Malignant hyperthermia (MH) [1] is a pharmacogenetic disorder of skeletal muscle that presents as an acute hypermetabolic response precipitated by exposure to potent volatile anesthetic gases including isoflurane, sevoflurane, desflurane, and halothane as well as to the depolarizing muscle relaxant succinylcholine. Early recognition and prompt diagnosis of MH in anesthesia patients are critical to prevent the life threatening consequences associated with the metabolic reactions associated with MH. Susceptibility to MH (MHS) is inherited as an autosomal dominant trait that is caused by mutations in the ryanodine receptor 1 (RYR 1) gene [1,2]. We present a case of an unexpected episode of MH following eight hours of sevoflurane anesthesia in an African American dialysis patient with a novel RYR1 gene mutation and no prior complications despite multiple previous anesthetics.
A 46-year-old African American female underwent resection of a recurrent ameloblastoma of the mandible, reconstruction with acrylic graft, cadaveric nerve reconstruction, and teeth extraction. Medical history included end-stage renal disease on hemodialysis, hypertension, gout, and hyperlipidemia. Medications included losartan and verapamil. The patient had three previous surgeries to resect the tumor, the last one in 1999. In addition to these surgeries, she had general anesthesia for hysterectomy and multiple vascular access procedures. She did not have any medication allergies. She underwent hemodialysis the day prior to surgery. Preoperative potassium on the day of surgery was 4.8 mEq/L.
Physical exam revealed a well-nourished female (75 kg, 165 cm) with vital signs: blood pressure 126/81 mm Hg, heart rate 70 bpm and temperature 36.8°C. Standard ASA monitors were attached in the operating room. Anesthetic induction consisted of intravenous (IV) lidocaine 100 mg, propofol 150 mg, fentanyl 250 mcg followed by cisatracurium 12 mg. Anesthesia was maintained with 2% sevoflurane in 50% oxygen/air mixture with intermittent fentanyl and cisatracurium. Initially, axillary skin temperature was used instead of esophageal because of the nature of the procedure.
Surgery was uneventful for hours with vital signs within acceptable ranges based on initial measurements. At approximately eight hours post- surgery start time, the ETCO2 was noted to have increased from 37 to 45 mm Hg despite unchanged ventilator settings and continued paralysis. The minute ventilation was increased, but over the next hour the ETCO2 continued to increase to 57 mm Hg, the heart rate to 107 bpm, and temperature to 39.4°C. After noticing increasing premature ventricular contractions, a potassium level was checked and found to be elevated at 6.3 mEq/L. Treatment for hyperkalemia was started concomitantly with a widening QRS including IV calcium chloride, sodium bicarbonate, insulin, and glucose. The QRS reverted to normal. However, approximately 20 minutes later, the patient became hemodynamically unstable with again widened QRS complexes noted on ECG. At this point the ETCO2 was 75 mm Hg and temperature 40.5°C. Treatment for MH was initiated consisting of stopping sevoflurane, removal from the ventilator, hyperventilating with Ambu bag on 100% oxygen, propofol infusion, and insertion of a radial arterial line. The temperature probe was moved to the esophagus and temperature confirmed. Dantrolene sodium was given as 2.5 mg/kg IV bolus followed by 20 mg boluses over the next hour totalling 280 mg. Twenty minutes after initiating dantrolene, the patient developed pulseless electrical activity (PEA). An arterial blood gas taken shortly before this showed pH 7.12, PaCO2 62 mm Hg, HCO3-1 15 mMol/L, base deficit 9.5 mMol/L, lactic acid 9.9 mMol/L and potassium 6.3 mEq/L. (Table 1) Cardiopulmonary resuscitation was started, 1 mg IV epinephrine and 1 mg IV atropine were given. Chest compressions were stopped approximately three minutes later following return of a palpable carotid pulse. After the patient was stabilized and dantrolene continued, the ETCO2 decreased from a maximum of 75 to around 30 mm Hg, temperature decreased to 37.7°C and the heart rate decreased from 115 to 90 bpm. In addition, the metabolic acidosis corrected. The graphic anesthetic record of vital signs is seen in Figure 1. The patient was packed with ice bags and transferred to the surgical intensive care unit (ICU) where dantrolene therapy was continued (1 mg/kg every 6hours for 2 days), hyperkalemia treated, and continuous renal replacement therapy initiated. Laboratory studies revealed creatinine phosphokinase (CPK) level of 2315 Units/L and positive urine myoglobin. The patient did well and was extubated on postoperative day two and was transferred to the inpatient ward on day three.
Table 1. Blood gas and electrolyte results
Tests |
|
|
Times/Sample Source |
|
|
|
|
11:35
Venous |
15:31 Venous |
17:01
Venous |
17:35 Venous |
17:45 Arterial |
17.59
Arterial |
K+mEq/L |
4.7 |
5.7 |
6.3 |
6.3 |
7.1 |
6.4 |
pH |
7.46 |
|
|
|
7.12 |
7.4 |
PaCO2 mmHg |
39 |
|
|
|
62 |
31 |
PaO2 mmHg |
49 |
|
|
|
276 |
353 |
HCO3 |
|
25 |
|
|
17.9 |
18 |
Base Excess |
3.4 |
0.3 |
|
|
-9.5 |
-4.3 |
Ca++ |
0.97 |
1.96 |
|
|
0.99 |
0.88 |
HCT |
33% |
29% |
|
|
25% |
25.50% |
Glu |
118 |
119 |
|
|
258 |
254 |
Mg |
|
+0.42 |
|
|
|
|
Na+ |
|
|
|
|
128 |
132 |
Lactic Acid |
|
3.7 |
|
|
|
|
|
|
|
|
|
|
|
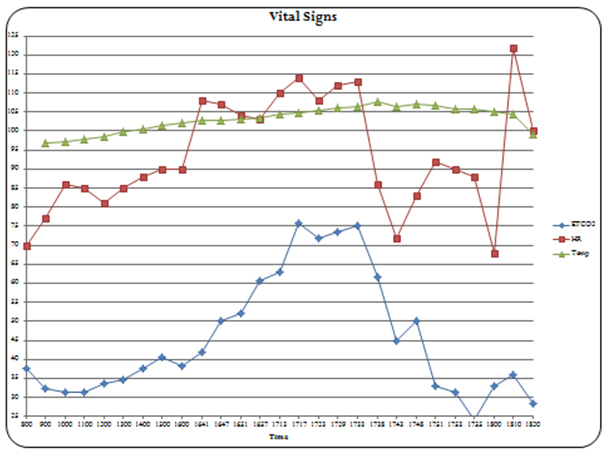
Figure 1. Intraoperative vital signs graphic record
During the initial postoperative discussion with the family, a brother of the patient was reported to have spent multiple days in ICU following one of his surgeries as well as having been “packed with ice” and having had extreme “soreness” for several days after the operation. The husband and son mentioned that the patient frequently complained of muscle spasms and soreness.
Two months later, the patient underwent arch-bar removal with a non-triggering anesthetic and did well. She was discharged the day of surgery. Several months later she underwent another mandibular surgery at which time, following obtained consent, blood samples and a muscle biopsy were taken for genetic analysis. Two years later the hardware had broken and she returned for placement of a titanium mandibular implant. During the intervening time, the patient was noted to have had frequent masseter spasms that were believed to have resulted in the breakage of the prior hardware placement. She was on “slow” hemodialysis at home because routine dialysis caused her face to “draw up” per her family. She continued to have frequent leg muscle spasms as well. Baseline CPK was 1653 units/L and on the first postoperative day was 2851 units/L. (normal value is less than 190 units/L). The patient underwent a non-triggering anesthetic, was stable throughout the entire perioperative period and discharged on the third postoperative day. In the holding area preoperatively, her husband related that approximately 30 years previously, when she had been participating in a high school track meet, she had “passed out” and was hospitalized for several days with “scars on her kidneys.” They were unaware of any high fever at that time.
Informed consent was obtained and the patient was enrolled in an IRB approved research study entitled “Genetic tests for Malignant Hyperthermia" at USUHS. Analysis of entire RYR1 gene was done as described previously [3]. Molecular genetic analysis of this patient revealed a novel nucleotide variant c.506T>A changing Isoleucine (Ile) to Asparagine (Asn) at 169 residue of RYR1 (Figure 2). This variant was located in within the mutational hot spot of RYR1 [1-3]. The Ile169Asn mutation was absent in publicly available genetic variant database of greater than 10,000 individuals of diverse ethnicity [4]. Mutation impact on protein function was predicted to be damaging by both PolyPhen (genetics.bwh.harvard.edu/pph2/) and SIFT (sift.jcvi.org/www/SIFT) analyses [5].
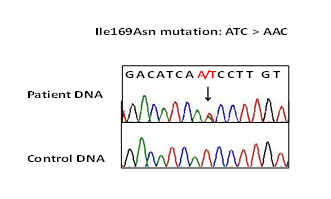
Figure 2. Ryanodine Receptor type 1 gene analysis of the patient. Mutation T to A changes Isoleucine, Ile: ATC to Asparagine, Asn: AAC at 169 residue of RYR1.
This report presents an unusual MH case in an African American dialysis patient. Several aspects of this case are worth noting: 1. The patient had an estimated ten prior exposures to general anesthesia without signs of MH. 2. The onset was delayed eight hours, the longest delay of onset yet reported with sevoflurane exposure. Previously 210 minutes was reported as the longest onset for sevoflurane inducing a MH episode [3,6].
Genetic analysis identified a novel mutation Ile169Asn in the RYR1 gene that encodes the skeletal muscle-specific intracellular calcium release channel. Autosomal dominant mutations in the RYR1 are known to be associated with MHS and central core disease [1-3,7]. MHS-associated mutations in the RYR1 result in excessive release of calcium from the sarcoplasmic reticulum causing increased free myoplasmic calcium when triggered by halogenated inhalation agents or succinylcholine. The increased myoplasmic calcium leads to sustained muscle contractions and activated glycogenolysis and cell metabolism with resultant heat production and excess lactate as depicted in Figure 3 [8]. Oxygen consumption is markedly increased as is carbon dioxide production.
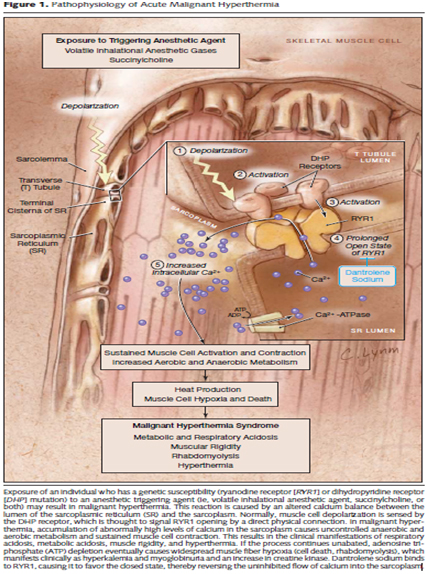
Figure 3. From Litman and Rosenberg JAMA 2005; 293: 2920 8 Used with permission of publisher, pending)
The absence of functional data and segregation analysis of extended family limits clinical significance of the Ile169Asn mutation. Ideally, finding a second MH family with the same mutation would be important to strengthen association of this mutation with MHS. However, this is not always possible as it is well known that many MH-associated RYR1 mutations are private and specific to a single family. This rarity of RYR1 mutation is further complicated by the fact that our case involves a patient of African American descent, a population that is not routinely screened for RYR1 gene in association with MHS. We do believe that the Ile169Asn mutation is of MH in the presented case. The mutation occurred within the RYR1 gene mutational hot spot [9] where the majority of genetic changes associated with MH are located. Second, this mutation is absent in the publicly available database that is comprised of more than 10,000 control individuals including more than 2000 African Americans. Thirdly, the mutation changes nonpolar Isoleucine into polar Asparagine. This type of change will likely have functional impact on protein structure of the RYR1.
Successful management of MH relies on early diagnosis and aggressive treatment as per the Malignant Hyperthermia Association of United States (MHAUS) guidelines available at http://www.mhaus.org. A clinical grading scale for MHS has been reported by Larach et al. [10] as seen in Table 2. After the MH episode while explaining the intraoperative events to the patient’s husband and son, the MHAUS website was used and given to them for future reference. Prior to the latest surgery, the husband was again instructed to encourage their sons to undergo genetic testing. Early diagnosis of MHS family members are critical as safe alternative anesthetic techniques are available. Meanwhile, should they require surgery, they should report family history of MHS to their anesthesia teams.
Table 2. Clinical indicators of malignant hyperthermia susceptibility [10]
Clinical Indicators |
Possible Points |
This Patient |
Masseter spasm shortly following succinylcholine |
15 |
0 |
Serum K+> 6.0 mEq.L-1 |
3 |
3 |
ETCO2 > 55 mmHg even after adequate minute ventilation |
15 |
15 |
Inappropriate rapid increase in temperature or |
15 |
0 |
Inappropriate increase in temperature > 38.8°C (101.8°F) |
10 |
10 |
Inappropriate unexplained arrhythmia |
3 |
3 (PEA) |
Arterial pH < 7.25 |
10 |
10 |
Rapid reversal with dantrolene |
5 |
5 |
Positive family history or own history of MH |
10 |
10 |
2021 Copyright OAT. All rights reserv
Resting elevated serum creatine kinase |
10 |
10 |
Total |
96 |
66 |
As described by Larach et al. [10] a score range of no points would be almost never MH, 3-9 points unlikely, 10-19 somewhat less than likely, 20=34 somewhat greater than likely, 35-49 very likely and 50+ almost certain. The score of 66 for this patient was calculated using the clinical data from the MH episode plus when she returned in 2015, it was known that she was MHS.
An additional unusual aspect of this report is the successful rescue from PEA. A recent series reported from MHAUS, [11] found no survivors with cardiac arrest during their episode of MH, despite being previously healthy and undergoing low to moderate risk surgery.
As more mutations are identified and reported as being associated with MHS, genetic testing may replace the gold standard caffeine halothane contracture test (CHCT) as the method of choice in confirming MHS due to the major limitations associated with CHCT, namely invasiveness and unavailability to patients that are unable to travel to testing sites.
As described by Larach et al. [10] a score range of no points would be almost never MH, 3-9 points unlikely,
10-19 somewhat less than likely, 20=34 somewhat greater than likely, 35-49 very likely and 50+ almost certain. The score of 66 for this patient was calculated using the clinical data from the MH episode plus when she returned in 2015, it was known that she was MHS.
An additional unusual aspect of this report is the successful rescue from PEA. A recent series reported from MHAUS, [11] found no survivors with cardiac arrest during their episode of MH, despite being previously healthy and undergoing low to moderate risk surgery.
As more mutations are identified and reported as being associated with MHS, genetic testing may replace the gold standard caffeine halothane contracture test (CHCT) as the method of choice in confirming MHS due to the major limitations associated with CHCT, namely invasiveness and unavailability to patients that are unable to travel to testing sites.
- Rosenberg H, Sambuughin N, Riazi S, Dirksen R (2013) Malignant Hyperthermia Susceptibility. Gene Reviews 2013; Pagon RA, Adam MP, Ardinger HH, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2014.
- Brandom BW, Bina S, Wong CA, Wallace T, Visoiu M, et al. (2013) Ryanodine receptor type 1 gene variants in the malignant hyperthermia-susceptible population of the United States. Anesth Analg 116: 1078-1086. [Crossref]
- Sambuughin N, Holley H, Muldoon S, Brandom BW, de Bantel AM, et al. (2005) Screening of the entire ryanodine receptor type 1 coding region for sequence variants associated with malignant hyperthermia susceptibility in the north american population. Anesthesiology 102: 515-521. [Crossref]
- Exome Variant Server. NHLBI Exome Sequencing Project. Seattle, WA, URL: http://evs.gs.washington.edu/EVS/ . Accessed Feb 12, 2014.
- Sambuughin N, Liu X, Bijarnia S, Wallace T, Verma IC, et al. (2013) Exome sequencing reveals SCO2 mutations in a family presented with fatal infantile hyperthermia. J Hum Genet 58: 226-228. [Crossref]
- Hopkins PM (2011) Malignant hyperthermia: pharmacology of triggering. Br J Anaesth 107: 48-56. [Crossref]
- Girard T, Treves S, Voronkov E, Siegemund M, Urwyler A (2004) Molecular genetic testing for malignant hyperthermia susceptibility. Anesthesiology 100: 1076-1080. [Crossref]
- Litman RS, Rosenberg H (2005) Malignant hyperthermia: update on susceptibility testing. JAMA 293: 2918-2924. [Crossref]
- Sei Y, Sambuughin N, Muldoon S (2004) Malignant hyperthermia genetic testing in North America Working Group Meeting. Bethesda, Maryland. September 4-5, 2002. Anesthesiology 100: 464-465. [Crossref]
- Larach MG, Localio AR, Allen GC, Denborough MA, Ellis FR, et al. (1994) A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology 80: 771-779. [Crossref]
- Larach MG, Brandom BW, Allen GC, Gronert GA (2007) Failure to rescue from fulminant malignant hyuperthermia: Deaths and their genetic variants. A 2052 ASA Annual Meeting 2013.