, Bronfman FC (2010) Axotomy-induced neurotrophic withdrawal causes the loss of phenotypic differentiation and downregulation of NGF signalling, but not death of septal cholinergic neurons. Mol Neurodegener 5: 5. [Crossref]
- Heimer L, Zahm DS, and Schmued LC (1990) The basal forebrain projection to the region of the nuclei gemini in the rat; a combined light and electron microscopic study employing horseradish peroxidase, fluorescent tracers and Phaseolus vulgaris-leucoagglutinin. Neuroscience 34: 707-731. [Crossref]
- Han F, Shioda N, Moriguchi S, Qin ZH, Fukunaga K (2008) The vanadium (IV) compound rescues septo-hippocampal cholinergic neurons from neurodegeneration in olfactory bulbectomized mice. Neuroscience 151: 671-679. [Crossref]
- Hozumi S, Nakagawasai O, Tan-No K, Niijima F, Yamadera F, et al. (2003) Characteristics of changes in cholinergic function and impairment of learning and memory-related behavior induced by olfactory bulbectomy. Behav Brain Res 138: 9-15. [Crossref]
- Nakajima A, Yamakuni T, Haraguchi M, Omae N, Song SY, et al. (2007) Nobiletin, a citrus flavonoid that improves memory impairment, rescues bulbectomy-induced cholinergic neurodegeneration in mice. J Pharmacol Sci 105: 122-126. [Crossref]
- Bobkova NV, Nesterova IV, Nesterov VV (2001) The state of cholinergic structures in forebrain of bulbectomized mice. Bull Exp Biol Med 131: 427-431. [Crossref]
- Avetisyan AV, Samokhin AN, Alexandrova IY, Zinovkin RA, Simonyan RA, et al. (2016) Mitochondrial Dysfunction in Neocortex and Hippocampus of Olfactory Bulbectomized Mice, a Model of Alzheimer's Disease. Biochemistry (Mosc) 81: 615-623. [Crossref]
2021 Copyright OAT. All rights reserv
- Stepanichev M, Markov D, Pasikova N, Gulyaeva N (2016) Behavior and the cholinergic parameters in olfactory bulbectomized female rodents: Difference between rats and mice. Behav Brain Res 297: 5-14. [Crossref]
- Stepanichev M, Nedogreeva OA, Gulyaeva N (2016) Mechanisms of impairments of functions of cholinergic neurons under the chronic neurodegenerative conditions. Fundamental and Applied Problems of Neurosciences: Functional Asymmetry, Neuroplasticity, and Neurodegeneration. Scientific Center of Neurology Moscow Pp: 892-897.
- Antunes MS, Jesse CR, Ruff JR, de Oliveira Espinosa D, Gomes NS, et al. (2016) Hesperidin reverses cognitive and depressive disturbances induced by olfactory bulbectomy in mice by modulating hippocampal neurotrophins and cytokine levels and acetylcholinesterase activity. Eur J Pharmacol 789: 411-420. [Crossref]
- Song C, Zhang XY, Manku M (2009) Increased phospholipase A2 activity and inflammatory response but decreased nerve growth factor expression in the olfactory bulbectomized rat model of depression: effects of chronic ethyl-eicosapentaenoate treatment. J Neurosci 29: 14-22. [Crossref]
- Hellweg R, Zueger M, Fink K, Hörtnagl H, Gass P (2007) Olfactory bulbectomy in mice leads to increased BDNF levels and decreased serotonin turnover in depression-related brain areas. Neurobiol Dis 25: 1-7. [Crossref]
- Hanisch UK, Seto D, Quirion R (1993) Modulation of hippocampal acetylcholine release: a potent central action of interleukin-2. J Neurosci 13: 3368-3374. [Crossref]
- Beck RD Jr, King MA, Huang Z, Petitto JM (2002) Alterations in septohippocampal cholinergic neurons resulting from interleukin-2 gene knockout. Brain Res 955: 16-23. [Crossref]
- Meola DM, Huang Z, King M, Petitto JM (2013) Loss of cholinergic phenotype in septohippocampal projection neurons: relation to brain versus peripheral IL-2 deficiency. Neurosci Lett 539: 60-64. [Crossref]
- Gulyaeva NV, Stepanichev MY (2010) Abeta(25-35) as proxyholder for amyloidogenic peptides: in vivo evidence. Exp Neurol 222: 6-9. [Crossref]
- Stepanichev M, Lazareva N, Tukhbatova G, Salozhin S, Gulyaeva N (2014) Transient disturbances in contextual fear memory induced by Aβ(25-35) in rats are accompanied by cholinergic dysfunction. Behav Brain Res 259: 152-157. [Crossref]
- Stepanichev MY, Tishkina AO, Lazareva NA, Mart’yanova EK, Tukhbatova GR (2015) The expression of the TrkA and TrkB high-affinity neurotrophin receptors in the rat hippocampus after intracerebroventricular administration of Aß(25–35) Neurochem J 9: 47-53.
- Uzakov SS, Ivanov AD, Salozhin SV, Markevich VA, Gulyaeva NV (2015) Lentiviral-mediated overexpression of nerve growth factor (NGF) prevents beta-amyloid [25-35]-induced long term potentiation (LTP) decline in the rat hippocampus. Brain Res 1624: 398-404. [Crossref]
- Ivanov AD, Tukhbatova GR, Salozhin SV, Markevich VA (2015) NGF but not BDNF overexpression protects hippocampal LTP from beta-amyloid-induced impairment. Neuroscience 289: 114-122. [Crossref]
- Colom LV, Castaneda MT, Hernandez S, Perry G, Jaime S, et al. (2011) Intrahippocampal amyloid-ß (1-40) injections injure medial septal neurons in rats. Curr Alzheimer Res 8: 832-840. [Crossref]
- Stepanichev MY, Zdobnova IM, Yakovlev AA, Onufriev MV, Lazareva NA (2003) Effects of tumor necrosis factor-alpha central administration on hippocampal damage in rat induced by amyloid beta-peptide (25-35). J Neurosci Res 71: 110-120. [Crossref]
- Durán-González J, Michi ED, Elorza B, Perez-Córdova MG, Pacheco-Otalora LF, et al. (2013) Amyloid ß peptides modify the expression of antioxidant repair enzymes and a potassium channel in the septohippocampal system. Neurobiol Aging 34: 2071-2076. [Crossref]
- Stepanichev MYu , Ivanov AD, Lazareva NA, Moiseeva YuV, Gulyaeva NV (2016) Neurodegenerative changes induced by injection of ß-amyloid peptide fragment (25-35) in hippocampus are associated with NGF-signalling activation. Vestnik RGMU 1: 13-18.
- Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, et al. (1999) Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging 20: 581-589. [Crossref]
- Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, et al. (2001) Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Pathol 158: 1345-1354. [Crossref]
- Casas C., Sergeant N., Itier J.M., Blanchard V., Wirths O., et al. (2004) Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 165: 1289-1300. [Crossref]
- Schmitz C, Rutten BP, Pielen A, Schafer S, Wirths O, et al. (2004) Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am J Pathol 164: 1495-1502. [Crossref]
- Boncristiano S, Calhoun ME, Kelly PH, Pfeifer M, Bondolfi L, et al. (2002) Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J Neurosci 22: 3234-3243. [Crossref]
- Lüth HJ, Apelt J, Ihunwo AO, Arendt T, Schliebs R (2003) Degeneration of beta-amyloid-associated cholinergic structures in transgenic APP SW mice. Brain Res 977: 16-22. [Crossref]
- Jones IW, Westmacott A, Chan E, Jones RW, Dineley K, et al. (2006) Alpha7 Nicotinic acetylcholine receptor expression in Alzheimer’s disease: Receptor densities in brain regions of the APP(SWE) mouse model and in human peripheral blood lymphocytes. J Mol Neurosci 30: 83-84. [Crossref]
- Wenk GL, McGann-Gramling K, Hauss-Wegrzyniak B (2004) The presence of the APPswe mutation in mice does not increase the vulnerability of cholinergic basal forebrain neurons to neuroinflammation. Neuroscience 125: 769-776. [Crossref]
- Foidl BM, Do-Dinh P, Hutter-Schmid B, Bliem HR, Humpel C (2016) Cholinergic neurodegeneration in an Alzheimer mouse model overexpressing amyloid-precursor protein with the Swedish-Dutch-Iowa mutations. Neurobiol Learn Mem 136: 86-96.
- Ubhi K, Rockenstein E, Vazquez-Roque R, Mante M, Inglis C, et al. (2013) Cerebrolysin modulates pronerve growth factor/nerve growth factor ratio and ameliorates the cholinergic deficit in a transgenic model of Alzheimer's disease. J Neurosci Res 91: 167-177. [Crossref]
- Devi L, Ohno M (2010) Phospho-eIF2Î ± level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS One 5: e12974. [Crossref]
- Christensen DZ, Bayer TA, Wirths O (2010) Intracellular Aß triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer's disease. Neurobiol Aging 31: 1153-1163. [Crossref]
- Jaffar S, Counts SE, Ma SY, Dadko E, Gordon MN, et al. (2001) Neuropathology of mice carrying mutant APPswe and/or PS1M146L transgenes: alterations in the p75NTR cholinergic basal forebrain septohippocampal pathway. Exp Neurol 170: 227-243. [Crossref]
- Nunes-Tavares N, Santos LE, Stutz B, Brito-Moreira J, Klein WL, et al. (2012) Inhibition of choline acetyltransferase as a mechanism for cholinergic dysfunction induced by amyloid-ß peptide oligomers. J Biol Chem 287: 19377-19385. [Crossref]
- Unterberger U, Höftberger R, Gelpi E, Flicker H, Budka H, et al. (2006) Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol 65: 348-357. [Crossref]
- Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397: 271-274. [Crossref]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, et al. (2012) Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485: 507-511. [Crossref]
- Ding Q, Markesbery WR, Chen Q, Li F, Keller JN (2005) Ribosome dysfunction is an early event in Alzheimer's disease. J Neurosci 25: 9171-9175. [Crossref]
- Ding Q, Markesbery WR, Cecarini V, Keller JN (2006) Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer's disease. Neurochem Res 31: 705-710. [Crossref]
- Ding Q, Zhu H, Zhang B, Soriano A, Burns R, et al. (2012) Increased 5S rRNA oxidation in Alzheimer's disease. J Alzheimers Dis 29: 201-209. [Crossref]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, et al. (1999) RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci 19: 1959-1964. [Crossref]
- Honda K, Smith MA, Zhu X, Baus D, Merrick WC, et al. (2005) Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol Chem 280: 20978-20986. [Crossref]
- Shan X, Chang Y, Lin CL (2007) Messenger RNA oxidation is an early event preceding cell death and causes reduced protein expression. FASEB J 21: 2753-2764. [Crossref]
- Stepanichev MY, Onufriev MV, Yakovlev AA, Khrenov AI, Peregud DI, et al. (2008) Amyloid-beta (25-35) increases activity of neuronal NO-synthase in rat brain. Neurochem Int 52: 1114-1124. [Crossref]
- Stepanichev MY, Zdobnova IM, Zarubenko II, Moiseeva YV, Lazareva NA, et al. (2004) Amyloid-beta(25-35)-induced memory impairments correlate with cell loss in rat hippocampus. Physiol Behav 80: 647-655. [Crossref]
- Gordon RY, Makarova EG, Podolski IY, Rogachevsky VV, Kordonets OL. (2012) Impairment of protein synthesis is an early effect of amyloid-ß in neurons. Neurochem J 6: 121-127.
- Virok DP, Simon D, Bozsó Z, Rajkó R, Datki Z, et al. (2011) Protein array based interactome analysis of amyloid-ß indicates an inhibition of protein translation. J Proteome Res 10: 1538-1547. [Crossref]
- Hernández-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I. (2016) Altered machinery of protein synthesis in Alzheimer's: from the nucleolus to the ribosome. Brain Pathol 26: 593-605. [Crossref]
- Di Domenico F, Barone E, Perluigi M, Butterfield DA. (2017) The triangle of death in Alzheimer's disease brain: the aberrant cross-talk among energy metabolism, mammalian target of rapamycin signaling, and protein homeostasis revealed by redox proteomics. Antioxid Redox Signal 26: 364-387. [Crossref]
- Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, et al. (2016) Pathological tau promotes neuronal damage by impairing ribosomal function and decreasing protein synthesis. J Neurosci 36: 1001-1007. [Crossref]
- Abisambra JF, Jinwal UK, Blair LJ, O'Leary JC 3rd., Li Q, et al. (2013) Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci 3: 9498-9507. [Crossref]
- Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D (2006) Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J Cell Biol 72: 201-209. [Crossref]
- Devi L, Ohno M (2014) PERK mediates eIF2a phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer's disease. Neurobiol Aging 35: 2272-2281. [Crossref]
- Santamaria J, Khalfallah O, Sauty C, Brunet I, Sibieude M, et al. (2009) Silencing of choline acetyltransferase expression by lentivirus-mediated RNA interference in cultured cells and in the adult rodent brain. J Neurosci Res 87: 532-544. [Crossref]