In medicine, the search for the cause of a disease has been critical to understanding the nature of the disorder, and an important step towards the discovery of effective therapies and prevention. The search for a cause is more difficult than it may seem at first. For example, even if we find the mechanism by which the disease progresses, the questions would be: what started the process; then, if we have found the factors that initiated the process that will lead to questions as to what happened to this person, and at this very moment. The answer to research questions often raises more questions, and that is how research progresses.
Communication between the immune system and the Central nervous system (CNS), essential for maintaining homeostasis, is exemplified by cross-talk between glia and neurons. While actively microglia cells are modulated by neurons in the healthy brain, little is known about the cross-talk between oligodendrocytes and neurons. Oligodendrocytes, the myelin-forming cells in the CNS, are essential for the propagation of action potentials along axons, and additionally they serve to support neurons by neurotropic factors. In demyelinating diseases, such as multiple sclerosis, oligodendrocytes are thought to be the victims. Also they have strong immune functions, express a wide variety of innate immune receptors, produces and respond to the chemokines and cytokines and modulate immune responses. In Addition, they elicit responses that cause progressive neurodegeneration. Under certain circumstances some cells cross the blood brain barrier and reach the parenchyma, activating a cascade of events culminating in an inflammatory lesion and demyelization. The main participants of these attacks are the CD4+T cells, antigen presenting glia (microglia and astrocytes), macrophages and B cells. On the other hand further evidence support that the beginning of autoimmune response is initiated within the CNS; we should consider other theories to explain not only multiple sclerosis as an autoimmune disease, and that starts outside the central nervous system. There is an intimate relationship that we must pay more attention in our research: the dialogue neuron-glia; the cross-talk between microglia and neuron help us to uncover novel pathways in the brain.
axon loss, cross-talk, glial cells, multiple sclerosis, myelin-forming cells
The brain is composed of two main classes of cells: neurons and glial cells. Both of them are key components of the nervous system. There are various kind of glial cells; astrocytes, oligodendroglia and microglia; and each of them have a variety of functions; capacitate neurons to establish connections and signal transmission, modulate the speed of nerve impulses and interneuron connections, called synapses, control neurotransmitter uptake and stimulate neuronal injury recovery. Glial cells and neurons act differently because the former cells communicate through chemical signals that are much slower than the electric signals emitted by neurons.
Communication between neurons and glial cells is not limited to the synapses; axonal high frequency stimuli cause phosphorylation of myelin basic protein (MBP), a component of myelin in the white matter tracts. The signaling directed toward oligodendrocytes, dependent axonal activity, is mediated by nitric oxide, which is released from axons and stimulates the phosphorylation of MBP in oligodendrocytes through a mechanism dependent of kinase mitogen-activated protein (MAP-K) [1,2]. Moreover, studies in both the Central Nervous System (CNS) and Peripheral Nervous System (PNS) activity indicate that neuronal impulses in fetuses (as well as in early postnatal stage) influence the development of myelin forming glia [3]. This communication is mediated by ionic changes in the extracellular space and influences neuronal activity; as well as non-synaptic release of neurotransmitters, growth factors or signaling molecules specialized in axon-glia communication. Blocking of K+ channels in oligodendrocyte precursor cells, by either depolarizing agents or ion channel specific toxins, prevents the proliferation and differentiation of this type of glial cell [2]. Thus, it can be observed that the large and complex communication network between axons and myelin-forming cells is part of the proper functioning of the CNS, which can be affected when disorders occur in myelin, and inevitably lead to demyelinating diseases such as multiple sclerosis [4].
Oligodendrocytes and Schwann Cells Myelin-Forming (SCMF) are the cells that myelinate the axons by a multilayered wrapping myelin sheath (Figure 1). Myelin is present throughout the CNS and is particularly concentrated in areas where axons are primarily exhibited (known as the white matter). White matter of mammalian brain contains about 50% to 60% of myelin, while myelinated peripheral nerves are even richer in myelin. Anatomically, myelinated axons from CNS and PNS have a similar ultrastructure when analyzed by electron microscopy; but there are significant differences between the two-myelin forming cells types. Morphologically; oligodendrocytes cells in CNS, extend and myelinate several axons processes simultaneously, while SCMF myelinate just one axon; Also, it is important to remember that the PNS nerves are wrapped by Schwann cells that do not form myelin (SCMF) which sheathe several axons (lower caliber 2 mm), each of which is located on a portion of the plasma membrane of the SCMF.
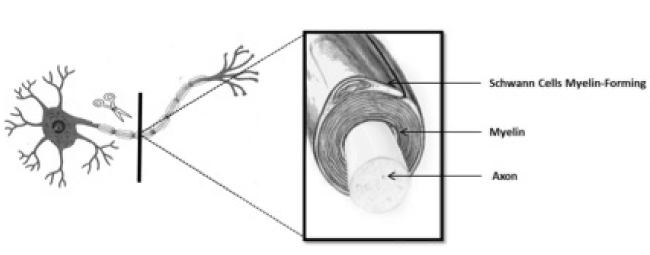
Figure 1. Schwann Cells Myelin-Forming (SCMF).
Cells that myelinate the axon by a multilayered wrapping myelin sheath.
Myelin is mainly composed of lipids that become approximately about 70% of dry weight in CNS while in the PNS are 90%. The major components of myelin are: cholesterol, glycerophospholipids and sphingolipids [5-9]. Myelin from CNS is different from that of PNS, for example, the amount of cholesterol and cerebroside in PNS myelin is lower and the sphingomyelin is greater than that found in the CNS. The glycosphingolipid cerebroside represents 20% of the dry weight of CNS myelin and 15% in the PNS. Another important difference between oligodendrocytes and SCMF is the protein composition of the myelin sheath where both types of cells express different proteins (Table 1) [10–12].
Table 1. Difference in the composition between oligodendrocytes and Schwann Cell Myelin-forming.
Protein |
30.0 |
24.7 |
29.5 |
39.0 |
39.5 |
55.3 |
56.9 |
Lipid |
70.0 |
75.3 |
70.5 |
54.9 |
55.0 |
32.7 |
37.0 |
Cholesterol |
27.7 |
28.1 |
27.3 |
27.5 |
23.6 |
22.0 |
23.0 |
Cerebroside |
22.7 |
24.0 |
23.7 |
19.8 |
22.5 |
5.4 |
14.6 |
Sulfatide |
3.8 |
3.6 |
7.1 |
5.4 |
5.0 |
1.7 |
4.8 |
Total galactolipid |
27.5 |
29.3 |
31.5 |
26.4 |
28.6 |
7.3 |
21.3 |
Ethanolamine
Phosphatides |
15.6 |
17.4 |
16.7 |
14.9 |
13.6 |
22.7 |
19.8 |
Lecithin |
11.2 |
10.9 |
11.3 |
12.8 |
12.9 |
26.7 |
22.0 |
Sphingomyelin |
7.9 |
7.1 |
3.2 |
7.7 |
6.7 |
6.9 |
3.8 |
Phosphatidylserine |
4.8 |
6.5 |
7.0 |
7.9 |
11.4 |
8.7 |
7.2 |
Phosphatidylinositol |
0.6 |
0.8 |
1.2 |
0.9 |
0.9 |
2.7 |
2.4 |
Plasmalogens |
12.3 |
14.1 |
14.1 |
11.2 |
12.2 |
8.8 |
11.6 |
Total phospholipid |
43.1 |
43.0 |
44.0 |
45.9 |
46.3 |
69.5 |
57.0 |
(Siegel GJ, Agranoff BW, Albers RW, et al., editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition. Philadelphia: Lippincott-Raven; 1999.
Studying Oligodendrocytes and SCMF arouse great interest because of its close links with demyelinating neurological disorders such as multiple sclerosis, which is one of the leading causes of neurological disability of CNS in young adults, whose etiology is multifactorial and unknown [13,14]. The myelin sheaths are a fundamental adaptation of vertebrates, where myelinated axons of the CNS and PNS are covered almost entirely by the myelin sheath, except the Ranvier´s nodes; small areas (approximately 1 mm) relatively exposed to the extracellular medium. The nodes of Ranvier are in contact with fine processes of the myelin-forming cells (paranodal handles) and with microvilli of SCMF and basal lamina in the PNS, while CNS is covered by cellular processes of astrocytes [12].
The organization of the axon union with the oligodendrocytes or SCMF is very special and almost similar. Yuxtaparanodal region in axonal groups have type K+ channels (shaker) Kv1.1, Kv1.2 and Kvb2 and are located besides the adhesion protein Caspr-2 belonging to the neurexins superfamily. In axonal membrane K+ channels SCMF type (shaker) Kv1.5 and Ca++ activated K+ are located. Moreover, the adherens junctions connect handles of paranodal myelin (not campacta) of regions and in the notches. These joints are made of E-cadherin and catenin a and b. The MAG protein is enriched in this area and it is suggested that participates in cell adhesion. Gap junctions are also among adherens junctions in the paranodal region as well in the membrane [15–17].
In the paranodal axonal region it is found the glycoprotein called Caspr-1 or paranodin also belonging to the superfamily neurexins [18,19]. Cell adhesion protein, neurofascin 155, (NF155) has been found in paranodal area in oligodendrocytes and SCMF Caspr-1. NF155 localized with Confocal microscopy, so it has been suggested that NF155 establishes binding with Caspr-1 and either directly or through binding proteins, such as contactin, Caspr-1 and 2 binds to cytoskeletal adapter proteins 4.1R and 4.1B, which in turn anchors to the cytoskeleton neurexins [20–23].
When a compression or transection injury of peripheral nerves is caused; a process called Wallerian degeneration begins; axons and myelin sheath are destroyed at the site of the lesion and the distal region degenerates (the proximal region of the nerve containing the cell bodies, in the case of the sciatic nerve, dorsal root ganglia). Following neuronal injury, the Schwann cells survive; macrophages enter to the site of injury cleaning the cellular debris produced by nerve damage allowing conditions of regeneration [24]. In order to establish contact with the proximal region and regenerate the nerve, the Schwann cells of the distal region begin to proliferate and synthesize basal lamina forming endoneural tubes or Bunger´s bands. Precursors of Schwann cells in culture cannot survive when they are devoid of axonal contact; while immature Schwann cells have the ability to survive, independently of axon contact, by autocrine signaling mechanisms [25]. Studies in vitro and in vivo, in Schwann cell precursors show that neurons or signals derived from neurons, not only rescue Schwann’s cells precursors from cell death by apoptosis, but also, allow these precursors to differentiate into Schwann cells according to the normal development program. So, the survival of Schwann´s cells precursor depends on signals derived from the axon, where neuroregulin (NRG) and endothelin emerge as the main candidates in this process [3,26–29].
The ability of adult Schwann cells to survive, in the absence of nerve axons, for several months after injury is crucial for regeneration as Schwann cells provide adherent substrates and trophic factors that promote axonal growth [25,30–32]. The survival of Schwann cells in the absence of axons is due, in part, to the existence of signaling from autocrine cells circuits that enable Schwann cells to survive. This represents a fundamental difference between Schwann cells and their precursors, which are completely dependent of axonal factors to survive. Tests in vitro of Schwann cells wherein (medium conditioned) cells were cultured to high confluence and subsequently transferred to primary cultures of Schwann cells, allowed the survival of Schwann cells, but when the culture media were transferred to precursors of Schwann cells, these were not able to survive. This medium was not mitogenic for both cell types. Thus, autocrine signaling components of Schwann cells have been identified and these include the IGF-2, platelet-derived growth factor (PDGF), neurotrophin-3 (NT3) and leukemia inhibitory factor. Moreover, laminin cooperates with these components as a promoter to prolong neuron survival [33,34].
Axon signals are involved in the differentiation of immature Schwann cells; again, the contact of Schwann cells with the appropriate axons provides the necessary stimulus to myelinization. Using transplantation techniques, in which nerves and crosslinking between myelinated nerves and not myelinated ones reveals that cells from immature Schwann receive signals from the axons to differentiate into SCMF. When you place together myelinated nerve grafts with unmyelinated nerve, the later is capable of inducing the production of myelin. However, a dispute arise as a main point of discussion, if there is a threshold myelination related to axon diameter, because only larger diameter axons get myelinated or if there are specific signaling molecules, including those that arise from SCMF, that induce axon myelinization [35,36]. Glial cells are interactive companions of neuronal activity, regulating many processes that allow greater plasticity of the nervous system, resulting in a complex web of cooperation and cellular interactions.
In diseases such as multiple sclerosis (MS), in which the myelin sheath in the CNS axons is dramatically altered, and eventually degenerated, it is not known if this is a secondary effect due to conduction disturbances imposed to nervous system caused by abnormalities in myelin or other mechanisms induced damage and atrophy of the axon, such as inflammatory signals [37,38]. However, it is possible that the inefficiency of the axon to conduct the nerve impulse is due to a lack of communication glia-axon to generate adequate conduction of the nerve. In the past, the involvement of neurons and axons was thought to be unimportant in MS (today, we know the great importance), and when it occurs is a result of inflammation or loss of myelin. However, in recent years it has been shown that axonal injury is very important in multiple sclerosis and is responsible for the failure experienced by patients [39,40].
This degeneration of axons can be effectively related to inflammation and demyelination, but the possibility that other causes take place is currently unknown. Also, it has been observed remyelination in atrophied areas; however, this is insufficient or absent in chronic conditions. The remyelination is carried out by the oligodendrocytes precursor cells (OPC) which must migrate to the site of injury, differentiate into oligodendrocytes, and finally form myelin; those processes are directed by signals from both axons and astrocytes. However, deregulation of extracellular signals lead to failure of adequate remyelination. Moreover, autoimmune etiology is one of the most accepted and studied factor in multiple sclerosis, in which lymphocytes T cells, macrophages and microglia destroy oligodendrocytes in the brain and spinal cord are involved in this pathology [41].
A postmortem study of 12 MS patients with MS shows that T lymphocytes and macrophages infiltrate damaged areas in both the brain and spinal cord, but, if you can see vast areas indicating apoptosis of oligodendrocytes, it suggest that the root of the neurological disorder is not only due to the immune response. This leads us to reconsider the participation of glial cells in various neurological demyelinating disorders where the nervous system leaves the normal context and begins to have a malfunction; that is pathological, and is in this condition (pathology) that glial cells may play a major role in the progress of such disorders.
Microglia represents between 10% and 20% of non-neuronal cell population, they are monocytes migrating in early stages of development to the central nervous system. These cells are in constant motion and perform immune surveillance. When damage at neuronal level or drastic changes of local environment occurs, different substances are released; such as neurotransmitters, growth factors, interleukins, etc. Microglia, in the brain, is characterized by a branched morphology, what is known as microglia at rest; occupy a defined territory, without physical contact, with another microglial cell. When activated, cellular morphology changes and finally, a hyperbranched amoeboid morphology develop (Figure 2). Those changes facilitate their migration from the parenchyma to the site of injury. Under stress conditions, the microglia cells secrete inflammatory cytokines and growth factors, whereas in infectious conditions, overactivation is induced; microglia cells respond releasing nitric oxide, superoxide radicals, proinflammatory cytokines such as interleukin 1β, tumor necrosis factor- α (TNF-α) and chemokines, in order to amplify the inflammatory response necessary to eliminate the pathogen, which contributes to neuronal death. This activation alters the permeability of the blood brain barrier allowing the entry of immune mediators; natural killer cells (NK) and lymphocytes.
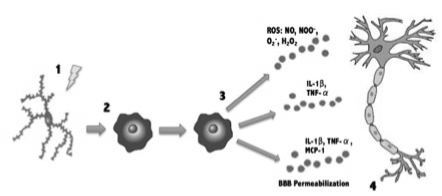
Figure 2. Microglial activation.
- Microglia at rest becomes activated by insults (injury, infection, stress, etc.)
- Upon activation Microglia turns into an amoeboid form.
- Activated Microglia secretes multiple pro-inflammatory, excitatory and neurotoxic factors. (PGE2, IL-1β, TNF-α, NO, NOO-, O2-, H2O2).
- Secreted factors interact with the neuron amplifying the inflammatory response trying to eliminate the injury factor, which causes neuronal damage and death.
The immune response to the central nervous system damage promotes both: a) microglial activation and b) tissue regeneration. It has been observed both in active demyelinating lesions and in areas of inflammation in the myelinating brain, events involved in demyelinating event. Immune cells such as macrophages and CD4 + T cells migrate to the central nervous system through the expression of chemokines and their receptors, releasing cytokines (IL-1β, TNF, IFN-γ, IL-12) leading to progressive demyelination. Demyelinating glia in patients with multiple sclerosis, processes are bonded to damage myelin sheaths leading to sclerosis; however, its contribution to the development of the disease is controversial, since it can also stimulate neuroregeneration in areas of damage during the relapse periods of MS [42,43].
Astrocytes are the most abundant cells in the central nervous system, surpassing neurons in a ratio of 10:1. This type of glial cells are involved in the homeostasis of the central nervous system; regulate neuronal function by releasing neurotropic factors, guide neuronal development, express receptors for neurotransmitters such as AMPA, GABA, NMDA and glutamate, among others, that contribute to their metabolism, and regulation of the extracellular pH and the concentration of potassium ions. Astrocytes also, are involved in the functional and structural integrity of the blood brain barrier (BBB). These cells respond strongly to the central nervous system infections and inflammatory pathologies inducing demyelination by immune auto reactive cells; process known as astrocytosis or reactive astrocytes, characterized by an increase in the size and number of cells expressing GFAP, reflecting predominantly a phenotypic change in astrocytes which involves the expression of new molecules that are not detectable on resting astroglia [44].
Activated microglia express, during the development of experimental allergic encephalitis and multiple sclerosis (mediator expression function), MHC class I antigens and activation of MHC class II. In general, activated microglial cells, in response to cell damage, are involved in autoimmune T cell mediated microglial reaction in nervous system. These reactions can be observed even if the initial injury is distant (such as in the peripheral nervous system) and start after a few hours of brain damage. Structural changes in the brain parenchyma, in all models, are preceded by early molecular alterations, such as induction of MHC antigen expression. In later stages of the disease, microglial response is maintained due to the presence of cellular debris associated with axonal degeneration and demyelination. Microglial activation appears to be independent of the kind of the pathological stimuli, as in all models, uniform changes occur, including: a) Proliferation; b) Transformation in phagocytic cells (macrophage morphology), and c) Stimulation of the expression of cellular molecules such as the receptor for complement C3 or the determining MUC102 surface; or MHC antigens (Figure 2). Interesting is the fact that an increase in MHC antigen expression indicates a state of microglial activation and does not mean itself that it is conducting an immune response in the tissue. The role of the expression of MHC class I antigen (Ia) in non-immune functions is unclear [45]. However, several studies have suggested that an activation molecule can signal through protein kinase C, resulting in the proliferation and differentiation of glial cells.
Multiple sclerosis (MS) is a chronic inflammatory and demyelinating disease of the central nervous system (CNS). Three forms of evolution of MS are broadly defined: relapsing-remitting, secondary progressive and primary progressive (Figure 3). The etiology of MS is unknown but there is sufficient epidemiological studies suggesting that the risk of suffering the disease would be determined by a combination of genetic and environmental factors. At pathological level, inflammatory infiltrates in the CNS of these patients are constituted mainly by T lymphocytes and macrophages, with selective destruction of the myelin lamellae, although axonal injury may be evident from the early stages of disease [46]. In recent decades it has been shown that the brain does not have an absolute absence of immune components, but rather an elaborate regulation therefore. Today it is known that the brain has its own immune response, which is crucial in the process of neurodegeneration because it can promote or inhibit, as an appropriate response, regeneration, while a failure in this response increases neuronal death. In demyelinating diseases of the central nervous system there is a loss of myelin sheaths, damage and axonal degeneration, characterized by leukocyte infiltration with few T cells and mononuclear cells, accompanied by a severe glial response to the central nervous system. These diseases are characterized by intermittent processes of demyelination followed by periods of remyelination, which are determined by activation cycles of neuroinflammatory processes.
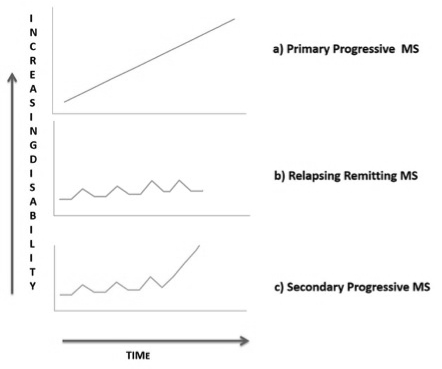
Figure 3.. Evolution forms of Multiple Sclerosis (MS).
- Primary Progressive Multiple Sclerosis (PPMS): Approximately 10% of patients with multiple sclerosis are diagnosed with PPMS. Characterized by progressive course and worsening of neurological functions from the beginning of clinically manifested MS with no distinct relapses or remissions. The rate of progression may vary over time, with occasional plateaus and temporary minor improvements.
- Relapsing-Remitting multiple sclerosis (RRMS): Is the most common form, affects about 85% of patients. Characterized by clearly defined attacks of worsening neurological function (relapses or exacerbations) followed by partial or complete recovery periods (remissions), during which no disease progression occurs and symptoms improve or disappear.
- Secondary-Progressive multiple sclerosis (SPMS): Approximately 50% of patients with relapsing-remitting MS developed SPMS within 10 years, for many patients, treatment with disease-modifying agents helps delay such progression. Characterized by an initial relapsing-remitting form, followed by secondary progressive disease course in which the disease continues to worsen with or without occasional periods of remission, minor recoveries or plateaus.
There are studies that support the evidence that autoimmune response initiate within the CNS [47,48]. Others, however, believe that the initial activation occurs outside the CNS. Within the CNS must exist a primary disorder of the neuron to develop MS. In an experimental system of mixed culture of neurons/glia, it was noted that only glia expresses MHC-II molecules after stimulation with IFN-γ when was physically distant from electrically active neurons. By adding tetrodotoxin (TTX) to the cultures to inhibit the action potential of neurons, glial cells expressed MHC-II in the entire crop culture. While it has been shown, the production of MBP by oligodendrocytes (OLD) depends on the neuronal electrical activity, as treatment with TTX inhibited myelination. It has also been postulated that the first event that occurs in MS is a functional interference and/or damage on the ability of myelination. Initially, this was not associated with morphological alterations, but later, myelin degeneration occurs. As a result, antigenic epitopes that were not accessible to the immune system (cryptic epitopes) may be presented by the microglia, astrocytes and endothelial cells in the context of certain HLA molecules. Upon entry into the CNS, T lymphocytes capable of recognizing these cryptic antigens initiate autoimmune reaction. Cytotoxic T lymphocytes, macrophages, and cytokines increase the pathologic process and/or inhibit the function of naked axons (Figure 4). Outside the CNS, the CNS has been considered a privileged system from the immunological point of view, since it tends to reduce and limit the local immune reactions. This is because an important inflammatory response in CNS may secondarily produce irreversible destruction of nerve cells. This effect is achieved in several ways: very low expression of HLA-I and II, low efficiency (under normal conditions) in antigen presentation and the presence of soluble factors in CSF that neutralize or counteract the effect of certain pro-inflammatory cytokines. In this context, the CNS would not be an appropriate place to start an autoimmune response that requires great efficiency; so many authors suggest that the demyelination process would begin after the entry into the CNS of activated lymphocytes. In this regard, we found that lymphocytes are activated in the peripheral blood of patients with MS outbreak similar to encephalitogenic cells with phenotypic characteristics of lymphocytes. As a reconciliation of both scenarios, it is postulated that, as a result of an initial involvement in CNS, antigen output through certain (primarily olfactory) cranial nerves through the cervical lymph nodes where efficient antigen presentation would take place.
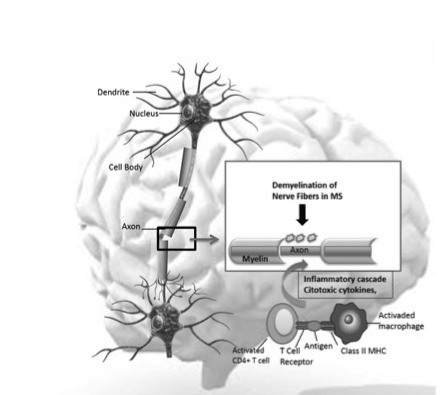
Figure 4. Demyelination and Axonal Damage in Multiple Sclerosis.
In multiple sclerosis, demyelination and axonal damage are critical processes in the pathogenesis of the disease. The lesions are composed of areas of myelin and oligodendrocyte loss, along with infiltrates of inflammatory cells, including activated lymphocytes, macrophages, cytotoxic cytokines and other inflammatory components that destroy selectively myelin sheaths resulting in axonal demyelination, which leads to the slowing or blockade of conduction.
Once the autoimmune response is triggered, the chain of events and the elements involved are basically the same as in any other non-autoimmune response, although with the peculiarities of the target organ: the CNS, and the existence of the BBB. There are a lot of studies that support this evidence, and analyzes each of the elements of this chain of events to assess the relative importance of certain cell types or certain cytokines which block the immune response as specifically as possible [46,49,50].
Researchers have described many potential antigens involved in the pathogenesis of EAE/MS. Until recently, studies were focused on two antigens: MBP and PLP, the two most abundant proteins of CNS myelin. However, despite the large number of papers found in the literature, no one has been able to clearly demonstrate its role as target antigens of the disease. This has led, in recent years, to look for other possible candidates. The most studied are the myelin protein antigens. Among them, apart from the MBP and the PLP, is the transaldolase 2'3'cyclic nucleotide 3'phosphodiesterase (CNPase), two members of the immunoglobulin superfamily oligodendroglial myelin glycoprotein (MOG) and associated glycoprotein including the myelin (MAG) and aB-cristallin hsp not myelin specific antigen [51]. Another candidate antigen is the S100-ß protein, a widely expressed in glia but not in oligodendrocytes star. Not other myelin antigens, different from protein, such as glucocerebrosides and ganglioside have also been considered as candidates in MS. Each antigen induces a clinical course, topography and composition of the different injuries and found differences in susceptibility to these antigens in different EAE models (Table 2).
Table 2. Models of induction of Experimental autoimmune encephalomyelitis (EAE).
MODELS |
STRAIN |
ADMINISTRATION |
Murine model |
SJL/J |
Homogenate in CFA |
|
|
Proteolipid protein (PLP) |
|
|
Myelin basic protein (MBP) |
|
C57BL/6 |
Myelin oligodendrocyte glycoprotein (MOG) |
Description of the antigens in different EAE murine models involving two strains of animals.
Abundant myelin antigens such as MBP or PLP is located in the thick layers of myelin; MOG antigen is located on the surface of myelin, in areas where the layers of myelin are very thin; passive transfer of S100-beta to specific cells induces lesions in the cerebral cortex, retina and uvea, plus white matter. The cellular composition of the infiltrate also varies. For example, after immunization with MBP, lesions are elicited mainly in macrophages and S100-ß-induced. T lymphocytes are antigens that induce a high inflammatory response (S100-ß) while others induce large demyelination (MOG) (Figure 5). PMB immunization in an experimental model induces an acute clinical course, the PLP 135-155 peptide un-SJL mouse relapsing course while immunization with MOG in susceptible H-2b mice induces a chronic non-remitting disease. Furthermore, S-100ss, yet a large inflammation inducing in the CNS, does not elicit a clinical EAE. Transferred to the pathogenesis of MS, these observations may help to explain the heterogeneity of the disease (Figure 6). As in EAE, this variability may reflect individual patterns in response to the same antigen, a variable response to different antigens, or both. Moreover, as discussed above, different autoantigens, by increasing the number of antigenic determinants, could be involved in various stages of disease.
MS is considered an autoimmune disease of the CNS. However, the above points, exhibited that there are still many outstanding questions about its pathogenesis. It has not shown a clear genetic predisposition; they are not set the key target antigens and it is unknown: 1) what triggers the autoimmune reaction; and 2) where starts. Furthermore, there are conflicting reports on the role of autoantibodies in demyelination and/or remyelination. On the other hand, the results obtained from different models of EAE and patients begin to reveal a large heterogeneity within the disease, we now known as MS and that may partly explain the conflicting data between different authors Multiple sclerosis (MS) is a chronic, inflammatory, and demyelinating disease of central nervous system (CNS). Under certain circumstances these cells cross the blood brain barrier and reach the parenchymal activating a cascade of events culminating in an inflammatory lesion (Figure 6). The main participants of this attack to the central nervous system are the CD4 + T cell, antigen presenting glia (microglia and astrocytes), macrophages and B cells. The BBB regulates the entry of cells and biomolecules such as antibodies, system factors, complement and cytokines into the central nervous system [49].
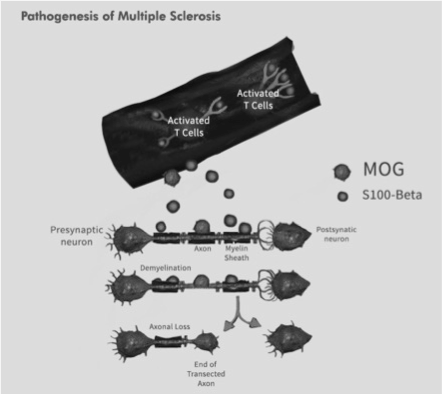
Figure 5. Antigens in Multiple Sclerosis.
Myelin Oligodendrocyte Glycoprotein (MOG) is located in the surface of myelin and passive transfer of S100-β induces damage at cortical level. Activated T-cells induce high inflammatory response leading to axonal loss in the presynaptic neuron.
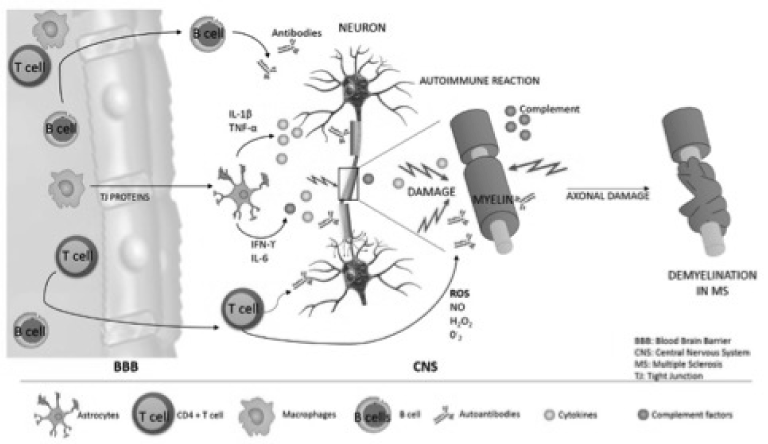
Figure 6. Multiple Sclerosis, an autoimmune disease of the Central Nervous System (CNS).
There are key target antigens that trigger an autoimmune response, this cellular components have the ability to enter the brain parenchyma and this microenvironment may lead to a neuroinflammatory state in the Central Nervous System that shed light in the pathogenesis of Multiple Sclerosis.
The anatomical basis of the selectivity of the barrier lies in the established tight junctions between endothelial cells, and low pinocytic activity in the microenvironment that surrounds; their integrity may be compromised in pathological conditions; inflammation of brain endothelium promotes migration of leukocytes into the CNS. In the brain of a healthy individual there is minimal immune surveillance; however, when the peripheral immune system is activated by specific antigenic stimulus, cellular components have the ability to enter the brain parenchyma. In vitro glial cultures have contributed to understanding the interactions established between glial and neuronal cells with the maintenance of brain homeostasis. It is likely that in the future, the study of the physiology of the glial system in central nervous system shed light on the pathogenesis of neuroinflammatory diseases and neurodegenerative and neuropsychiatric disorders, in particular multiple sclerosis that daily affects a growing number of human beings.
Finally, it is undeniable that glial cells should be considered as one of the main targets for the development of new drugs, diagnostic methods and clinical treatment of multiple sclerosis.
2021 Copyright OAT. All rights reserv
- Atkins CM, Yon M, Groome NP, Sweatt JD (1999) Regulation of myelin basic protein phosphorylation by mitogen-activated protein kinase during increased action potential firing in the hippocampus. J Neurochem 73: 1090–1097. [Crossref]
- Fields RD, Stevens-Graham B (2002) New insights into neuron-glia communication. Science 298: 556-562. [Crossref]
- Jessen KR, Mirsky R (2002) Signals that determine Schwann cell identity. J Anat 200: 367-376. [Crossref]
- Franklin RJ (2002) Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci 3: 705-714. [Crossref]
- Norton WT, Cammer W (1984) Isolation and Characterization of Myelin. In: Morell P (Ed.), Myelin. Boston: Springer US. 147–195.
- Norton WT, Autilio LA (1966) The lipid composition of purified bovine brain myelin. J Neurochem 13: 213-222. [Crossref]
- Linington C, Waehneldt TV, Neuhoff V (1980) The lipid composition of light and heavy myelin subfractions isolated from rabbit sciatic nerve. Neurosci Lett 20: 211-215. [Crossref]
- O'Brien JS, Sampson EL, Stern MB (1967) Lipid composition of myelin from the peripheral nervous system. Intradural spinal roots. J Neurochem 14: 357-365. [Crossref]
- DeVries GH, Zetusky WJ, Zmachinski C, Calabrese VP (1981) Lipid composition of axolemma-enriched fractions from human brains. J Lipid Res 22: 208-216. [Crossref]
- Horrocks LA (1967) Composition of myelin from peripheral and central nervous systems of the squirrel monkey. J Lipid Res 8: 569-576. [Crossref]
- Spritz N, Singh H, Geyer B (1973) Myelin from human peripheral nerves. Quantitative and qualitative studies in two age groups. J Clin Invest 52: 520-523. [Crossref]
- Monuki E, Lemke G (1995) Molecular Biology of Myelination. In: Waxman SG, Kocsis JD (Ed.), The Axon: Structure, Function and Pathophysiology. New York: Oxford University Press. 92–124.
- Bjartmar C, Yin X, Trapp BD (1999) Axonal pathology in myelin disorders. J Neurocytol 28: 383-395.
- Myrna A. R. Dent (2003) Daño y reparación del sistema nervioso. Cienc Ergo Sum 10:68–79.
- Scherer SS (1999) Nodes, paranodes, and incisures: from form to function. Ann N Y Acad Sci 883: 131-142. [Crossref]
- Trapp BD, Kidd GJ (2000) Axo-glial septate junctions. The maestro of nodal formation and myelination? J Cell Biol 150: F97-97F100. [Crossref]
- Chittajallu R, Chen Y, Wang H, Yuan X, Ghiani CA, et al. (2002) Regulation of Kv1 subunit expression in oligodendrocyte progenitor cells and their role in G1/S phase progression of the cell cycle. Proc Natl Acad Sci U S A 99: 2350-2355. [Crossref]
- Scherer SS, Arroyo EJ (2002) Recent progress on the molecular organization of myelinated axons. J Peripher Nerv Syst 7: 1-12. [Crossref]
- Poliak S, Peles E (2003) The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci 4: 968-980. [Crossref]
- Peles E, Nativ M, Lustig M, Grumet M, Schilling J, et al. (1997) Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J 16: 978–88. [Crossref]
- Einheber S, Zanazzi G, Ching W, Scherer S, Milner TA, et al. (1997) The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J Cell Biol 139:1495–506. [Crossref]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, et al. (2000) An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol 150: 657-666. [Crossref]
- Denisenko-Nehrbass N, Oguievetskaia K, Goutebroze L, Galvez T, Yamakawa H, et al. (2003) Protein 4.1B associates with both Caspr/paranodin and Caspr2 at paranodes and juxtaparanodes of myelinated fibres. Eur J Neurosci 17: 411-416. [Crossref]
- Scherer SS, Salzer JL (1997) Axon-Schwann cell interactions during peripheral nerve degeneration and regeneration. In: Jessen KR, Richardson WD (Eds.), Glial Cell Development: basic principles and clinical relevance. Oxford: Bios Scientific.
- Fernandez-Valle C, Bunge RP, Bunge MB (1995) Schwann cells degrade myelin and proliferate in the absence of macrophages: evidence from in vitro studies of Wallerian degeneration. J Neurocytol 24: 667–79. [Crossref]
- Fawcett JW, Keynes RJ (1990) Peripheral nerve regeneration. Annu Rev Neurosci 13: 43-60. [Crossref]
- Dugandzija-Novakovic S, Koszowski AG, Levinson SR, Shrager P (1995) Clustering of Na+ channels and node of Ranvier formation in remyelinating axons. J Neurosci 15: 492-503. [Crossref]
- Garratt AN, Voiculescu O, Topilko P, Charnay P, Birchmeier C (2000) A dual role of erbB2 in myelination and in expansion of the schwann cell precursor pool. J Cell Biol 148: 1035-1046. [Crossref]
- Jessen KR, Mirsky R (2004) The Schwann Cell Lineage. In: Kettenmann H, Ranson B (Eds.), Neuroglia, (2ndedn), New York: Oxford University Press., 85–100.
- Allodi I, Udina E, Navarro X (2012) Specificity of peripheral nerve regeneration: interactions at the axon level. Prog Neurobiol 98: 16-37. [Crossref]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, et al. (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485: 517-521. [Crossref]
- Lewis GM, Kucenas S2 (2014) Perineurial glia are essential for motor axon regrowth following nerve injury. J Neurosci 34: 12762-12777. [Crossref]
- Meier C, Parmantier E, Brennan A, Mirsky R, Jessen KR (1999) Developing Schwann cells acquire the ability to survive without axons by establishing an autocrine circuit involving insulin-like growth factor, neurotrophin-3, and platelet-derived growth factor-BB. J Neurosci 19: 3847–59. [Crossref]
- Lobsiger CS, Schweitzer B, Taylor V, Suter U (2000) Platelet-derived growth factor-BB supports the survival of cultured rat Schwann cell precursors in synergy with neurotrophin-3. Glia 30: 290-300. [Crossref]
- Heinen A, Lehmann HC, Küry P (2013) Negative regulators of schwann cell differentiation-novel targets for peripheral nerve therapies? J Clin Immunol 33: S18-26. [Crossref]
- Woszczycka-Korczyńska I, Olakowska E, Marcol W, Lewin-Kowalik J, Jędrzejowska-Szypułka H (2013) [Schwann cells in therapy of spinal cord injuries]. Postepy Hig Med Dosw (Online) 67: 680-689. [Crossref]
- Hirano A, Llena J (1995) Morphology of Central Nervous System Axons. In: Waxman SG, Kocsis JD (Eds.), The Axon: Structure, Function and Pathophysiology. New York: Oxford University Press., 180–189.
- Barnett MH, Prineas JW (2004) Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 55: 458-468. [Crossref]
- Bando Y, Nomura T, Bochimoto H, Murakami K, Tanaka T, et al. (2015) Abnormal morphology of myelin and axon pathology in murine models of multiple sclerosis. Neurochem Int 81: 16-27. [Crossref]
- Recks MS, Stormanns ER, Bader J, Arnhold S, Addicks K, et al. (2013) Early axonal damage and progressive myelin pathology define the kinetics of CNS histopathology in a mouse model of multiple sclerosis. Clin Immunol 149:32–45. [Crossref]
- Pohl HB, Porcheri C, Mueggler T, Bachmann LC, Martino G, et al. (2011) Genetically induced adult oligodendrocyte cell death is associated with poor myelin clearance, reduced remyelination, and axonal damage. J Neurosci 31: 1069-1080. [Crossref]
- Aschner M (1998) Astrocytes as mediators of immune and inflammatory responses in the CNS. Neurotoxicology 19: 269-281. [Crossref]
- Claycomb KI, Johnson KM, Winokur PN, Sacino AV, Crocker SJ (2013) Astrocyte regulation of CNS inflammation and remyelination. Brain Sci 3: 1109-1127. [Crossref]
- Nave KA, Werner HB (2014) Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol 30: 503-533. [Crossref]
- Gehrmann J, Matsumoto Y, Kreutzberg GW (1995) Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 20: 269-287. [Crossref]
- Ortiz GG, Pacheco-Moisés FP, Bitzer-Quintero OK, Ramírez-Anguiano AC, Flores-Alvarado LJ, et al. (2013) Immunology and oxidative stress in multiple sclerosis: clinical and basic approach. Clin Dev Immunol 2013: 708659. [Crossref]
- Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM (2008) Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med 14: 337-342. [Crossref]
- Racke MK (2009) Immunopathogenesis of multiple sclerosis. Ann Indian Acad Neurol 12: 215-220. [Crossref]
- Ortiz GG, Pacheco-Moisés FP, Macías-Islas MÁ, Flores-Alvarado LJ, Mireles-Ramírez MA, et al. (2014) Role of the blood-brain barrier in multiple sclerosis. Arch Med Res 45: 687-697. [Crossref]
- di Penta A, Moreno B, Reix S, Fernandez-Diez B, Villanueva M, et al. (2013) Oxidative Stress and Proinflammatory Cytokines Contribute to Demyelination and Axonal Damage in a Cerebellar Culture Model of Neuroinflammation. PLoS One 8: e54722. [Crossref]
- Jana M, Pahan K (2013) Down-regulation of Myelin Gene Expression in Human Oligodendrocytes by Nitric Oxide: Implications for Demyelination in Multiple Sclerosis. J Clin Cell Immunol 4. [Crossref]