Human c-ErbB2 (HER2) has long been used as a marker of breast cancer (BC) for sub-categorization for the prediction of prognosis, and determination of therapeutic strategies. HER2 overexpressing BCs are more invasive/metastatic; but patients respond to monoclonal antibody therapy with trastuzumab or tyrosine kinase inhibitors, at least at early stages. To date, numerous mouse models that faithfully reproduce HER2(+) BCs have been created in mice. We recently reviewed different mouse models of BC overexpressing wild type or mutant neu driven by MMTV, neu, or doxycycline-inducible promoters. These mice have been used to demonstrate the histopathology, oncogenic signaling pathways initiated by aberrant overexpression of HER2 in the mammary epithelium, and interaction between oncogenes and tumor suppressor genes at molecular levels. In this review, we focus on their clinical applications. They can be used to test the efficacy of HER(2) inhibitors before starting clinical trials, characterize the tumor-initiating cells that could be the cause of relapse after therapy as well as to analyze the molecular mechanisms of resistance targeting HER2. MMTV-HER2 mouse models have recently been established since the monoclonal antibody to HER2 (trastuzumab; Herceptin®) does not recognize the rat neu protein. An early study demonstrated that early intervention with HER2 monoclonal antibody would be beneficial for preventing mammary carcinogenesis. MDA-7/IL-24 as well as naturally-occurring chemicals have also been tested using MMTV-neu models. Recent studies have shown that MMTV-neu models are useful to develop vaccines to HER2 for immunotherapy. The mouse models employing HER2/neu will be essential for future antibody or drug screenings in order to overcome resistance to trastuzumab or HER(2)-specific tyrosine kinase inhibitors.
MMTV, HER2, neu, Dmp1 (Dmtf1), breast cancer, transgenic mice, trastuzumab, lapatinib, vaccination, MDA-7/IL-24, therapy
Breast cancer (BC) is the second most frequent cause of death from cancer and thus is one of the largest public health issues in the United States and the industrialized nations [1,2]. Numerous molecular markers have been used in BC to predict the prognosis for patients [3-5]. Human BCs have been subcategorized into 5 groups dependent on the expression of markers for hormone-receptors (estrogen receptor [ER] and progesterone receptor [PR]), human ErbB2 [HER2], and Ki67 [3,6-10]. This sub-classification of BC has been used to predict the prognosis and decide the treatment modalities. ER-negative BCs (luminal B, HER2-type, and basal-type) are often associated with aggressive disease, including amplification of HER2 or c-Myc oncogenes and mutation of the p53 gene [2,11] The use of monoclonal antibody anti-HER2 (trastuzumab, Herceptin®) has been deployed to treat HER2(+) BC, but the prognosis of such patients is poor since >60% of them experience relapse during the first year due to HER2 modifications, defects in the antibody dependent cellular cytotoxicity, or alterations in HER2 signaling pathways [11-13].
The genomic locus for HER2 is amplified and the protein is overexpressed in 20-30 % of human BCs [14, 15]. The HER/HER family (HER1-HER4) is made up of four structurally related receptor tyrosine kinases (RTKs) with the EGFR as the founding member of the family [16-20] (Figure 1). Activation of the HER/HER family receptors other than HER2 (an orphan receptor) requires binding of a soluble, growth factor-ligand located in the receptor that triggers receptor dimerization and phosphorylation, and activation of downstream pathways to elicit response inside the cell. EGFR (HER1) is activated by growth factor-ligands such as epidermal growth factor (EGF), heparin binding EGF (HB-EGF), amphiregulin (Areg), or transforming growth factor alpha (TGF-α) [16-19]. The product of the human c-ErbB-2 gene (HER2) is a 185-kilodalton glycoprotein with protein-tyrosine kinase activity [21-23]. HER3 and HER4 are activated by the heregulin or neuregulin family of growth factors [17-19]. Each HER receptor, upon activation by growth factor binding, initiates hetero- or homo-dimerization of receptors [15-19]. HER receptor dimerization stimulates auto-phosphorylation of the molecule, followed by trans-phosphorylation of the heterodimerization partner. HER phosphorylation occurs at specific tyrosine residues within the intracellular domain of the HER receptors. These phosphorylated tyrosine residues recruit adaptor proteins such as Grb2 and the p85 subunit of the phosphatidylinositol 3 kinase (PI3K) complex, which initiate the activation of several downstream pathways such as protein kinase B (AKT/PKB) and the mitogen activated protein kinase (MAPK) pathways [24] (Figure 1). HER receptor-mediated activation of downstream pathways enables a growth factor to elicit proliferation, survival, or migration of the cancer cell [15-19].
The transforming potential of HER2/neu has been demonstrated in a number of ways. In vitro transformation assays have been utilized to show that high levels of expression of c-ErbB-2 mediate transformation in NIH 3T3 cells [25], as determined by tumor formation after injection into athymic mice and in anchorage - independent growth assays. Mouse mammary tumor virus (MMTV) - based expression vectors with neu have been shown to achieve transformation in the NIH 3T3 cell line [26,27]. We recently reviewed the effect of overexpression in murine mammary epithelia was analyzed through construction of MMTV-neu/HER2 transgenic mice [15,28] through a variety of approaches, i.e., wild type or mutant, rat neu or human ERBB2, constitutive or drug inducible/de-inducible. Multiparous female mice with the oncogenic rat neu (MMTV-neuNT) develop mammary adenocarcinomas at an average age of six months [29] (Table 1). MMTV promoter-based overexpression of the proto-oncogenic form of rat neu (MMTV-ErbB2) also results in focal breast tumors but with a longer latency [30] (Table 1). Later mouse models for BC induced by amplification and overexpression of the activated neu gene driven by the neu promoter was generated [31], which displayed stronger, but similar phenotypes as MMTV-neu or ErbB2 models. A knock-in model for rat neu was also established at the mouse neu locus, which was not sufficient for the development of mammary tumors [32]. A doxycycline (dox) - inducible model for neu overexpression was created by Moody et al. with successful transformation of mammary epithelial cells with lung metastasis [33]. More recently, MMTV-driven overexpression of human ERBB2 (HER2) was generated to test the therapeutic effects of monoclonal antibodies to HER2 since the rat neu protein is not recognized by trastuzumab [34-37].
Although overexpression of HER2 stimulates cell proliferation in transformed cells, it induces cell cycle arrest in wild type murine embryonic fibroblasts (MEFs) [38]. Our recent studies indicate that the Dmp1-Arf-p53 tumor suppressor pathway [39-56,57-64] plays a critical role in quenching hyperproliferative signaling stemming from c-Myc [43], oncogenic Ras [44,47], HER2/neu [38], or cyclin D1 [50] overexpression. Consistently, all oncogene-driven carcinogenesis was significantly accelerated in Dmp1-deficient mice, and the presence of Dmp1α significantly delayed the appearance of neuNT-driven mammary tumors [51].
IL-24 is a cytokine that belongs to the IL-10 family of interleukins that signal through IL-20R1/IL-20R2 and IL-22R1/IL-20R2 [65,66]. This interleukin is also known as melanoma differentiation-associated 7 (mda-7) due to its discovery as a tumor suppressing protein. IL-24 controls cell survival and proliferation by inducing rapid activation of transcription factors linked STAT1 and STAT3. IL-24 is predominantly released by activated monocytes, macrophage sand T helper 2 cells and acts on non-hematopoietic tissues such as skin, lung and reproductive tissues [65,66]. IL-24 performs important roles in wound healing, psoriasis and cancer. Importantly, cell death occurs in cancer cells following exposure to IL-24. IL-24-mediated tumor suppressor pathway was recently characterized using the MMTV-neu as a model [67]. Numerous labs are now developing vaccines against HER2 using MMTV-neu models. The use of MMTV-neu models for novel HER2 target therapies through a variety of approaches is reviewed.
Table 1. Use of transgenic mouse models engaging HER2/neu in clinical applications.
Mouse model |
Mean latency of tumors |
Pathology |
Clinical Use |
References |
MMTV-neuNT (mutant), FVB or Balb/c |
197 days |
Rapidly growing, invasive mammary tumors, metastatic* |
17-AAG, MoAb, Vac |
[29,100,103,139,149] |
MMTV-neuNT (mutant), Balb/c |
21 weeks |
Rapidly growing, multifocal mammary tumors* |
Cytokine therapy; Vac |
[122,123-126,135-138,140] |
MMTV-neu (wild type) |
205 - 367 days |
Focal Adca, lung metastasis after long latency |
TIC, Cytokine therapy, TKI |
[30,84-88,67,123,124] |
MMTV-NIC (neu-IRES-Cre) |
146 days |
Aggressive mammary tumors |
|
[91,95] |
MMTV-NIC;Ptenflox/flox |
43 vs. 198 days |
|
PI3KI |
[96] |
MMTV-NIC;p110aflox/flox |
95% survival at 225 days |
|
PI3KI |
[106] |
MMTV-NIC;p110bflox/flox |
125 vs. 135 days |
|
|
[106] |
MMTV-HER2* |
28 weeks |
Adca with areas of solid, tubular, and papillary growth cellular polymorphism with mitosis and metastasis |
MoAb |
[35] |
MMTV-HER2*; MMTV-rtTA; TetOp-HA-PIKCAH1047R |
76 days |
Rapidly growing, large, multiple tumors; increased angiogenesis, lung metastasis; EMT; mammospheres |
MoAb, PI3KI |
[97] |
MMTV-HER2*D16 |
17 weeks |
Tubular adca consisting of outer pale cells, an intermediate darker fusiform cells, and an inner zcells with pinkish cytoplasm |
MoAb |
[37] |
WAP-HER2* |
no tumor |
None of the tissues expressing HER2 demonstrated obvious abnormality. The mice were healthy and have a normal life span. |
Vac |
[135] |
|
neuNT: mutant rat neu |
Adca: Adenocarcinoma |
TKI: tyrosine kinase |
|
|
MMTV-NIC; bicistronic vector |
*independent of pregnancy |
inhibitor screening |
|
|
expressing rat neu and cre |
|
Vac: vaccine development |
|
|
recombinase |
|
TIC: tumor initiating cells |
|
|
HER2*: human ErbB2 |
|
MoAb: monoclonal antibody |
|
|
wild type |
|
screening |
|
Gene-engineered mice that overexpress HER2/neu (wild type or mutant) in mammary glands, and their clinical uses are summarized. The MMTV promoter has been used in all the cases except for ref. 135 (WAP-HER2), which did not produce spontaneous mammary tumors, but were tolerant for HER2. The MMTV-NIC is a unique model that expresses neuNT and cre recombinase simultaneously to study the collaboration between oncogenic new expression and oncogene/tumor suppressor gene inactivation. These mice have been used in characterization of tumor initiating cells (TIC), testing the HSP90 inhibitor tanespimycin (17-AAG), anti-tumor cytokines (IL-12, IL-24), HER2 vaccines (DNA, peptide), HER(2) kinase inhibitors (lapanitib, erlotinib, and others), PI3K inhibitors, and monoclonal antibodies (trastuzumab, pertuzumab).
neuNT: mutant rat neu, Adca: Adenocarcinoma, TKI: tyrosine kinase inhibitor screening, MMTV-NIC: bicistronic vector expressing rat neu and cre recombinase, Vac: vaccine development, TIC: tumor initiating cells
HER2*: human ErbB2 wild type, MoAb: monoclonal antibody screening, *independent of pregnancy
A variety of human malignancies, including breast cancers, brain tumors colon cancers, prostate cancers, and melanomas are thought to be organized in a hierarchy, whereby a relatively minor population of tumor initiating cells (TIC) or cancer stem cells (CSCs) is responsible for tumor growth and the vast majority of remaining cells is non-tumorigenic [68-70]. Treatment resistance for cancer can be either de novo – because of traits that tumor cells possess before treatment – or acquired – because of properties that tumor cells gain in response to treatment. TICs have the capacity to self-renew and generate new tumors that consist entirely of clonally derived cell types present in the parental tumor. TICs are resistant to many conventional cancer therapies, and can survive treatment in spite of dramatic shrinkage of the tumor. Residual TICs can eventually regrow, which results in disease relapse. It has also been hypothesized that TIC may be responsible for metastatic disease. If these hypotheses are correct, targeting TICs are imperative for achieving a cure of malignancy.
Markers specific for normal stem cells are commonly used for isolating CSCs from solid tumors and hematological tumors. Cell surface markers have proved useful for isolation of CSC-enriched populations including CD24, CD44, CD133, epithelial cell adhesion molecule (EpCAM), ATP-binding cassette B5 (ABCB5), ABCG2, and CD200 [71-74]. ALDH (aldehyde dehydrogenase), which catalyzes the oxidation of aromatic aldehydes to carboxyl acids, has also been used as a marker of CSCs. It has a role in conversion of retinol to retinoic acid, which is essential for cell survival.
Metastasis is the major cause of tumor-related lethality. This potential depends on factors that determine cell growth, angiogenesis, invasion and other basic processes. In epithelial tumors, the epithelial-mesenchymal transition (EMT) [20,75-77] is considered to be a crucial event. EMT and the reverse transition from mesenchymal to an epithelial phenotype (named MET) are involved in embryonic development, which involves disruption of epithelial cell homeostasis and the acquisition of a migratory mesenchymal phenotype. EMT appears to be controlled by canonical pathways such as WNT and transforming growth factor β (TGFβ), and a number of transcription factors [77-80]. Translocation of β-catenin from adherens junctions to the nucleus may lead to a loss of E-cadherin, and subsequently to EMT. Nuclear β-catenin apparently can directly, transcriptionally activate EMT-associated target genes, such as the E-cadherin gene repressor SLUG [81]. Mechanical properties of the tumor microenvironment, or hypoxia, can influence CSC properties and metastatic behavior [82]. Tumor cells undergoing an EMT may be precursors for metastatic cancer cells, or even metastatic CSCs [83].
Analysis of TICs in model systems of BC would offer uniform and accessible source of tumor cells and the power of mouse genetics to dissect these rare cells. The HER2/neu proto-oncogene is overexpressed in an aggressive form of human breast cancer. MMTV-neu models [wild type neu] [30] have often been used to characterize the TICs in BC (mammary tumors) since the tumor cells have a clear marker, and are sensitive to a variety of anti-HERR2 therapies [84-88] (Table 1). Liu et al. [84] report on the functional identification of mouse HER2/neu TICs that can induce tumors after transplantation into the mammary gland of recipient mice. Secondary tumors formed after injecting MMTV-neu TICs resemble primary tumors in the original transgenic mice and are organized in a hierarchy containing TICs as well as their non-tumorigenic descendants. To study MMTV-neu TICs in vitro, they grew tumorspheres under non-adherent culture conditions [84]. Tumorsphere forming units (TFU) capable of producing tumorspheres retained tumorigenic potential and were indistinguishable by several criteria from TICs [84]. Interestingly, MMTV-neu TICs and TFUs were committed to the luminal cell fate when induced to differentiate in vitro. Their data defined the characteristics of the MMTV-neu TIC and TFU, which helps to explain marker expression profiles of HER2(+) BC. In addition, the similarity between TICs and TFUs in this system provides a rationale for TFU-based screens to target tumor-initiating cells in HER2(+) BC.
Then the same group used serial dilution and single-cell transplantation assays to identify MMTV-neu [30] mouse mammary TICs as CD24(+):JAG1(−) at a frequency of 2-4.5 % [85]. A 17-gene Her2/neu-TIC enriched signature (HTICS), generated on the basis of differentially expressed genes in TIC versus non-TIC fractions and trained on one HER2+ BC cohort, predicted clinical outcome on multiple independent HER2(+) cohorts. HTICS consists of eight up-regulated (Aurkb, Ccna2, Scrn1, Npy, Atp7b, Chaf1b, Ccnb1, Cldn8) and nine down-regulated genes (Nrp1, Ccr2, C1qb, Cd74, Vcam1, Cd180, Itgb2, Cd72, St8sia4). The up-regulated subset includes genes associated with passage through the S/G2/M phase of the cell cycle (Aurkb, cyclinB1, Cyclin A2; Chaf1b) [85]. Down-regulated in HTICS are genes involved in cell adhesion, angiogenesis. HTICS’ prognostic power was independent of other predictors, divided lymph node(+)HER2(+) BC into low and high-risk subgroups: the 10 year overall survival of 84 % for HTICS(−) and 24 % for HTICS(+) tumors (p = 0.002) in HER2(+)ERα(−) patients. Retrospective analyses revealed that patients with HTICS(+)HER2(+)ERα(−) tumors resisted chemotherapy, but responded to chemotherapy plus trastuzumab. HTICS is, therefore, a powerful prognostic signature for HER2(+)ERα(−) BC that can be used to identify high risk patients that would benefit from anti-HER2 therapy [85]. Finally, they described the rationale behind development of this prognosticator and present new features of the signature, including elevated PI3K pathway activity and low TNFα and IFNγ signaling in high-risk mammary tumors [85].
Lo et al. used different cell surface markers to characterize TICs in MMTV-neu mice [86]. They showed that surface markers CD49f (integrin α6), CD61 (integrin β3), and epithelial-specific antigen (Ep-CAM) were aberrantly overexpressed in HER2(+) mammary tumor cells. Analysis of these three markers and CD24 detected abnormal proliferation of the luminal progenitor cells in pre-neoplastic mammary glands of MMTV-neu-transgenic mice [30], indicating that HER2-driven mammary tumors originated from aberrant luminal progenitor cells. The combined markers for CD49f and CD61 further delineated the CD49fhighCD61high fraction as a TIC-enriched population, which displayed increased tumorsphere formation, enhanced tumorigenicity, and drug resistance to paclitaxel and doxorubicin [86]. Moreover, the TIC-enriched population manifested increased TGFβ signaling and exhibited gene expression signatures of stem cell-ness and EMT, where self-renewal and clonogenicity of TICs were suppressed by the drug that inhibit the TGFβ signaling. Finally, they showed that the CD61 signaling pathway was required for sustaining active TGFβ signaling and self-renewal of TICs. Therefore, the cooperative integrin-β3-TGFβ signaling axis was identified as a potential therapeutic target for HER2-induced TICs [87].
Since many HER2(+) BC patients do not respond to therapy or relapse with aggressive metastatic disease, there is an urgent need for new therapeutics that can target HER2(+) BC and potentiate the effect of anti-HER2 inhibitors, in particular those that can target TICs. Deng et al. showed that MMTV-neu [30] mammary tumor cells cultured as non-adherent spheres or adherent monolayer cells select for mutations for p53 that caused immortalization of the cultures. After serial passages, only sphere conditions maintained TICs [88], which was consistent with the Liu study [84]. Using tumorsphere formation as surrogate for TICs, they screened p53 mutant HER2/neu(+) tumorspheres and monolayer cells with a lentivirus shRNA kinome library and identified kinases such as the mitogen-activated protein kinase and the TGFb receptor protein family as well as autophagy factor ATG1/ULK1 and the non-canonical IkB kinase (IKK), TANK-binding kinase 1 (TBK1). Depletion of TBK1 or pharmacologic inhibition of TBK1 and its related protein IKKe suppressed growth of both human and mouse HER2/neu(+) BC (mammary tumor) cells. TBK1/IKKe inhibition accelerated cellular senescence by suppressing the p65 subunit of NF-kB and inducing p16Ink4a. In addition, TBK1/IKKe inhibition cooperated with lapatinib to accelerate the apoptosis of HER2/neu(+) BC (mammary tumor) cells. Their results suggest that patients with HER2(+) BC may benefit from anti-TBK1/IKKe plus anti-HER2 combination therapies [88].
Using a mouse model of recurrent mammary tumorigenesis in combination with bioinformatics analyses of BC patients, Abravanel et al. identified the role for Notch signaling in mammary tumor dormancy and recurrence using MMTV-rtTA; TetO-HER2/neu mice [89]. They found that 1) Notch signaling was acutely upregulated in tumor cells following HER2/neu pathway inhibition, 2) Notch signaling remained activated in a subset of dormant residual tumor cells that persist following HER2/neu downregulation, 3) activation of Notch signaling accelerates tumor recurrence, and 4) the inhibition of Notch signaling impairs recurrence in mice. Consistent with these findings, meta-analysis of microarray data from over 4,000 BC patients revealed that elevated Notch pathway activity is independently associated with an increased rate of recurrence. Together, these results demonstrate the role of Notch signaling in tumor recurrence from dormant residual tumor cells, and provide evidence that dormancy is a targetable stage of BC progression [89].
HER2/neu transgenic mouse models have also been used to test the therapeutic efficacy of the erlotinib (an EGFR inhibitor) [90], HER1/2 inhibitor lapatinib (a dual tyrosine kinase inhibitor that interrupts the HER2/neu and EGFR pathways) [90], and pan-HER family tyrosine kinase inhibitor AZD8931 [91]. The former two drugs have been used alone or in combination with the drugs carboplatin and paclitaxel.
Usary et al. [90] examined the efficacy of four chemotherapeutic or targeted anti-cancer drugs, alone and in combination, using mouse models representing three distinct BC subtypes, i.e., basal-like (C3(1)-T-antigen transgenic) [92], luminal B (MMTV-neu transgenic) [30], and Claudin-low (T11/TP53−/− orthotropic syngeneic murine transplants [OST]) [93,94]. Lapatinib or erlotinib alone exhibited exceptional efficacy in the MMTV-neu model [30] in short term responses and long term survival [90]; some combination therapies (i.e., carboplatin plus paclitaxel in C3(1)-T-antigen transgenic and carboplatin plus erlotinib in T11/TP53−/− OST) were more active and life-prolonging than those by a single agent. Through analysis of mRNA expression in chemotherapy-treated murine tumors, they identified a pair of gene expression signatures (i.e., RESP-HUM [12 genes, e.g. glutathione S-transferase, complement component 4B, apolipoprotein D, acetyl CoA carboxylase b] and UNTREATED-HUM [30 genes, e.g. DEK, E2F8, high-mobility group box2, zinc finger protein 131/367]) that predicted pathological complete response (pCR) to neoadjuvant anthracycline/taxane therapy in human patients with BC. Of note, the RESP-HUM signature predicted pCR more strongly in the normal-like and Claudin-low subtypes whereas the UNTREATED-HUM signature better tracked response within the basal-like subtype [90]. In conclusion, mouse - derived gene signatures can predict treatment response suggesting that mice can be used to identify new biomarkers for human cancer patients [90].
Although a number of patients with HER2(+) BCs have been treated with HER2-targeted therapies, the disease often relapses due to resistance, which is a major clinical problem. Creedon et al. showed the utility of a genetically engineered mouse model of HER2-driven BC (MMTV-Neu-IRES-Cre, MMTV-NIC) (Table 1) [95,96] to define the mechanisms of resistance to the pan-HER family tyrosine kinase inhibitor AZD8931 (Figure 1) [91]. Genetic manipulation of MMTV-NIC mice demonstrated that loss of PTEN was associated with de novo resistance to AZD8931. Using a tumor fragment transplantation model, 50% of tumors developed resistance to AZD8931. Analysis of the resistant tumors showed two distinct patterns: 1) tumors in which reduced membranous HER2 expression was associated with an EMT characteristic [75-77], and 2) those retained HER2 expression with an epithelial morphology. Further AZD8931 treatment resulted in the generation of secondary resistant tumors that again showed EMT or epithelial morphology [91]. Because the induction of EMT in HER2-driven mouse mammary tumors via expression of an activating PI3K mutation was associated with de novo resistance to HER2-targeted therapies (explained in detail later in the resistance section) [97] and because the expression of transcription factors that drive EMT (e.g. TWIST, SNAIL, ZEB) [20] was related to de novo trastuzumab resistance [98], these molecules can be targeted in the therapy of BC that are resistant to AZD8931.
HER2/neu is an oncoprotein client that is highly dependent on HSP90. This and other oncogenic client proteins (e.g. B-RAF, C-RAF, ALK and CDK4) are depleted by 17-AAG (tanespimycin) (Figure 1) [99] in both animal tumors and patients. Rodrigues et al. investigated the metabolic response of 17-AAG in spontaneous HER2/neu-driven mammary tumors in MMTV-neuNT mice [29] by Magnetic Resonance Spectroscopy (MRS) and in cells isolated and cultured from these tumors. The mammary tumors were very sensitive to 17-AAG and responded in a dose-dependent manner to 17-AAG, all of which caused significant regression. At the higher doses, the tumor extracts showed significant decreases in phosphocholine and phosphoethanolamine. Extracts of isolated cells cultured from the mammary carcinomas showed a significant decrease in viable cell number and total phosphomonoesters after 17-AAG treatment. Western blots confirmed the expected action of 17-AAG in inducing HSP72 and significantly depleting HSP90 client proteins, including HER2/neu both in tumors and in isolated cells. The data demonstrate the high degree of sensitivity of this clinically relevant HER2/neu-driven tumor model to HSP90 inhibition by 17-AAG and suggest that the metabolic signature of choline phospholipids obtained by MRS could be useful both as a preclinical and clinical tool for investigating surrogate markers of response to treatment [100].
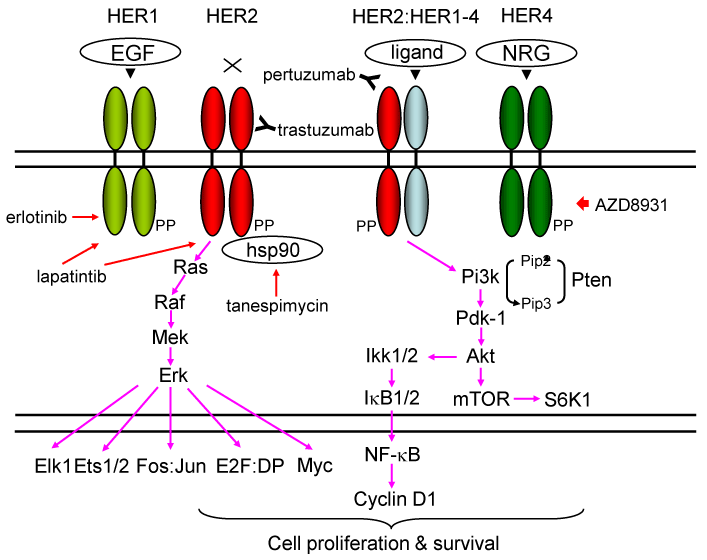
Figure 1. . Signaling pathways stimulated by HER protein expression stimulated by ligands or overexpression
The genomic locus for HER2 is amplified in ~20 % and the protein is overexpressed in ~30 % of human BCs, and is associated with aggressive disease with shorter overall and disease-free survival [15]. EGFR (HER1) is activated by growth factor-ligands such as epidermal growth factor (EGF), heparin binding EGF (HB-EGF), amphiregulin (Areg), transforming growth factor alpha (TGF-α), betacellulin, epiregulin, and epigen [16-19,153,154]. Heregulins/neuregulins (HRGs/NRGs) are the ligands for HER3 and HER4 [155]. Currently there are four known NRG genes: NRG1, NHRG2, NRG3 and NRG4 [156,157]. Following ligand engagement, HER1-4 engages and allosterically activates its kinase partner, in this case HER2. Although HER2 molecules make both homo-and hetero-dimers, HER3 do not form homodimers, and does not have protein-tyrosine kinase activity. Phosphorylation of its C-terminal tail leads to recruitment of adapter proteins leading to activation of Pi3k and Ras pathways [15-19,24]. Activation of Pi3k leads to phosphorylation of membrane phosphoinositides producing PIP3, which in turn docks the PH domain-containing proteins Pdk1 and Akt [24]. Membrane - bound Akt is phosphorylated and activated by Pdk1. Activated Akt proceeds to phosphorylate a plethora of cellular substrates involved in diverse biological processes. These processes include accelerated G1-S progression as demonstrated by increased cyclin D1 and decreased p27Kip1 levels, and enhanced cell survival through increased phosphorylation of Bad and increased NF-kB levels [15-19,24]. Indeed aberrant overexpression of cyclin D1 is frequently observed in human cancers, caused by different mechanisms [62]. Thus deregulation of HER2 leads to tumorigenesis. Aberrant overexpression of HER2 activates the Dmp1 promoter to stimulate the Arf-Mdm2-p53 self-autonomous tumor surveillance pathway through Pi3k-Akt-NF-kB and Ras-Raf-Mek-Erk-Jun cascades to eliminate incipient cancer cells by cell cycle arrest or apoptosis [10,15,57,63].
Trastuzumab (Herceptin®) binds the extracellular domain of HER2. Pertuzumab (Perjeta®) binds another HER2 domain, preventing dimerization. Tanespimycin (17-AAG) inhibits HSP90, causing HER2 conformation change. Lapatinib is a small-molecule tyrosine kinase that inhibits HER1 and HER2. Erlotinib is a specific inhibitor for HER1. AZD8931 is a pan-HER family tyrosine-kinase inhibitor.
MMTV-driven transgenic mouse models for HER2 [35,37] have been used to test the efficacy of monoclonal antibody to HER2: trastuzumab (Herceptin®) [101,102] in vivo. Finkle et al. created MMTV-HER2 (human wild type ERBB2 transgenic) mice [35] since the monoclonal antibody to HER2 (trastuzumab) does not recognize the rat neu protein [34]. The transgenic mice were randomly assigned to one of three treatment groups and received i.p. injections of mu4D5 (the murine version of trastuzumab) from 17 weeks of age until either 52 weeks or death. High levels of HER2 expression were detectable in mammary glands of young virgin founder mice. Mammary adenocarcinomas were frequently found in female founders and progeny at an average age of 28 weeks, with some progressing to metastatic disease. The incidence of mammary tumors was significantly reduced, and tumor growth inhibition was observed in mice receiving mu4D5 compared with control mice [35]. Early intervention with mu4D5 was beneficial in MMTV-HER2 transgenic mice albeit at high risk for developing mammary adenocarcinomas. This study suggests a potential benefit of early treatment with Herceptin® in HER2-positive primary BCs.
A splice isoform of the HER2 receptor that lacks exon 16 (Δ16HER2: this is coding exon 16, exon 20 in 31 exons for HER2) is expressed in many HER2(+) BCs [36], and has been linked with resistance to the HER2-targeting antibody trastuzumab [103]. Castagnoli et al. reported that Δ16HER2 was sufficient to accelerate mammary tumorigenesis and improve the response to trastuzumab [37]. A comparative analysis of effector signaling pathways activated by Δ16HER2 and wild type HER2 revealed that Δ16HER2 was optimally functional through a link to SRC activation (pSRC). Clinically, HER2-positive BCs from patients who received trastuzumab exhibited a positive correlation in Δ16HER2 and pSRC abundance, consistent with the mouse genetic results. Moreover, patients expressing high pSRC or an activated "Δ16HER2 metagene" were found to receive the greatest benefit from trastuzumab treatment.
This report [37] seems to contrast directly with those of Zhang et al. [103], who reported a lower clinical response rate and a higher progressive disease rate after trastuzumab treatment in HER2-positive BC patients with high pSRC expression. However, it should be noted that Zhang study consisted of 57 BC patients who received first-line trastuzumab-based therapy in metastatic BC whereas the Castagnoli report [37] includes BC patients treated with trastuzumab-based regimens in an adjuvant setting [104]. They speculate that although HER2-positive primary BC expressing high levels of pSRC are initially dependent on HER2 and all its potential driver isoforms and are, thus, responsive to trastuzumab; the progression of such BCs due to a high HER2-dependent growth rate might lead to accumulation of genetic alterations that result in less HER2 dependency, which, in turn, results in significantly less or even no responsiveness to trastuzumab as reported by Zhang et al. [103]. In conclusion, activated HER2 signaling is indicative of benefits from the addition of trastuzumab to chemotherapy and that Δ16HER2 expression is not a reliable indicator of trastuzumab resistance but instead a mirror of pSRC activity, reflecting Δ16HER2 homodimer-mediated driver activity leading to high responsiveness to trastuzumab.
The involvement of HER4 (Figure 1) in acquired resistance to HER2-targeted therapies remains controversial. Canfield et al. [105] demonstrated that genetic ablation of HER4, but not HER1-3, led to apoptosis in lapatinib - resistant cells, suggesting that the efficacy of pan-HER inhibitors –(afatinib, canertinib, dacomitinib, naratinob: irreversible, and valitinib - reversible) was, at least in part, mediated by the inhibition of HER4. Moreover, HER4 was upregulated at the protein level in HER2(+) BC cell lines selected for acquired lapatinib resistance in vitro and in MMTV-neu mice following prolonged lapatinib treatment. Depletion of HER4 caused a decrease in AKT phosphorylation in resistant cells, but not in sensitive cells, suggesting that HER4 activated the PI3K/AKT pathway in lapatinib - resistant cells. Importantly, HER4 knockdown triggered apoptosis not only in lapatinib - resistant cells, but also in trastuzumab-resistant cells. Thus although HER4 is dispensable for HER2(+) BC cells, it may play a key role in the survival of HER2(+) cancer cells after they develop resistance to HER2 inhibitors: lapatinib or trastuzumab [105].
Class Ia PI3K (Figure 1) is required for oncogenic receptor-mediated transformation; however, the individual roles of the two commonly expressed class Ia PI3K isoforms in oncogenic receptor signaling have not been elucidated in vivo. Utermark et al. showed that depletion of p110α blocks tumor formation in both polyoma middle T antigen- and HER2/neu (MMTV-NIC) -induced transgenic models of BC [106]. Interestingly, p110α ablation resulted in nearly complete protection in mammary tumor development (95% survival at 225 days in NIC;p110αL/L in vs. 135 days in NIC) while p110β ablation resulted in both increased ductal branching and tumorigenesis (125 days in NIC;p110βL/L vs. 135 days in NIC) [106]. Biochemical analyses suggest a competition model in which the less active p110β competes with the more active p110α for common receptor binding, thereby modulating the level of PI3K activity associated with activated receptors. They demonstrated a novel p110β-based regulatory role in receptor-mediated PI3K activity, and identify p110α as an important target for the treatment of HER2(+) BC [106].
HER2 amplification and PI3K catalytic subunit alpha (PIK3CA) mutations often co-exist in BCs. Aberrant activation of the PI3K pathway has been shown to correlate with a diminished response to HER2-directed therapies as described above. Hanker et al. generated a mouse model of HER2-overexpressing, PIK3CAH1047R - mutant BC by crossing MMTV-HER2 with MMTV-rtTA together with TetOp-HA-PIKCAH1047R-IRES-luciferase mice (Table 1) [35,97,107]. Mice expressing both HER2 and mutant PIK3CA in the mammary epithelium developed tumors with shorter latencies compared with those expressing either one of these. HER2 and mutant PIK3CA also cooperated to promote lung metastases. By microarray analysis, HER2-driven tumors clustered with luminal BCs, whereas PIK3CAH1047R tumors were associated with Claudin-low BCs [93,108]. PIK3CA and HER2(+);PIK3CAH1047R tumors expressed elevated transcripts encoding markers of EMT and stem cells [97]. Cells from HER2(+); PIK3CAH1047R tumors more efficiently formed mammospheres and developed lung metastases. Of note, HER2(+);PIK3CAH1047R tumors were resistant to trastuzumab alone and in combination with lapatinib (HER1/2 dual kinase inhibitor) or pertuzumab (monoclonal antibody to HER2, Perjeta®) (Figure 1) [109]. Both drug resistance and enhanced mammosphere formation were reversed by treatment with a PI3K inhibitor BKM120 [110]. In summary, PIK3CAH1047R accelerates HER2-mediated breast epithelial transformation and metastasis, alters the phenotype of HER2-overexpressing BCs, and generates resistance to approved combinations of anti-HER2 therapies [97].
Although hundreds of knockout mouse models have been created so far to demonstrate the biological activities of candidate tumor suppressor genes (TSGs) in vivo [111-114], there are only a couple of overexpression models for TSGs to demonstrate their activities for tumor prevention. Super p53 mice [115], super Ink4a/Arf mice [116,117], MMTV-Dmp1α [51], MMTV-p16Ink4a [118], are typical examples for these. Of note, the former three models exhibited signs of accelerated aging, which happen as a result of increased tumor-suppressive activities in vivo for long period of time.
Melanoma differentiation associated gene-7/interleukin-24 (mda-7/IL-24) encodes a tumor suppressor gene implicated in the growth of various tumor types including BC [65]. Dr. Fisher’s group reported that recombinant adenovirus-mediated mda-7/IL-24 expression in the mammary glands of methylnitrosourea (MNU) - treated rats suppressed mammary tumor development [67,119-121]. Since most MNU-induced tumors in rats contain activating mutations in Ha-ras, they later examined the effect of MDA-7/IL-24 on HER2/neu-induced mammary tumors, in which the RAS pathway is induced. Li et al. [67] generated doxycycline (dox)-inducible MDA-7/IL-24-transgenic mice and crossed them with HER2/neu-transgenics [30]. Triple-transgenic mice treated with doxycycline (dox) exhibited a strong inhibition of tumor development, demonstrating the tumor suppressor activity by MDA-7/IL-24 in immune-competent mice. MDA-7/IL-24 induction also inhibited the growth of tumors generated by injection of HER2/neu tumor cells isolated from triple-transgenic mice that had not been treated with dox into the mammary fat pads of isogenic FVB mice. Despite initial growth suppression, tumors in triple compound mice lost mda-7/IL-24 expression and grew after longer latency, indicating that continuous presence of this cytokine within the tumor microenvironment is crucial to sustain the tumor-inhibitory activity, which mimic the MDTG model that had been reported from our lab [51]. Mechanistically, MDA-7/IL-24 exerted its tumor-suppressive effect on HER2/neu mammary tumor cells, at least in part, through PERP (p53 apoptosis effector related to PMP-22) with growth arrest and apoptosis-inducing capacity. Overall, their results established mda-7/IL-24 as a suppressor of mammary tumor development and provided a rationale for using this cytokine in the prevention/treatment of human BC [67,119-121].
Other cytokines, such as IL-12 and IL-15 have also been used in MMTV-neu or WAP-HER2 transgenic mice in Balb/c [122-126]. IL-12 is a cytokine that stimulates IFN-γ production from NK cells and T cells. Boggio et al. showed the efficacy of administration of mouse recombinant IL-12 (rIL-12) in counteracting the occurrence and progression of spontaneous mammary carcinomas in the females inbred mice transgenic for the rat HER-2/neu oncogene [29,123]. Tumor inhibition in both lines was associated with deficient peri- and intra-tumoral angiogenesis, infiltration of reactive cells, production of pro-inflammatory cytokines, and inducible nitric oxide synthase activation [123]. Vascular damage resulted in a high degree of hemorrhagic necrosis of established tumor masses. In both Balb-neuT [122] and FVB-MMTV-neuNT mice [29], 5-day i.p. courses of 50/100 ng of IL-12/day inhibited mammary carcinogenesis in early pre-neoplastic lesions. Inhibition appears to depend primarily on the ability of IL-12 to interfere with early tumor angiogenesis; however later treatments were much less effective. The efficacy of early IL-12 courses suggests that they could be used to prevent mammary tumors in individuals at risk, whereas their lower efficacy in later stages of carcinogenesis put some constraints on their use in the management of overt neoplastic lesions. Nonetheless, the efficacy of IL-12 points to enhancement of non-specific immunity as an effective way to prevent mammary tumors in individuals at risk [124].
Sonoda et al. studied whether Gd-DTPA-Gel-Cis, a conjugate of gadolinium (Gd), cis diamminedichloroplatinum (Cis), diethylenetriaminepentaacetic acid (DTPA)-dianhydride, and bovine gelatin (Gel) can be used as an intravascular contrast agent at MRI and as an antitumor cell proliferation agent in vitro [127]. They injected Gd-DTPA-Gel-Cis into the caudal vein of female HER2/neu-transgenic mice [30] with spontaneous mammary tumors. On T1-weighted MRI scans of mice injected with Gd-DTPA-Gel-Cis, they observed a 23 % increase in signal intensity. The survival rates of cells exposed to Gd-DTPA-Gel-Cis or Cis were 71 % and 58 %, respectively, of the survival rates observed after treatment with Gel alone. Hence, Gd-DTPA-Gel-Cis can be used as an MRI contrast medium as well as an anti-tumor agent.
Berberine (BBR), natural isoquinoline alkaloid and its synthetic derivative 13-(4-chlorophenylethyl) berberine iodide, NAX014 significantly reduced neu tumor formation. Immunohistochemical study showed that CD34 expression was subverted indicating reduced vessel density, decrease in perforin and granzyme activity in HER2/neu mammary tumors by NAX014 [128,129].
Brassinin is a phytoalexin, a class of natural products derived from plants that includes the widely known compound resveratrol. Brassinin has been shown to have chemopreventive activity in preclinical models, but the mechanisms underlying its anti-cancer properties are unknown. Banerjee et al. showed that brassinin and a synthetic derivative 5-bromo-brassinin (5-Br-brassinin) are bioavailable inhibitors of indoleamine 2,3-dioxygenase (IDO), a pro-toleragenic enzyme that drives immune escape in cancer [130]. Like other known IDO inhibitors, both of these compounds combined with chemotherapy to elicit regression of mammary gland tumors in MMTV-neu mice (wild type) [30]. This response to treatment was lost in athymic mice and Ido-null mice, indicating a requirement for active host T-cell immunity, and in providing direct genetic evidence that IDO inhibition is essential to the anti-tumor mechanism of action of 5-Br-brassinin. The natural product brassinin thus provides the structural basis for a new class of compounds with in vivo anticancer activity that is mediated through the inhibition of IDO.
It is increasingly acknowledged that BC is an immunogenic disease. Because the HER2 gene is amplified in 20% of BCs and the protein is overexpressed in nearly half of BCs [15], it has been a target for DNA and peptide vaccine development [131-134]. Transgenic mice that express HER2/neu in mammary glands have been used for vaccine developments. Most experiments had been conducted in DNA vaccination using MMTV-neu-transgenic mice in Balb/c mouse model [122,125,135-138] although viruses [139,140] have also been used for immunization. The former approach using virus-like replication particles of Venezuelan equine encephalitis virus containing VRP-neu was especially encouraging since vaccination completely prevented tumor development in MMTV-neuNT mice [139]. Clinical testing of this vaccine in patients with HER2(+) BC was warranted from their study. Later, the usefulness of chemokines CCL4, CCL9, CCL21 as an adjuvant for DNA vaccination has been shown in Balb/c mice through intramuscular immunization of plasmid DNAs followed by tumor challenge of syngeneic HER2/neu(+) D2F2 cells [141-143].
Although other investigators used MMTV-neu models for vaccine development, Piechocki et al. created WAP-HER2 (wild type) mice to study the immunological response (Table 1) [135]. The mice were tolerant to HER2 and hence appropriate for testing HER2 vaccines. The HER2 protein was expressed in the secretory mammary epithelia during pregnancy and lactation without tumor development in C57BL/6 strain. Tolerance to HER2 was demonstrated by inoculating mice with a syngeneic tumor expressing high levels of HER2. Tolerance to HER2 was demonstrated by inoculating mice with a syngeneic ovarian tumor expressing high levels of HER2 (ID8/E2). Tumors grew exclusively in the HER2-transgenic mice without inducing an antibody response, while the non-transgenic littermates remained tumor-free for 10 months and exhibited a robust anti-ErbB-2 antibody response. When immunized five times with plasmid DNA encoding ErbB2 and GM-CSF, 33% of the HER2-transgenic mice rejected a lethal challenge of EL-4/E2 tumor cells whereas all immunized littermates rejected the tumor. In summary, WAP-HER2-transgenic mice demonstrated tolerance to ErbB2 which was partially overcome by DNA vaccination. The breakable tolerance of HER2-transgenic mice is particularly suited for testing human ErbB2-based vaccines [135].
Peptide vaccines have also been developed in HER2-transgenic mice [144-146]. Several genetic and non-genetic vaccination strategies targeted against HER2 exhibited various degrees of success in animal models. Peptide vaccines like E75, GP2, AE37, and Ii-Key [147] and peptide-pulsed autologous dendritic cell vaccine (NCT00923143) are in clinical trials (www.clinicaltrials.gov). These peptides evoke cytotoxic T cell mediated immunity and are restricted by the requirement for HLA matching.
HER2/neu-targeted vaccines raise strong CD8(+) T cell responses to a dominant peptide (rat neu [420-429]) in WT FVB/N mice, and protect them from a neu-expressing tumor challenge, which does not happen in HER2/neu-transgenic mice. However, treatment of neu-transgenic mice with vaccine and cyclophosphamide containing chemotherapy resulted in tumor protection in a proportion of mice [148]. This effect was specifically abrogated by the transfer of neu-derived CD4(+)CD25(+) T cells. Rat neu [420-429] - specific CD8(+) T cells were identified only in MMTV-neu (wild type) mice given vaccine and cyclophosphamide chemotherapy, which rejected tumor challenge. It was considered that cyclophosphamide inhibited regulatory T cells by selectively depleting the cycling population of CD4(+)CD25(+) T cells in MMTV-neu mice. These findings demonstrate that MMTV-neu mice possess latent pools of high-avidity neu-specific CD8(+) T cells that can be recruited to produce an effective anti-tumor response if regulatory T cells are blocked or removed by a drug before vaccination [148].
Though anti-HER2 antibody appears to play a significant role in prevention and therapy, naturally occurring anti-HER2 antibody associated with the cleaved ectodomain of overexpressed HER2 self-antigen is detectable in patients. 100 % of MMTV-neuNT mice [29] develop mammary carcinomas. It has been shown that vaccination with HER2-associated with a T helper cell epitope P30 can offer protection against transplantable tumors [149], but it is unclear whether the same vaccine protects against naturally developing tumors. 100 % protection against tumor development was observed subsequent to prophylactic immunization; however high circulating antibody titers were unable to prevent disease progression once tumor was established, suggesting the importance of early immunization in BC prevention [149].
Finally it should be noted that the combination of DNA vaccination plus IL-12 [125], monoclonal antibody to HER2/neu [150], chemotherapy [151], or adenovirus [152] in a murine adenocarcinoma model. These combination therapies will be widely used in clinics after confirmation of results in murine mammary carcinoma models overexpressing neu.
We have reviewed mouse models of human breast cancers overexpressing HER2. The mean latency of mammary tumor development, the pathological findings, and their clinical uses have been summarized in Table 1. MMTV-neu (wild type) models have been most frequently used for translational studies for the characterization of TICs/CSCs, testing TKIs, and to conduct cytokine therapies. MMTV-neuNT mouse models in FVB or Balb/c backgrounds have been used for vaccine development for HER2 and to test the HSP inhibitor 17-AAG in vivo. MMTV-HER2 (wild type or Δ16) models have been used to test monoclonal antibodies in mice before clinical application in humans.
The major problem of HER2-targeting therapy of BC is the development of therapy resistance. MMTV-neu mouse models have often been used to characterize the TICs/CSCs for BC (mammary tumors) since the tumor cells have a marker, and are sensitive to a variety of anti-HER2 therapies. Since the aberrant activity of the PI3K-Akt pathway caused by 1) loss of PTEN, 2) activation by HER4 expression, and 3) PI3CA mutation all correlate with diminished response to HER2 therapies, PI3K and Akt inhibitors are promising chemotherapeutic agents to overcome the problem. Likewise, the increased activity of integrin-β3-TGFβ signaling and aberrant Notch signaling was reportedly associated with therapy resistance to HER2. Detailed characterization and simultaneous inactivation of these signaling cascades will be essential to overcome the resistance to HER2-target therapies using TKIs or monoclonal antibodies.
We thank all members of Dr. Inoue’s lab for sharing unpublished research data.
K. Inoue was supported by NIH/NCI 2R01CA106314, ACS RSG-07-207-01-MGO, and KG080179.
- Siegel RL, Miller KD, Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66: 7-30. [Crossref]
- Ross JS, Hortobagyi GN (2005) Molecular Oncology of Breast Cancer. Publishers: Sudbury, Massachusetts.
- Taneja P, Maglic D, Kai F, Zhu S, Kendig RD, et al. (2010) Classical and Novel Prognostic Markers for Breast Cancer and their Clinical Significance. Clin Med Insights Oncol 4: 15-34. [Crossref]
- Kos Z, Dabbs DJ (2016) Biomarker assessment and molecular testing for prognostication in breast cancer. Histopathology 68: 70-85. [Crossref]
- Inoue K, Fry EA (2016) Novel biomarkers for breast cancer. Biomark Cancer 8: 1-18.
- Patani N, Martin LA, Dowsett M (2013) Biomarkers for the clinical management of breast cancer: international perspective. Int J Cancer 133: 1-13. [Crossref]
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. (2000) Molecular portraits of human breast tumours. Nature 406: 747-752. [Crossref]
- Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, et al. (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98: 10869-10874. [Crossref]
- Kittaneh M, Montero AJ, Glück S (2013) Molecular profiling for breast cancer: a comprehensive review. Biomark Cancer 5: 61-70. [Crossref]
- Maglic D, Zhu S, Fry EA, Taneja P, Kai F, et al. (2013) Prognostic value of the hDMP1-ARF-Hdm2-p53 pathway in breast cancer. Oncogene 32: 4120-4129. [Crossref]
- Guestini F, McNamara KM, Ishida T, Sasano H (2015) Triple Negative Breast Cancer Chemosensitivity and Chemoresistance: Current Advances in Biomarkers Indentification. Expert Opin Ther Targets 20: 705-720. [Crossref]
- Madrid-Paredes A, Cañadas-Garre M, Sánchez-Pozo A, Calleja-Hernández MÁ (2015) Non-HER2 signaling pathways activated in resistance to anti-HER2 therapy in breast cancer. Breast Cancer Res Treat 153: 493-505. [Crossref]
- Patel TA, Dave B, Rodriguez AA, Chang JC, Perez EA, et al. (2014) Dual HER2 blockade: preclinical and clinical data. Breast Cancer Res 16: 419. [Crossref]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, et al. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177-182. [Crossref]
- Fry EA, Taneja P, Inoue K (2016) Oncogenic and tumor-suppressive mouse models for breast cancer engaging HER2/neu. Int J Cancer . [Crossref]
- Howe LR, Brown PH (2011) Targeting the HER/EGFR/ErbB family to prevent breast cancer. Cancer Prev Res (Phila) 4: 1149-1157. [Crossref]
- Appert-Collin A, Hubert P, Crémel G, Bennasroune A (2015) Role of ErbB Receptors in Cancer Cell Migration and Invasion. Front Pharmacol 6: 283. [Crossref]
- Wilson KJ, Mill C, Lambert S, Buchman J, Wilson TR, Hernandez-Gordillo V, Gallo RM, Ades LM, Settleman J, Riese DJ 2nd (2012) EGFR ligands exhibit functional differences in models of paracrine and autocrine signaling. Growth Factors 30: 107-116. [Crossref]
- Roskoski R Jr (2014) The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 79: 34-74. [Crossref]
- Inoue K, Fry EA (2015) Aberrant Splicing of Estrogen Receptor, HER2, and CD44 Genes in Breast Cancer. Genet Epigenet 7: 19-32. [Crossref]
- Yamamoto T, Ikawa S, Akiyama T, Semba K, Nomura N, et al. (1986) Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature 319: 230-234. [Crossref]
- Akiyama T, Sudo C, Ogawara H, Toyoshima K, Yamamoto T. The product of the human c-erbB-2 gene: a 185-kilodalton glycoprotein with tyrosine kinase activity. Science 1986;232:1644-1646. [Crossref]
- Schneider MR, Yarden Y (2016) The EGFR-HER2 module: a stem cell approach to understanding a prime target and driver of solid tumors. Oncogene 35: 2949-2960. [Crossref]
- Citri A, Skaria KB, Yarden Y (2003) The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res 284: 54-65. [Crossref]
- Hudziak RM, Schlessinger J, Ullrich A (1987) Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc Natl Acad Sci U S A 84: 7159-7163. [Crossref]
- Hung MC, Schechter AL, Chevray PY, Stern DF, Weinberg RA (1986) Molecular cloning of the neu gene: absence of gross structural alteration in oncogenic alleles. Proc Natl Acad Sci U S A 83: 261-264. [Crossref]
- Di Marco E1, Pierce JH, Knicley CL, Di Fiore PP (1990) Transformation of NIH 3T3 cells by overexpression of the normal coding sequence of the rat neu gene. Mol Cell Biol 10: 3247-3252. [Crossref]
- Taneja P1, Frazier DP, Kendig RD, Maglic D, Sugiyama T, et al. (2009) MMTV mouse models and the diagnostic values of MMTV-like sequences in human breast cancer. Expert Rev Mol Diagn 9: 423-440. [Crossref]
- Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P (1988) Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 54: 105-115. [Crossref]
- Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, et al. (1992) Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A 89: 10578-10582. [Crossref]
- Weinstein EJ, Kitsberg DI, Leder P (2000) A mouse model for breast cancer induced by amplification and overexpression of the neu promoter and transgene. Mol Med 6: 4-16. [Crossref]
- Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, et al. (2000) Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci U S A 97: 3444-3449. [Crossref]
- Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, et al. (2002) Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell 2: 451-461. [Crossref]
- Schwall RH, Hollingshead P, Erickson S, Winer J, Williams M, et al. (2003) Herceptin-sensitivity of HER2-transgenic mouse mammary tumors. Breast Cancer Res 5: 14. [Crossref]
- Finkle D, Quan ZR, Asghari V, Kloss J, Ghaboosi N, et al. (2004) HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clin Cancer Res 10: 2499–2511. [Crossref]
- Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, et al. (2006) Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer 13: 221-232. [Crossref]
- Castagnoli L, Iezzi M, Ghedini GC, Ciravolo V, Marzano G, et al. (2014) Activated d16HER2 homodimers and SRC kinase mediate optimal efficacy for trastuzumab. Cancer Res 74: 6248-6259. [Crossref]
- Taneja P, Maglic D, Kai F, Sugiyama T, Kendig RD, et al. (2010) Critical roles of DMP1 in human epidermal growth factor receptor 2/neu-Arf-p53 signaling and breast cancer development. Cancer Res 70: 9084-9094. [Crossref]
- Inoue K, Sherr CJ (1998) Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol Cell Biol 18: 1590-1600. [Crossref]
- Inoue K, Sherr CJ, Shapiro LH (1998) Regulation of the CD13/aminopeptidase N gene by DMP1, a transcription factor antagonized by D-type cyclins. J Biol Chem 273: 29188-29194. [Crossref]
- Inoue K, Roussel MF, Sherr CJ (1999) Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc Natl Acad Sci U S A 96: 3993-3998. [Crossref]
- Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, et al. (2000) Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev 14: 1797-1809. [Crossref]
- Inoue K, Zindy F, Randle DH, Rehg JE, Sherr CJ (2001) Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev 15: 2934-2939. [Crossref]
2021 Copyright OAT. All rights reserv
- Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue K (2005) Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol Cell Biol 25: 220-232. [Crossref]
- Mallakin A, Taneja P, Matise, LA, Willingham MC, Inoue K (2006) Expression of Dmp1 in specific differentiated, nonproliferating cells and its repression by E2Fs. Oncogene 25: 7703-7713. [Crossref]
- Taneja P, Mallakin A, Matise LA, Frazier DP, Choudhary M, et al. (2007) Repression of Dmp1 and Arf transcription by anthracyclins: critical roles of the NF-kappaB subunit p65. Oncogene 26: 7457-7466. [Crossref]
- Mallakin A, Sugiyama T, Taneja P, Matise LA, Frazier DP, et al. (2007) Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell 12: 381-394. [Crossref]
- Mallakin A, Sugiyama T, Kai F, Taneja P, Kendig RD, et al. (2010) The Arf-inducing transcription factor Dmp1 encodes transcriptional activator of amphiregulin, thrombospondin-1, JunB and Egr1. Int J Cancer 126: 1403-1416. [Crossref]
- Frazier DP, Kendig RD, Kai F, Maglic D, Sugiyama T, et al. (2012) Dmp1 physically interacts with p53 and positively regulates p53’s stabilization, nuclear localization, and function. Cancer Res 72: 1740-1750. [Crossref]
- Zhu S, Mott RT, Fry EA, Taneja P, Kulik G, et al. (2013) Cooperation between Dmp1 loss and cyclin D1 overexpression in breast cancer. Am J Pathol 183: 1339-1350. [Crossref]
- Fry EA, Taneja P, Maglic D, Zhu S, Sui G, et al. (2013) Dmp1α inhibits HER2/neu-induced mammary tumorigenesis. PLoS One 8: e77870. [Crossref]
- Maglic D, Stovall DB, Cline JM, Fry EA, Mallakin A, et al. (2015) DMP1ß, a splice isoform of the tumor suppressor DMP1 locus, induces proliferation and progression of breast cancer. J Pathol 236: 90-102. [Crossref]
- Tschan MP, Gullberg U, Shan D, Torbett BE, Fey MF, et al. (2008) The hDMP1 tumor suppressor is a new WT1 target in myeloid leukemias. Leukemia 22: 1087-1090. [Crossref]
- Kobayashi M, Srour EF (2011) Regulation of murine hematopoietic stem cell quiescence by Dmtf1. Blood 118: 6562-6571. [Crossref]
- Tschan MP, Federzoni EA, Haimovici A, Britschgi C, Moser BA, et al. (2015) Human DMTF1ß antagonizes DMTF1a regulation of the p14(ARF) tumor suppressor and promotes cellular proliferation. Biochim Biophys Acta 1849: 1198-1208. [Crossref]
- Peng Y, Dong W, Lin TX, Zhong GZ, Liao B, et al. (2015) MicroRNA-155 promotes bladder cancer growth by repressing the tumor suppressor DMTF1. Oncotarget 6: 16043-16058. [Crossref]
- Inoue K, Mallakin A, Frazier DP (2007) Dmp1 and tumor suppression. Oncogene 26: 4329-4335. [Crossref]
- Sugiyama T, Frazier DP, Taneja P, Kendig RD, Morgan RL, et al. (2008) Signal transduction involving the dmp1 transcription factor and its alteration in human cancer. Clin Med Oncol 2: 209-219. [Crossref]
- Sugiyama T, Frazier DP, Taneja P, Morgan RL, Willingham MC, et al. (2008) Role of DMP1 and its future in lung cancer diagnostics. Expert Rev Mol Diagn 8: 435-447. [Crossref]
- Inoue K, Sugiyama T, Taneja P, Morgan RL, Frazier DP (2008) Emerging roles of DMP1 in lung cancer. Cancer Res 68: 4487-4490. [Crossref]
- Inoue K, Fry EA, Frazier DP (2015) Transcription factors that interact with p53 and Mdm2. Int J Cancer 138: 1577-1585. [Epub ahead of print] [Crossref]
- Inoue K, Fry EA (2015) Aberrant expression of Cyclin D1 in cancer. Signal Transduction Insights 4: 1-13.
- Inoue K, Fry EA (2016) Aberrant splicing of the DMP1-INK4a/ARF-MDM2-p53 pathway in cancer. Int J Cancer.139: 33-41.
- Inoue K, Fry EA (2016) Haplo-insufficient tumor suppressor genes. A book chapter in ‘Advances in Genetics Research’, Volume 17, Nova Science Publishers, Inc.
- Menezes ME, Bhatia S, Bhoopathi P, Das SK, Emdad L, et al. (2014) MDA-7/IL-24: multifunctional cancer killing cytokine. Adv Exp Med Biol 818: 127-153. [Crossref]
- Whitaker EL, Filippov VA, Duerksen-Hughes PJ (2012) Interleukin 24: mechanisms and therapeutic potential of an anti-cancer gene. Cytokine Growth Factor Rev 23: 323-331. [Crossref]
- Li YJ, Liu G, Xia L, Xiao X, Liu JC, et al. (2015) Suppression of Her2/Neu mammary tumor development in mda-7/IL-24 transgenic mice. Oncotarget 6: 36943-36954. [Crossref]
- Izrailit J, Reedijk M (2012) Developmental pathways in breast cancer and breast tumor-initiating cells: therapeutic implications. Cancer Lett 317: 115-126. [Crossref]
- Wei W, Lewis MT (2015) Identifying and targeting tumor-initiating cells in the treatment of breast cancer. Endocr Relat Cancer 22: R135-155. [Crossref]
- Rangel MC, Bertolette D, Castro NP, Klauzinska M, Cuttitta F, et al. (2016) Developmental signaling pathways regulating mammary stem cells and contributing to the etiology of triple-negative breast cancer. Breast Cancer Res Treat 156: 211-226. [Crossref]
- Marhaba R, Klingbeil P, Nuebel T, Nazarenko I, Buechler MW, et al. (2008) CD44 and EpCAM: cancer-initiating cell markers. Curr Mol Med 8: 784-804. [Crossref]
- Ranji P, Salmani Kesejini T, Saeedikhoo S, Alizadeh AM (2016) Targeting cancer stem cell-specific markers and/or associated signaling pathways for overcoming cancer drug resistance. Tumour Biol 37: 13059-13075. [Crossref]
- Burger PE, Gupta R, Xiong X, Ontiveros CS, Salm SN, et al. (2009) High aldehyde dehydrogenase activity: a novel functional marker of murine prostatestem/progenitor cells. Stem Cells 27: 2220-2228. [Crossref]
- La Porta C (2009) Cancer stem cells: lessons from melanoma. Stem Cell Rev 5: 61-65. [Crossref]
- Diepenbruck M, Christofori G (2016) Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol 43: 7-13. [Crossref]
- Ren H, Du P, Ge Z, Jin Y, Ding D, et al. (2016) TWIST1 and BMI1 in Cancer Metastasis and Chemoresistance. J Cancer 7: 1074-1080. [Crossref]
- Smith BN, Bhowmick NA (2016) Role of EMT in Metastasis and Therapy Resistance. J Clin Med 5. [Crossref]
- Liu X, Yun F, Shi L, Li ZH, Luo NR, et al. (2015) Roles of Signaling Pathways in the Epithelial-Mesenchymal Transition in Cancer. Asian Pac J Cancer Prev 16: 6201-6206. [Crossref]
- Cantelli G, Crosas-Molist E, Georgouli M, Sanz-Moreno V (2016) TGFβ -induced transcription in cancer. Semin Cancer Biol.
- Asem MS, Buechler S, Wates RB, Miller DL, Stack MS (2016) Wnt5a Signaling in Cancer. Cancers (Basel) 8. [Crossref]
- Conacci-Sorrell M, Simcha I, Ben-Yedidia T, Blechman J, Savagner P, et al. (2003) Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J Cell Biol 163: 847-857. [Crossref]
- Spill F, Reynolds DS, Kamm RD, Zaman MH (2016) Impact of the physical microenvironment on tumor progression and metastasis. Curr Opin Biotechnol 40: 41-48. [Crossref]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, et al. (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438: 820-827. [Crossref]
- Liu JC, Deng T, Lehal RS, Kim J, Zacksenhaus E (2007) Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res 67: 8671-8681. [Crossref]
- Liu JC, Voisin V, Bader GD, Deng T, Pusztai L, et al. (2012) Seventeen-gene signature from enriched Her2/Neu mammary tumor-initiating cells predicts clinical outcome for human HER2+:ERa- breast cancer. Proc Natl Acad Sci U S A 109: 5832-5837. [Crossref]
- Lo PK, Kanojia D, Liu X, Singh UP, Berger FG, et al. (2012) CD49fand CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFß signaling. Oncogene 31: 2614-2626. [Crossref]
- Liu JC, Egan SE, Zacksenhaus E (2013) A tumor initiating cell-enriched prognostic signature for HER2+:ERa- breast cancer; rationale, new features, controversies and future directions. Oncotarget 4: 1317-1328. [Crossref]
- Deng T, Liu JC, Chung PE, Uehling D, Aman A, et al. (2014) shRNA kinome screen identifies TBK1 as a therapeutic target for HER2+ breast cancer. Cancer Res 74: 2119-2130. [Crossref]
- Abravanel DL, Belka GK, Pan TC, Pant DK, Collins MA, et al. (2015) Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J Clin Invest 125: 2484-2496. [Crossref]
- Usary J, Zhao W, Darr D, Roberts PJ, Liu M, et al. (2013) Predicting drug responsiveness in human cancers using genetically engineered mice. Clin Cancer Res 19: 4889-4889. [Crossref]
- Creedon H, Balderstone LA, Muir M, Balla J, Gomez-Cuadrado L, Tracey N, et al. (2016) Use of a genetically engineered mouse model as a preclinical tool for HER2 breast cancer. Dis Model Mech 9: 131-140. [Crossref]
- Hoenerhoff MJ, Shibata MA, Bode A, Green JE (2011) Pathologic progression of mammary carcinomas in a C3(1)/SV40 T/t-antigentransgenic rat model of human triple-negative and Her2-positive breast cancer. Transgenic Res 20: 247-259. [Crossref]
- Prat A, Perou CM (2011) Deconstructing the molecular portraits of breast cancer. Mol Oncol 5: 5-23. [Crossref]
- Song G, Darr DB, Santos CM, Ross M, Valdivia A, et al. (2014) Effects of tumor microenvironment heterogeneity on nanoparticle disposition and efficacy in breast cancer tumor models. Clin Cancer Res 20: 6083-6095. [Crossref]
- Ursini-Siegel J, Hardy WR, Zuo D, Lam SH, Sanguin-Gendreau V, et al. (2008) ShcA signalling is essential for tumour progression in mouse models of human breast cancer. EMBO J 27: 910-920. [Crossref]
- Schade B, Rao T, Dourdin N, Lesurf R, Hallett M, et al. (2009) PTEN deficiency in a luminal ErbB-2 mouse model results in dramatic acceleration of mammary tumorigenesis and metastasis. J Biol Chem 284: 19018-19026. [Crossref]
- Hanker AB, Pfefferle AD, Balko JM, Kuba MG, Young CD, et al. (2013) Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc Natl Acad Sci U S A 110: 14372-14377. [Crossref]
- Cristina Oliveras-Ferraros, Bruna Corominas-Faja, Sílvia Cufí, Alejandro Vazquez-Martin, Begoña Martin-Castillo, et al. (2012) Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin). Cell Cycle 11: 4020-4032. [Crossref]
- Hong DS, Banerji U, Tavana B, George GC, Aaron J, et al. (2013) Targeting the molecular chaperone heat shock protein 90 (HSP90): lessons learned and future directions. Cancer Treat Rev 39: 375-387. [Crossref]
- Rodrigues LM, Chung YL, Al Saffar NM, Sharp SY, Jackson LE, et al. (2012) Effects of HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG) onNEU/HER2 overexpressing mammary tumours in MMTV-NEU-NT mice monitored by Magnetic Resonance Spectroscopy. BMC Res Notes 5: 250.
- Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, et al. (2011) Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci U S A 108: 7142-7147. [Crossref]
- Junttila TT, Li J, Johnston J, Hristopoulos M, Clark R, et al. (2014) Antitumor efficacy of a bispecific antibody that targets HER2 and activates T cells. Cancer Res 74: 5561-5571. [Crossref]
- Zhang S, Huang WC, Li P, Guo H, Poh SB, et al. (2011) Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med 17: 461-469. [Crossref]
- Campiglio M, Bufalino R, Sasso M, Ferri E, Casalini P, et al. (2013) Effect of adjuvant trastuzumab treatment in conventional clinical setting: an observational retrospective multicenter Italian study. Breast Cancer Res Treat 141: 101-110. [Crossref]
- Canfield K, Li J, Wilkins OM, Morrison MM, Ung M, et al. (2015) Receptor tyrosine kinase ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells. Cell Cycle 14: 648-655. [Crossref]
- Utermark T, Rao T, Cheng H, Wang Q, Lee SH, et al. (2012) The p110a and p110ß isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev 26: 1573-1586. [Crossref]
- Hanker AB, Cook RS, Arteaga CL (2013) Mouse models and anti-HER2 therapies. Oncotarget 4: 1866-1867. [Crossref]
- Ma CX, Luo J, Ellis MJ (2010) Molecular profiling of triple negative breast cancer. Breast Dis 32: 73-84. [Crossref]
- O'Sullivan CC, Swain SM (2013) Pertuzumab: evolving therapeutic strategies in the management of HER2-overexpressing breast cancer. Expert Opin Biol Ther 13: 779-790. [Crossref]
- Maira SM, Pecchi S, Huang A, Burger M, Knapp M, et al. (2012) Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther 11: 317-328. [Crossref]
- Meuwissen R, Jonkers J, Berns A (2001) Mouse models for sporadic cancer. Exp Cell Res 264: 100-110. [Crossref]
- Jonkers J, Berns A (2002) Conditional mouse models of sporadic cancer. Nat Rev Cancer 2: 251-265. [Crossref]
- Taneja P, Zhu S, Maglic D, Fry EA, Kendig RD, et al. (2011) Transgenic and knockout mice models to reveal the functions of tumor suppressor genes. Clin Med Insights Oncol 5: 235-257. [Crossref]
- Inoue K, Fry EA, Taneja P. Recent progress in mouse models for tumor suppressor genes and its implications in human cancer. Clin Med Oncol 2013;7:103-122. [Crossref]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, et al. (2002) p53 mutant mice that display early ageing-associated phenotypes. Nature 415: 45-53. [Crossref]
- Matheu A, Pantoja C, Efeyan A, Criado LM, Martín-Caballero J, et al. (2004) Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev 18: 2736-2746. [Crossref]
- Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, et al. (2007) Delayed ageing through damage protection by the Arf/p53 pathway. Nature 448: 375-379. [Crossref]
- Yang C, Ionescu-Tiba V, Burns K, Gadd M, Zukerberg L, et al. (2004) The role of the cyclin D1-dependent kinases in ErbB2-mediated breast cancer. Am J Pathol 164: 1031-1038. [Crossref]
- Sarkar D, Su ZZ, Vozhilla N, Park ES, Gupta P, et al. (2005) Dual cancer-specific targeting strategy cures primary and distant breast carcinomas in nude mice. Proc Natl Acad Sci U S A 102: 14034-14039. [Crossref]
- Sauane M, Su ZZ, Gupta P, Lebedeva IV, Dent P, et al. (2008) Autocrine regulation of mda-7/IL-24 mediates cancer-specific apoptosis. Proc Natl Acad Sci U S A 105: 9763-9768. [Crossref]
- Dash R, Bhoopathi P, Das SK, Sarkar S, Emdad L, et al. (2014) Novel mechanism of MDA-7/IL-24 cancer-specific apoptosis through SARI induction. Cancer Res 74: 563-574. [Crossref]
- Lucchini F, Sacco MG, Hu N, Villa A, Brown J, et al. (1992) Early and multifocal tumors in breast, salivary, harderian and epididymal tissues developed in MMTV-Neu transgenic mice. Cancer Lett 64: 203-209. [Crossref]
- Boggio K, Nicoletti G, Di Carlo E, Cavallo F, Landuzzi L, et al. (1998) Interleukin 12-mediated prevention of spontaneous mammary adenocarcinomas in two lines of Her-2/neu transgenic mice. J Exp Med 188: 589-596. [Crossref]
- Boggio K, Di Carlo E, Rovero S, Cavallo F, Quaglino E, et al. (2000) Ability of systemic interleukin-12 to hamper progressive stages of mammary carcinogenesis in HER2/neu transgenic mice. Cancer Res 60: 359-364. [Crossref]
- Di Carlo E, Rovero S, Boggio K, Quaglino E, Amici A, et al. (2001) Inhibition of mammary carcinogenesis by systemic interleukin 12 or p185neu DNA vaccination in Her-2/neu transgenic BALB/c mice. Clin Cancer Res 7: 830s-837s. [Crossref]
- Croci S, Nanni P, Palladini A, Nicoletti G, Grosso V, et al. (2015) Interleukin-15 is required for immunosurveillance and immunoprevention of HER2/neu-driven mammary carcinogenesis. Breast Cancer Res 17: 70. [Crossref]
- Sonoda A, Nitta N, Ohta S, Nitta-Seko A, Murata S, et al. (2009) Investigation using an HER-2/neu transgenic mouse model of a newly developed MR contrast agent with the effect of an antitumor drug. J Magn Reson Imaging 30: 907-910. [Crossref]
- Pierpaoli E, Arcamone AG, Buzzetti F, Lombardi P, Salvatore C, et al. (2013) Antitumor effect of novel berberine derivatives in breast cancer cells. Biofactors 39: 672-679. [Crossref]
- Pierpaoli E, Damiani E, Orlando F, Lucarini G, Bartozzi B, et al. (2015) Antiangiogenic and antitumor activities of berberine derivative NAX014 compound in a transgenic murine model of HER2/neu-positive mammary carcinoma. Carcinogenesis 36: 1169-1179. [Crossref]
- Banerjee T, Duhadaway JB, Gaspari P, Sutanto-Ward E, Munn DH, et al. (2008) A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene 27: 2851-2857. [Crossref]
- Criscitiello C, Curigliano G (2015) Immunotherapy of Breast Cancer. Prog Tumor Res 42: 30-43. [Crossref]
- van Rooijen JM, Stutvoet TS, Schröder CP, de Vries EG (2015) Immunotherapeutic options on the horizon in breast cancer treatment. Pharmacol Ther 156: 90-101. [Crossref]
- Peres Lde P, da Luz FA, Pultz Bdos A, Brígido PC, de Araújo RA, et al. (2015) Peptide vaccines in breast cancer: The immunological basis for clinical response. Biotechnol Adv 33: 1868-1877. [Crossref]
- Wei WZ, Jones RF, Juhasz C, Gibson H, Veenstra J (2015) Evolution of animal models in cancer vaccine development. Vaccine 33: 7401-7407. [Crossref]
- Piechocki MP, Ho YS, Pilon S, Wei WZ (2003) Human ErbB-2 (Her-2) transgenic mice: a model system for testing Her-2 based vaccines. J Immunol 171: 5787-5794. [Crossref]
- Wei H, Wang S, Zhang D, Hou S, Qian W, et al. (2009) Targeted delivery of tumor antigens to activated dendritic cells via CD11c molecules induces potent antitumor immunity in mice. Clin Cancer Res 15: 4612-4621. [Crossref]
- Conti L, Ruiu R, Barutello G, Macagno M, Bandini S, et al. (2014) Microenvironment, oncoantigens, and antitumor vaccination: lessons learned from BALB-neuT mice. Biomed Res Int 2014: 534969. [Crossref]
- Lamolinara A, Stramucci L, Hysi A, Iezzi M, Marchini C, et al. (2015) Intradermal DNA Electroporation Induces Cellular and Humoral Immune Response and Confers Protection against HER2/neu Tumor. J Immunol Res2015: 159145. [Crossref]
- Wang X, Wang JP, Maughan MF, Lachman LB (2005) Alphavirus replicon particles containing the gene for HER2/neu inhibit breast cancer growth and tumorigenesis. Breast Cancer Res 7: R145-155. [Crossref]
- Gallo P, Dharmapuri S, Nuzzo M, Maldini D, Cipriani B, et al. (2007) Adenovirus vaccination against neu oncogene exerts long-term protection from tumorigenesis in BALB/neuT transgenic mice. Int J Cancer 120: 574-584. [Crossref]
- Nguyen-Hoai T, Baldenhofer G, Sayed Ahmed MS, Pham-Duc M, Vu MD, et al. (2012) CCL21 (SLC) improves tumor protection by a DNA vaccine in a Her2/neu mouse tumor model. Cancer Gene Ther 19: 69-76. [Crossref]
- Nguyen-Hoai T, Baldenhofer G, Ahmed MS, Pham-Duc M, Gries M, et al. (2012) CCL19 (ELC) improves TH1-polarized immune responses and protective immunity in a murine Her2/neu DNA vaccination model. J Gene Med14: 128-137. [Crossref]
- Nguyen-Hoai T, Pham-Duc M, et al. (2016) CCL4 as an adjuvant for DNA vaccination in a Her2/neu mouse tumor model. Cancer Gene Ther 23: 162-167. [Crossref]
- Maha Zohra Ladjemi, William Jacot, Thierry Chardès, André Pèlegrin, Isabelle Navarro-Teulon (2010) Anti-HER2 vaccines: new prospects for breast cancer therapy. Cancer Immunol Immunother 59: 1295–1312. [Crossref]
- Pravin TP Kaumaya, Kevin Chu Foy. Peptide vaccines and peptidomimetics targeting HER and VEGF proteins may offer a potentially new paradigm in cancer immunotherapy. Future Oncol 2012; 8: 961–987. [Crossref]
- Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK (2014) Peptide Vaccine: Progress and Challenges. Vaccines (Basel) 2: 515-536. [Crossref]
- Mittendorf EA, Holmes JP, Murray JL, von Hofe E, Peoples GE (2009) CD4+ T cells in antitumor immunity: utility of an li-key HER2/neu hybrid peptide vaccine (AE37). Expert Opin Biol Ther 9: 71-78. [Crossref]
- Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, et al. (2005) Recruitment of latent pools of high-avidity CD8(+) T cells to the antitumor immune response. J Exp Med 201: 1591-1602. [Crossref]
- Mukhopadhyay A, Dyring C, Stott DI (2014) Comparison of prophylactic and therapeutic immunisation with an ErbB-2 (HER2) fusion protein and immunoglobulin V-gene repertoire analysis in a transgenic mouse model of spontaneous breast cancer. Vaccine 32: 1012-1018. [Crossref]
- Orlandi F, Guevara-Patiño JA, Merghoub T, Wolchok JD, Houghton AN, et al. (2011) Combination of epitope-optimized DNA vaccination and passive infusion of monoclonal antibody against HER2/neu leads to breast tumor regression in mice. Vaccine 29: 3646-3454. [Crossref]
- Eralp Y, Wang X, Wang JP, Maughan MF, Polo JM, et al. (2004) Doxorubicin and paclitaxel enhance the antitumor efficacy of vaccines directed against HER 2/neu in a murine mammary carcinoma model. Breast Cancer Res 6: R275-R283. [Crossref]
- Wang X, Wang JP, Rao XM, Price JE, Zhou HS, et al. (2005) Prime-boost vaccination with plasmid and adenovirus gene vaccines control HER2/neu+ metastatic breast cancer in mice. Breast Cancer Res 7: R580-588. [Crossref]
- Mohamed A, Krajewski K, Cakar B, Ma CX (2013) Targeted therapy for breast cancer. Am J Pathol 183: 1096-1112. [Crossref]
- Singh B, Carpenter G, Coffey RJ (2016) EGF receptor ligands: recent advances. F1000Res. [Crossref]
- Memon AA, Sorensen BS, Melgard P, Fokdal L, Thykjaer T, et al. (2004) Expression of HER3, HER4 and their ligand heregulin-4 is associated with better survival in bladder cancer patients. Br J Cancer 91: 2034-2041. [Crossref]
- Breuleux M (2007) Role of heregulin in human cancer. Cell Mol Life Sci 64: 2358-2377. [Crossref]
- Khurana A, Gonzalez-Guerrico A, Lupu R (2014) Heregulin in breast cancer: old story, new paradigm. Curr Pharm Des 20: 4874-4878. [Crossref]