Although each cancer type is individually distinct, most cancers initially occur due to genomic mutations of oncogenes and tumor suppressor genes, leading to enhancement or disruption of specific cellular processes, including mitochondrial-mediated events. As an organelle necessary for both cell survival and cell death, the mitochondrion is involved in a variety of diseases, including cancer. Specific alterations to mitochondrial DNA in cancer can result in increased proliferation and avoidance of cell death pathways. Since cancer cells utilize mitochondria to enhance disease progression, specific targeting of mitochondrial-regulated processes and pathways may present advantageous anticancer treatments.
Mitochondria, cancer development, mitochondrial permeability transition pore
Although overall cancer death rates have declined by 20% since climaxing in 1991 [1], this group of diseases is still of major importance regarding treatment and prevention. As cancer cells are continuously replicating, treatment regimens often fail to completely rid a patient of malignant cells, resulting in repopulation of the tumor [2]. Thus, ongoing research is necessary for continued observance and management of this complicated group of diseases.
Cancer is composed of a vast group of diseases characterized by unchecked cellular proliferation and metastasis of abnormal cells throughout the body. The development of cancer is initiated by three known factors. Firstly, genomic mutations constitutively activate oncogenes to promote uncontrolled cell growth [3]. Additionally, genetic modifications deactivate tumor suppressor genes so that they fail to inhibit the robust cell growth [4]. Last, stability genes necessary for genomic repair are inactivated, allowing for higher rates of mutations in the cellular genome [5]. All three genetic mutation types allow normal cells to progress to cancer. Once cells become cancerous they exhibit large metabolic imbalances [6] and increased resistance to cell death [7], two processes regulated by mitochondria.
These organelles are comprised of an Outer Membrane (OM), An Inner Membrane (IM), an intermembrane space, a cristae interior, and a matrix [8]. Mitochondria perform key roles in cellular function, including cell survival, energy generation, stabilization of Reactive Oxygen Species (ROS), and cell death pathway regulation. As mitochondria are largely involved in cell health, it is anticipated that they are involved in cancer development and progression [9-11].
Here, mitochondrial processes needed for cancer cell survival are discussed. Cell death pathways are examined in relation to enhanced tumor progression. Finally, presently available and potential treatment strategies are reviewed.
Otto Warburg was one of the first investigators to implicate mitochondria in cancer. His phenomenon, termed the Warburg effect, demonstrates that tumor cells exhibit increased glycolytic Adenosine Triphosphate (ATP) production and reduced Oxidative Phosphorylation (OXPHOS) [12]. Since then, every aspect of the mitochondrion has undergone thorough investigation to shed light on the alterations promoting this aggressive disease.
Mutations in oncogenes and tumor suppressor genes are leading factors promoting tumorigenesis and metastasis. These mutated genes encode transcription factors, which in turn control the gene expression patterns and signaling pathways that lead to cancer development [13]. Several transcription factor pathways are involved in tumorigenesis.
Nuclear Factor-κB (NF-κB) is involved in the regulation of inflammatory response genes, cell cycle regulatory genes, and anti-apoptotic genes. In a variety of human cancer cells, NF-κB activation inhibits apoptosis, leading to enhanced tumor resistance to chemotherapeutic agents [14]. Numerous studies have demonstrated that suppression of NF-κB leads to apoptosis induction and tumor regression [15-18]. Next, Activator Protein 1 (AP-1) regulates many cellular processes, including proliferation, differentiation, survival, and death. Overexpression of the AP-1 family has been demonstrated to induce tumor formation [19]. Similar to NF-κB, inhibition of AP-1 decreased cell viability [20] and reduced tumor volume [21] in numerous studies, suggestive of the potential of utilizing AP-1 in anticancer treatments. An additional transcription factor family involved in cancer is Signal Transducer and Activator of Transcription (STAT), which has roles in cell differentiation, development, proliferation, inflammation, and apoptosis. In many human cancer cell lines, STAT proteins often become constitutively activated, which promotes oncogenic transformation by regulating cell proliferation [22] and cell death pathways [23]. Blockade of STAT proteins, especially STAT3, resulted in decreased proliferation and increased cell death in several studies [24,25]. In breast and prostate cancer, steroid hormone receptors play a key role in mediating the tumorigenic effects of testosterone, androgen, progesterone, and estradiol [26]. Finally, manipulation of steroid receptors, including inhibition of androgen receptors in prostate cancer cells [27] and silencing of estrogen-regulated genes in breast cancer cells [28], have provided novel strategies for the treatment of hormone-specific tumors.
These transcription factors, as well as others, propel cancer development through a variety of pathways, such as altering the cell cycle to enhance proliferation rate, blocking apoptosis to promote malignant cell survival, and changing gene expression patterns which affect cellular and mitochondrial processes.
Mitochondria contain a DNA molecule (mtDNA) that encodes genes essential for normal mitochondrial function. Human mtDNA contains 37 genes that code for 13 polypeptides involved in respiration and OXPHOS, and two ribosomal RNAs in addition to 22 transfer RNAs important for protein synthesis [29]. Furthermore, mtDNA has a noncoding region with a displacement loop for control of mtDNA replication and transcription [30]. As mtDNA regulates vital processes, modifications to its genomic material can profoundly impact healthy cells.
Numerous studies have reported the effects of mtDNA mutations in tumors. To begin, mtDNA copy number has been examined in diverse cancers, indicating that changes in mtDNA content may be regulated in a tumor-specific manner [31,32]. Moreover, studies have shown that mutations in both the coded [33,34] and non-coded [35-37] mtDNA regions are associated with cancer growth. Interestingly, these alterations strongly correlate with patient outcome [38], as displacement loop mutations typically correlate with lower survival rates outcome [39]. Thus, the severity of mtDNA alterations in cancer cells and patient survival seem to depend on both mtDNA content deviations and mutation locations.
In healthy cells, ATP manufacturing is dependent upon oxygen uptake, the Tricarboxylic Acid (TCA) cycle, and the Electron Transport Chain (ETC). Dehydrogenases of the TCA cycle are stimulated by mitochondrial calcium (Ca2+) intake (Figure 1.1), which propels the reduction of Nicotinamide Adenine Dinucleotide (oxidized NAD+ to reduced NADH) [40]. In addition, succinate dehydrogenase, also known as complex II (succinate-ubiquinone oxidoreductase) of the ETC, catalyzes the oxidation of succinate to fumarate in the TCA cycle [41]. ETC complex I (NADH-ubiquinone oxidoreductase) receives electrons from NADH [42], while complex II gains electrons from succinate. Next, the accepted electrons are transfered to complex III (ubiquinol-cytochrome c oxidoreductase) and incorporated into Cytochrome c (cyto c) for delivery to complex IV (cytochrome c oxidase) [42], which are then used in the reduction of oxygen to water [43]. Protons are generated during the ETC process and used by ATP synthase (often referred to as complex V) in the OXPHOS pathway to generated ATP from Adenosine Diphosphate (ADP) and inorganic phosphate [44]. Once produced, ATP propels many activities in the cell, including signal transduction, active transport, and DNA synthesis. Consequently, changes in energy production due to mtDNA mutations can affect these processes.
Warburg initially discovered that tumor cells display increased glucose uptake, enhanced glycolytic ATP generation, and diminished OXPHOS [6]. Essentially, instead of utilizing pyruvate molecules in the TCA cycle to power the OXPHOS pathway for generation of ATP as is done by normal cells (Figure 1.2-1.4), cancer cells convert pyruvate into lactic acid for energy generation (Figure 2.1). A possible explanation for Warburg’s findings involves ATP synthase malfunctioning as hyperglycemia in hepatocarcinoma cells reduced ATP synthase dimer stability in a new study [45]. Also, Isidoro et al. found that expression of the β-catalytic subunit of ATP synthase is decreased in cancer cells [46], further implicating the involvement of complex V.
Although the Warburg effect is widely recognized, several groups have challenged it, revealing that mitochondria in tumors are able to operate OXPHOS at lower capacities along with glycolysis [47]. In fact, malignant cells can switch from a glycolytic state to OXPHOS under glucose-limiting conditions to adjust to changes in the cellular environment [48]. Utilizing both respiration systems under diverse settings is important for tumor survival. For instance, glucose deprivation elevates OXPHOS in breast tumor cells while control cells remain unaffected [49]. Conversely, hypoxia improves respiration in control cells whereas it is impaired in breast cancer cells [50], signaling for tumor cells to switch to glycolysis. Therefore, cancer cells can direct the energy metabolism systems according to their specific needs under a variety of conditions; this likely aids in cancer cell growth and resistance to cell death.
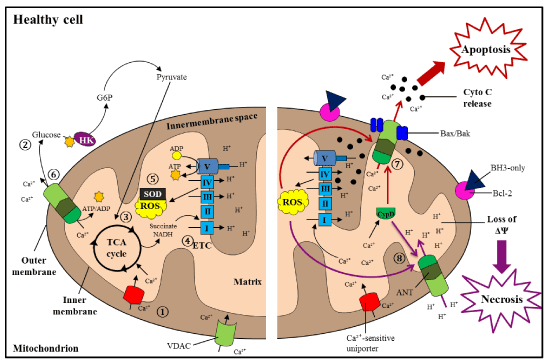
Figure 1. Mitochondrial processes and pathways in healthy cells
During regular conditions, healthy cells chiefly use mitochondrial OXPHOS for generation of ATP. 1) Ca2+is readily taken up into mitochondria through VDAC and Ca2+-sensitive uniporter channels on the OM and IM, respectively; these ions go on to stimulate ATP production. 2) During glycolysis, glucose molecules are converted into pyruvate molecules, 3) which are taken up into mitochondria for use in the TCA cycle. 4) Reduced forms of NADH and succinate are used by the ETC to drive ATP assembly. 5) Anti-oxidants (superoxide dismutase, SOD) maintain the levels of ROS generated during the ETC process. 6) The MPTP, comprised of VDAC, ANT, and CypD, expels surplus Ca2+ions from mitochondria. 7) During cell injury, apoptotic cell death (red pathway) is instigated as a result of uncontrolled Ca2+and/or ROS levels. Anti-apoptotic Bcl-2 is repressed by BH3-only proteins, which allows pro-apoptotic Bax and Bak to interact. This induces opening of the MPTP for release of cyto c and Ca2+into the cytosol, propelling the organized collapse of the cell. 8) Necrosis (purple pathway) occurs due to excessive Ca2+ and/or ROS accumulation and reduction of ATP stores, leading to loss of ΔΨ and the unplanned rupture of the cell [123].
Under homeostatic conditions, enhanced ATP production via enhanced ETC activity propels the reduction of oxygen to water. Consequently, this promotes leakage of free electrons from the ETC complexes, leading to the creation of superoxides, free radicals, and peroxides jointly known as ROS [51]. To compensate, ROS scavenging enzymes are also activated to neutralize oxidants [52]. Healthy cells tightly regulate the balance between oxidants and anti-oxidants to prevent destructive consequences (Figure 1.5). Nevertheless, ETC complex activity deficits are linked with reduced energy and heightened ROS generation [53]. Chiefly, reduced complex I, II, and IV respiratory capacities lead to increased risk of dysfunction [54]. Due to insufficient ROS scavenger levels, damaging oxidants accumulate within the cell and cause damage, such as mtDNA mutations [55]. Thus, unrestrained oxidative stress can propel cancer initiation and metastasis [56].
Elevated oxidative stress has been observed in many different tumors, with persistently high ROS levels seen in malignant cells (Figure 2.2) compared with paired controls [57]. Accordingly, cells are able to use mitochondrial ROS as a mechanism to increase their chance of cancer development through a kind of pro-cancer feedback loop. Also, extra-mitochondrial ROS production can affect cancer behavior, correlating with enhanced tumor growth and invasiveness [58,59]. Thus, cancer cells can utilize enhanced intra- and extra-mitochondrial ROS generation to increase tumor developmental and metastatic abilities.
However, elevated ROS levels can cause substantial damage to normal and cancerous cells, alike. As a result, tumor cells are capable of rebalancing ROS production and elimination by activating antioxidants (Figure 2.3) for restoration of an optimal redox state necessary for continuous proliferation [60]. For example, a study found that increasing ROS levels caused inhibition of glycolytic enzyme pyruvate kinase M2, which in turn activated the antioxidant systems required for detoxifying ROS in cancer cells [61]. Therefore, tumor cells regulate ROS levels for initial development and long-term management of cancer progression.
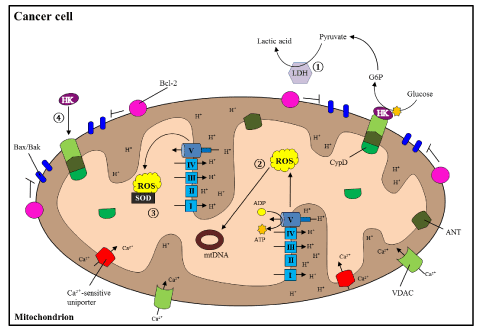
Figure 2. Altered mitochondrial processes during cancer
In cancer cells, there is a shift from OXPHOS to glycolytic ATP production, 1) producing large amounts of lactic acid via Lactate Dehydrogenase (LDH). 2) High levels of ROS induce carcinogenic mutations in mtDNA, but are 3) balanced by activated antioxidants (such as superoxide dismutase, SOD). 4) HK inhibits opening of the MPTP, and as a result blocks cell death induction. Thus, the combination of increased carcinogenicity and decreased cell death promote cancer cell growth [123].
A nonselective mitochondrial Ca2+-activated pore was initially discovered to take in and extrude ions during membrane permeability oscillations [62]. Currently called the mitochondrial permeability transition pore (MPTP, Figure 1.6), this pore is often connected with cell death provoked by stress and Ca2+ overload [63]. Under stress conditions in healthy cells, the MPTP forms in the IM where its induction can lead to mitochondrial swelling, loss of transmembrane potential (ΔΨ), apoptotic mediator release, and eventual cell death [64]. However, MPTP-mediated cell death is suppressed in tumor cells, rendering them resistant to therapies [65]. Many factors participate in protecting cancer cells from MPTP-regulated membrane disruption and cell death induction, a few of which are presently discussed.
Cyclophilin D (CypD) is a prolyl isomerase located within the mitochondrial matrix and has been established as a modulatory component of the MPTP [66-68]. During oxidative stress or Ca2+ overload in healthy cells, CypD translocation to the IM activates the MPTP, inducing cell death [69]. Interestingly, cancer cells overexpress CypD, leading to the suppression of cell death by interacting with anti-apoptotic Bcl-2 to inhibit the release of cyto c [70]. CypD-mediated inhibition of cell death also correlates with mitochondrial-bound hexokinases (HK, Figure 2.4). Inactivation of CypD results in the release of bound HK-II and enhances pro-apoptotic Bax/Bak-mediated apoptosis [71]. In addition, heat shock protein interactions with CypD inhibit normal CypD-dependent MPTP opening and cell death in some tumors [72]. Thus, CypD expression seems necessary for cancer growth, and is regulated by many molecular interactions.
The Translocator Protein (TSPO; initially known as the peripheral benzodiazepine receptor) is another recognized component, first linked to the MPTP due to its ligand interactions [73]. Moreover, the OM has been shown to have a regulatory role in MPTP formation, primarily through TSPO [74]. Similar to CypD, TSPO is elevated in many types of cancer, especially breast cancer [75]. Increased levels of TSPO are associated with enhanced invasiveness of breast cancer [76,77], and correlate with shortened disease-free survival in lymph node-negative patients [78]. It has been postulated that high TSPO levels may render malignant cells more resistant to MPTP formation and cell death, comparable to CypD. This seems plausible, though additionally research is needed to determine the function of elevated TSPO levels in cancer.
Two other mitochondrial membrane channels were proposed as MPTP components [79], but have since been shown to be unnecessary for MPTP formation.
First, mitochondria take up small molecules and ions through Voltage Dependent Anion Channels (VDAC) along the OM [80]. VDAC was originally hypothesized to play a role in MPTP configuration [81], however recent findings suggest otherwise [82,83]. In cancer cells, VDAC channels are significantly upregulated. As expected, downregulation of VDAC directly affects proliferation [84,85]. HK expression is also linked with VDAC quantities; in tumor cells, overexpression of HK-I and -II induces VDAC closure and prevents MPTP opening [86]. This HK-mediated closure of VDAC may allow for CypD-induced MPTP inhibition (Figure 2.4), thus enhancing tumor cell proliferative abilities.
Second, Adenine Nucleotide Translocase (ANT) was another postulated component of the MPTP [87], as it functions to catalyze the exchange of mitochondrial ATP for cytosolic ADP through the IM [88]. However, studies demonstrated it is not essential for MPTP induction [89]. Of the four isoforms of ANT, increased expression of ANT-1 and -3 promotes cancer cell death [90,91]. In contrast, enhanced expression of ANT-2 and -4 renders malignant cells more resistant [92,93]. Furthermore, ANT-2 seems to be critical for importation of glycolytic ATP in cancer cells [94]. Connecting ANT to the MPTP, overexpression of CypD inhibits ANT-1-mediated apoptosis in tumor cells [95]. Hence, the ANT isoforms oppositely participate in MPTP regulation in cancer, with CypD interactions vital in control cancer cell survival.
Collectively, this suggests that altered interactions between CypD, TSPO, VDAC, and ANT may play important roles in promoting cancer growth. Therefore, MPTP-induced cell death via manipulation of any of these factors may hold the key to future cancer treatment methods.
Cells often become damaged, infected, or malignant. When this happens, survival is no longer a priority. If this occurs, mitochondria have control of signal transduction pathways, such as apoptosis and necrosis, for proper destruction of the cell.
Two alternative pathways can initiate apoptosis within cells. Death receptors on the cell surface initiate apoptosis via the extrinsic path [96], while mitochondria activate the intrinsic route [97] (Figure 1.7). Apoptosis signaling stimulates initiator caspases [98], which propel the cleavage of death substrates through activation of executioner caspases [99]. Upon activation by caspases, anti-apoptotic proteins release pro-apoptotic proteins [100], driving the release of cyto c and other apoptotic factors from the opened MPTP [101]. The apoptotic factors exacerbate the process, causing the organized collapse and shrinkage of the cell until the cell body is engulfed by neighboring cells for removal [102]. Also known as programmed cell death as it can be purposefully initiated; apoptosis is a pathway that can be activated specifically by mitochondria.
Most often, cancer cells display increased inhibition of apoptosis due to mutations that either disrupt pro-apoptotic proteins [103,104] or elevate anti-apoptotic proteins [105]. On the contrary, a recent study found that enhanced levels of pro-apoptotic Bad promote prostate cancer growth [106]. This discrepancy illustrates that cancer cells tightly regulate all apoptotic protein levels in accordance with their proliferative needs. Additionally, in the event of apoptosis initiation, some cancer cells can reverse the process and survive [7]. Clearly, tumors manipulate many aspects of apoptosis to avoid death, which render cancer therapies ineffective. However, simultaneously targeting cancer cell-specific apoptotic inhibitory pathways while administering current anticancer agents is a novel avenue that may eliminate the resistance seen in tumors.
In the event of cellular injury, an unplanned cell death pathway called necrosis can be activated (Figure 1.8). Death receptor adaptor activation causes their translocation to the IM [107,108], which interferes with ANT-mediated ATP/ADP exchange. This disruption leads to ATP depletion and ROS accumulation [109]. Heightened ROS and Ca2+ levels stimulate mitochondrial uncoupling, swelling, and loss of ΔΨ due to opening of the MPTP [110,111]. These disparities initiate degradative enzymes which aggravate the process until plasma membrane rupture [112] and leakage of the intracellular contents in an unorganized manner.
Studies have shown that necrotic cell death is impaired in cancer cells, although the particular mechanisms are still being examined. Nakagawa and colleagues found that inhibiting CypD protects malignant cells from necrosis [113]. Also, although Leucine zipper/EF hand-containing Tansmembrane-1 (LETM1) induces necrosis in normal cells, LETM1 overexpression is common in many forms of cancer, leading to the inhibition of necrotic cell death [114]. Due to inconsistent outcomes with apoptosis-mediated treatments, the induction of necrosis may provide an alternative tool for treating cancer cells resistant to apoptosis-inducing methods.
As many cancers become resistant to chemotherapy and radiation regimens, targeting of mitochondrial-mediated processes is being investigated for enhancement of available anticancer agents. A large variety of drugs are approved for cancer treatment, such as cisplatin [115], paclitaxel [116], and trastuzumab [117]. However, due to cell repopulation and resistance, many antitumor agents fail to reduce cancer progression and metastasis in the long run. Concurrent treatment may aid the antitumor effects of available treatments.
Apoptosis manipulation is a popular target as programmed cell death is inhibited in cancer cells. Reducing the levels of antiapoptotic proteins in tumor cells using antisense constructs (Genasense) showed promising results for sensitizing cancer cells to apoptosis-mediated anticancer drugs in animals [118], but less desirable effects in clinical trials. Instead, small molecule mimetics that bind and inactivate antiapoptotic proteins have been proposed as an alternative method [119]. In essence, inhibition of antiapoptotic proteins could help sensitize malignant cells to available antineoplastic treatments (Figure 3).
Looking specifically at the MPTP, a few drugs are being tested for anticancer ability. As shown in Figure 3, targeting lonidamine to malignant cell mitochondria causes ΔΨ disruption and MPTP-induced apoptosis [120]. Furthermore, co-treatment with lonidamine and arsenic trioxide enhances ROS generation and leads to MPTP-mediated cell death in human leukemia cells [121]. Also, CypD seems to play a role in cisplatin-mediated pancreatic cancer cell death [122]. As CypD overexpression typically enhances cancer cell resistance, it will be interesting to see whether manipulation of CypD or other MPTP components has an effect on sensitizing tumor cells to antitumor agents.
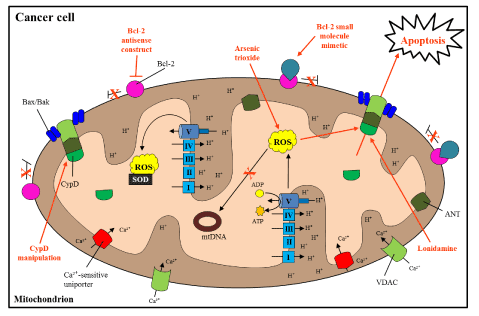
Figure 3. Mitochondrial targets for treatment of cancer
Since cancer mitochondria are vastly altered, specific targeting of dysfunctional processes and pathways may lead to novel treatment regiments (labeled in red). In cancer cells, manipulation of Bcl-2 antiapoptotic proteins can be used to induce apoptosis. Bcl-2 antisense constructs and Bcl-2 small molecular mimeticshave been used in animal and clinical trials to inactivate Bcl-2 specifically, with varied results. Lonidaminehas been observed to disrupt ΔΨ and propel MPTP-regulated cell death. Moreover, arsenic trioxide together with lonidamine further increases ROS output, leading to apoptosis. Manipulation of CypD may aid in the sensitization of cancer cells to anticancer therapeutics. Essentially, disrupting cancer cell resistance by inducing cell death using the indicated therapeutics/target areas could provide additional anticancer treatment options [123].
Mitochondrial dysfunction and tight regulation of cellular activities allows for cancer cell growth and resistance to cell death. With effective treatment options limited for cancer, research has shifted to target-specific and/or concurrent therapeutic regimens. Since mitochondrial dysfunction is highly implicated in cancer, targeting specific mitochondrial processes may enhance the susceptibility of diseased cells to available drugs. This novel approach toward disease treatment would increase the quantity and quality of therapeutic options.
This work was supported by grants from the National Institute of Aging (R37AG037319) and the National Institute of General Medical Science (R01GM095355).
- 1. Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA: a cancer journal for clinicians 63: 11-30.
- 2. Kim JJ, Tannock IF (2005) Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer 5: 516-525.[Crossref]
- 3. Land H, Parada LF, Weinberg RA (1983) Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304: 596-602.[Crossref]
- 4. Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, et al. (1994) Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. ProcNatlAcadSci USA 91: 9700-9704.[Crossref]
- 5. Masutani C, Kusumoto R, Yamada A, Yuasa M, Araki M, et al. (2000) Xerodermapigmentosum variant: from a human genetic disorder to a novel DNA polymerase. Cold Spring HarbSymp Quant Biol 65: 71-80.
- 6. Warburg O (1956) On respiratory impairment in cancer cells. Science 124: 269-270.[Crossref]
- 7. Tang HL, Yuen KL, Tang HM, Fung MC (2009) Reversibility of apoptosis in cancer cells. Br J Cancer 100: 118-122.[Crossref]
- 8. Perkins G, Renken C, Martone ME, Young SJ, Ellisman M, et al. (1997) Electron tomography of neuronal mitochondria: three-dimensional structure and organization of cristae and membrane contacts. JStructBiol 119: 260-272. [Crossref]
- 9. Imanishi H, Hattori K, Wada R, Ishikawa K, Fukuda S, et al. (2011) Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS One 6: e23401.[Crossref]
- 10. Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, et al. (2005) Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res 65: 1655-1663. [Crossref]
- 11. Ma Y, Bai RK, Trieu R, Wong LJ (2010) Mitochondrial dysfunction in human breast cancer cells and their transmitochondrialcybrids. BiochimBiophysActa 1797: 29-37.[Crossref]
- 12. WARBURG O (1956) On the origin of cancer cells. Science 123: 309-314.[Crossref]
- 13. Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411: 355-365.[Crossref]
- 14. Baldwin AS (2001) Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest 107: 241-246.[Crossref]
- 15. Nakanishi C, Toi M (2005) Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer 5: 297-309.[Crossref]
- 16. Ni J, Takayama K, Inoshima N, Uchino J, Harada A, et al. (2005) Gene transfer of inhibitor kappaB in human lung cancer cell line NCI-H460 inhibits tumorigenesis and angiogenesis in vivo. Anticancer Res 25: 69-77.[Crossref]
- 17. Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, et al. (2004) NF-kappa B-mediated repression of growth arrest- and DNA-damage-inducible proteins 45alpha and gamma is essential for cancer cell survival. ProcNatlAcadSci USA 101: 13618-13623.[Crossref]
- 18. Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, et al. (2012) NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol 13: 1272-1279. [Crossref]
- 19. Eferl R, Ricci R, Kenner L, Zenz R, David JP, et al. (2003) Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell 112: 181-192.[Crossref]
- 20. Bonovich M, Olive M, Reed E, O'Connell B, Vinson C (2002) Adenoviral delivery of A-FOS, an AP-1 dominant negative, selectively inhibits drug resistance in two human cancer cell lines. Cancer Gene Ther9: 62-70.[Crossref]
- 21. Suto R, Tominaga K, Mizuguchi H, Sasaki E, Higuchi K, et al. (2004) Dominant-negative mutant of c-Jun gene transfer: a novel therapeutic strategy for colorectal cancer. Gene Ther11: 187-193.[Crossref]
- 22. Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19: 2474-2488.[Crossref]
- 23. Niu G, Bowman T, Huang M, Shivers S, Reintgen D, et al. (2002) Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 21: 7001-7010.[Crossref]
- 24. Lee SO, Lou W, Qureshi KM, Mehraein-Ghomi F, Trump DL, et al. (2004) RNA interference targeting Stat3 inhibits growth and induces apoptosis of human prostate cancer cells. Prostate 60: 303-309.[Crossref]
- 25. Xi S, Gooding WE, Grandis JR (2005) In vivo antitumor efficacy of STAT3 blockade using a transcription factor decoy approach: implications for cancer therapy. Oncogene 24: 970-979.[Crossref]
- 26. Kumar R, Gururaj AE, Vadlamudi RK, Rayala SK(2005) The clinical relevance of steroid hormone receptor corepressors. Clin Cancer Res 11: 2822-2831. [Crossref]
- 27. Liao X, Tang S, Thrasher JB, Griebling TL, Li B (2005) Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Mol Cancer Ther 4: 505-515.[Crossref]
- 28. Buluwela L, Pike J, Mazhar D, Kamalati T, Hart SM, et al. (2005) Inhibiting estrogen responses in breast cancer cells using a fusion protein encoding estrogen receptor-alpha and the transcriptional repressor PLZF. Gene Ther 12: 452-460. [Crossref]
- 29. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, et al. (1981) Sequence and organization of the human mitochondrial genome. Nature 290: 457-465.[Crossref]
- 30. Chang DD, Clayton DA (1985) Priming of human mitochondrial DNA replication occurs at the light-strand promoter. ProcNatlAcadSci USA 82: 351-355.[Crossref]
- 31. Tseng LM, Yin PH, Chi CW, Hsu CY, Wu CW, et al. (2006) Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 45: 629-638.[Crossref]
- 32. Mambo E, Chatterjee A, Xing M, Tallini G, Haugen BR, et al. (2005) Tumor-specific changes in mtDNA content in human cancer. Int J Cancer 116: 920-924.[Crossref]
- 33. Chihara N, Amo T, Tokunaga A, Yuzuriha R, Wolf AM, et al. (2011) Mitochondrial DNA alterations in colorectal cancer cell lines. J Nippon Med Sch 78: 13-21.[Crossref]
- 34. Yin PH, Wu CC, Lin JC, Chi CW, Wei YH, et al. (2010) Somatic mutations of mitochondrial genome in hepatocellular carcinoma. Mitochondrion 10: 174-182.[Crossref]
- 35. Sanchez-Cespedes M, Parrella P, Nomoto S, Cohen D, Xiao Y, et al. (2001) Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer Res 61: 7015-7019.[Crossref]
- 36. Nomoto S, Yamashita K, Koshikawa K, Nakao A, Sidransky D(2002) Mitochondrial D-loop mutations as clonal markers in multicentric hepatocellular carcinoma and plasma. Clin Cancer Res 8: 481-487. [Crossref]
- 37. Miyazono F, Schneider PM, Metzger R, Warnecke-Eberz U, Baldus SE, et al. (2002) Mutations in the mitochondrial DNA D-Loop region occur frequently in adenocarcinoma in Barrett's esophagus. Oncogene 21: 3780-3783.[Crossref]
- 38. Bai RK, Chang J, Yeh KT, Lou MA, Lu JF, et al. (2011) Mitochondrial DNA content varies with pathological characteristics of breast cancer. J Oncol 2011: 496189.[Crossref]
- 39. Yu M, Zhou Y, Shi Y, Ning L, Yang Y, et al. (2007) Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 59: 450-457.[Crossref]
- 40. McCormack JG (1985) Characterization of the effects of Ca2+ on the intramitochondrial Ca2+-sensitive enzymes from rat liver and within intact rat liver mitochondria. Biochem J 231: 581-595.[Crossref]
- 41. Yankovskaya V, Horsefield R, Törnroth S, Luna-Chavez C, Miyoshi H, et al. (2003) Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299: 700-704.[Crossref]
- 42. McCormack JG, Halestrap AP, Denton RM (1990) Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70: 391-425.[Crossref]
- 43. Takehara Y, Kanno T, Yoshioka T, Inoue M, Utsumi K (1995) Oxygen-dependent regulation of mitochondrial energy metabolism by nitric oxide. Arch BiochemBiophys 323: 27-32.[Crossref]
- 44. NalinCM, Cross RL (1982) Adenine nucleotide binding sites on beef heart F1-ATPase. Specificity of cooperative interactions between catalytic sites. J BiolChem 257: 8055-8060. [Crossref]
- 45. Domenis R, Bisetto E, Rossi D, Comelli M, Mavelli I (2012) Glucose-modulated mitochondria adaptation in tumor cells: a focus on ATP synthase and inhibitor factor 1. Int J MolSci 13: 1933-1950.[Crossref]
- 46. Isidoro A, Martínez M, Fernández PL, Ortega AD, Santamaría G, et al. (2004) Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem J 378: 17-20.[Crossref]
- 47. Bellance N, Benard G, Furt F, Begueret H, Smolková K, et al. (2009) Bioenergetics of lung tumors: alteration of mitochondrial biogenesis and respiratory capacity. Int J BiochemCell Biol 41: 2566-2577. [Crossref]
- 48. Weber K, Ridderskamp D, Alfert M, Hoyer S, Wiesner RJ. (2002) Cultivation in glucose-deprived medium stimulates mitochondrial biogenesis and oxidative metabolism in HepG2 hepatoma cells. BiolChem 383: 283-290.[Crossref]
- 49. Smolková K, Bellance N, Scandurra F, Génot E, Gnaiger E, et al. (2010) Mitochondrial bioenergetic adaptations of breast cancer cells to aglycemia and hypoxia. J BioenergBiomembr 42: 55-67.[Crossref]
- 50. Rodríguez-Enríquez S1, Carreño-Fuentes L, Gallardo-Pérez JC, Saavedra E, Quezada H, et al. (2010) Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int J Biochem Cell Biol 42: 1744-1751. [Crossref]
2021 Copyright OAT. All rights reserv
- 51. Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552: 335-344.[Crossref]
- 52. McCord JM, Fridovich I (1969) Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J BiolChem 244: 6049-6055.[Crossref]
- 53. Mutisya EM, Bowling AC, Beal MF (1994) Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J Neurochem 63: 2179-2184.[Crossref]
- 54. Young-Collier KJ, McArdle M, Bennett JP (2012) The dying of the light: mitochondrial failure in Alzheimer's disease. J Alzheimers Dis 28: 771-781.[Crossref]
- 55. Sawyer DE, Roman SD, Aitken RJ (2001) Relative susceptibilities of mitochondrial and nuclear DNA to damage induced by hydrogen peroxide in two mouse germ cell lines. Redox Rep 6: 182-184.[Crossref]
- 56. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, et al. (2008) ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320: 661-664.[Crossref]
- 57. Toyokuni S, Okamoto K, Yodoi J, Hiai H (1995) Persistent oxidative stress in cancer. FEBS Lett 358: 1-3.[Crossref]
- 58. Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK (2008) Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res 68: 1777-1785.[Crossref]
- 59. Mochizuki T, Furuta S, Mitsushita J, Shang WH, Ito M, et al. (2006) Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Oncogene 25: 3699-3707. [Crossref]
- 60. Pelicano H, Lu W, Zhou Y, Zhang W, Chen Z, et al. (2009) Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res 69: 2375-2383. [Crossref]
- 61. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, et al. (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334: 1278-1283. [Crossref]
- 62. Al-Nasser, Crompton M (1986) The reversible Ca2+-induced permeabilization of rat liver mitochondria. Biochem J 239: 19-29.[Crossref]
- 63. Crompton M, Costi A, Hayat L (1987) Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem J 245: 915-918.[Crossref]
- 64. Ichas F, Mazat JP (1998) From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. BiochemBiophysActa 1366: 33-50.[Crossref]
- 65. Ling X, Zhou Y, Li SW, Yan B, Wen L (2010) Modulation of mitochondrial permeability transition pore affects multidrug resistance in human hepatocellular carcinoma cells. Int J BiolSci 6: 773-783. [Crossref]/li>
- 66. Tanveer A, Virji S, Andreeva L, Totty NF, Hsuan JJ, et al. (1996) Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur J Biochem 238: 166-172.[Crossref]
- 67. Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, et al. (2005) Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. ProcNatlAcadSci USA 102: 12005-12010. [Crossref]
- 68. Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, et al. (2005) Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J BiolChem 280: 18558-18561.[Crossref]
- 69. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, et al. (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658-662.[Crossref]
- 70. Eliseev RA, Malecki J, Lester T, Zhang Y, Humphrey J, et al. (2009) Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J BiolChem 284: 9692-9699.[Crossref]
- 71. Machida K, Ohta Y, Osada H (2006) Suppression of apoptosis by cyclophilin D via stabilization of hexokinase II mitochondrial binding in cancer cells. J BiolChem 281: 14314-14320.[Crossref]
- 72. Ghosh JC, Siegelin MD, Dohi T, Altieri DC (2010) Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res 70: 8988-8993.[Crossref]
- 73. Kinnally KW, Zorov DB, Antonenko YN, Snyder SH, McEnery MW, et al. (1993) Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. ProcNatlAcadSci USA 90: 1374-1378.[Crossref]
- 74. Sileikyte J, Petronilli V, Zulian A, Dabbeni-Sala F, Tognon G, et al. (2011) Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor). J BiolChem 286: 1046-1053.[Crossref]
- 75. Mukherjee S, Das SK (2012) Translocator protein (TSPO) in breast cancer. CurrMol Med 12: 443-457.[Crossref]
- 76. Hardwick M, Fertikh D, Culty M, Li H, Vidic B, et al. (1999) Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res 59: 831-842. [Crossref]
- 77. Wu X, Gallo KA (2013) The 18-kDa translocator protein (TSPO) disrupts mammary epithelial morphogenesis and promotes breast cancer cell migration. PLoS One 8: e71258.[Crossref]
- 78. Galiègue S, Casellas P, KramarA, Tinel N, Simony-Lafontaine J(2004) Immunohistochemical assessment of the peripheral benzodiazepine receptor in breast cancer and its relationship with survival. ClinCancer Res 10: 2058-2064. [Crossref]
- 79. Crompton M, Virji S, Ward JM (1998) Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. EurJBiochem 258: 729-735. [Crossref]
- 80. Gincel D, Zaid H, Shoshan-Barmatz V. (2001) Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J 358: 147-155. [Crossref]
- 81. Szabó I, Zoratti M (1993) The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Lett330: 201-205.[Crossref]
- 82. Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD (2007) Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9: 550-555.[Crossref]
- 83. Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P (2006) Properties of the permeability transition in VDAC1(-/-) mitochondria. BiochimBiophysActa 1757: 590-595.[Crossref]
- 84. Abu-Hamad S, Sivan S, Shoshan-Barmatz V (2006) The expression level of the voltage-dependent anion channel controls life and death of the cell. ProcNatlAcadSci USA 103: 5787-5792.[Crossref]
- 85. Koren I, Raviv Z, Shoshan-Barmatz V (2010) Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo. Cancer BiolTher 9: 1046-1052.[Crossref]
- 86. Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V(2004) In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J 377: 347-355.[Crossref]
- 87. Halestrap AP, Brenner C (2003) The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem 10: 1507-1525. [Crossref]
- 88. Klingenberg M (1975) Energetic aspects of transport of ADP and ATP through the mitochondrial membrane. Ciba Found Symp: 105-124.[Crossref]
- 89. Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, et al. (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427: 461-465.[Crossref]
- 90. Bauer MK, Schubert A, Rocks O, Grimm S (1999) Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol 147: 1493-1502.[Crossref]
- 91. Zamora M, Granell M, Mampel T, Viñas O (2004) Adenine nucleotide translocase 3 (ANT3) overexpression induces apoptosis in cultured cells. FEBS Lett 563: 155-160.[Crossref]
- 92. Chevrollier A, Loiseau D, Chabi B, Renier G, Douay O, et al. (2005) ANT2 isoform required for cancer cell glycolysis. J BioenergBiomembr 37: 307-316.[Crossref]
- 93. Gallerne C, Touat Z, Chen ZX, Martel C, Mayola E, et al. (2010) The fourth isoform of the adenine nucleotide translocator inhibits mitochondrial apoptosis in cancer cells. Int J Biochem Cell Biol 42: 623-629.[Crossref]
- 94. Chevrollier A, Loiseau D, Reynier P, Stepien G (2011) Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. BiochimBiophysActa 1807: 562-567.[Crossref]
- 95. Schubert A, Grimm S (2004) Cyclophilin D, a component of the permeability transition-pore, is an apoptosis repressor. Cancer Res 64: 85-93.[Crossref]
- 96. Tartaglia LA, Ayres TM, Wong GH, Goeddel DV (1993) A novel domain within the 55 kd TNF receptor signals cell death. Cell 74: 845-853.[Crossref]
- 97. Fulda S, Scaffidi C, Susin SA, Krammer PH, Kroemer G, et al. (1998) Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J BiolChem 273: 33942-33948.[Crossref]
- 98. Chen M, Wang J (2002) Initiator caspases in apoptosis signaling pathways. Apoptosis 7: 313-319.[Crossref]
- 99. Slee EA, Adrain C, Martin SJ (2001) Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J BiolChem 276: 7320-7326.[Crossref]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, et al. (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8: 705-711. [Crossref]
- 101. Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, et al. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727-730.[Crossref]
- 102. Nagata S, Hanayama R, Kawane K (2010) Autoimmunity and the clearance of dead cells. Cell 140: 619-630.[Crossref]
- 103. Meijerink JP, Mensink EJ, Wang K, Sedlak TW, Slöetjes AW, et al. (1998) Hematopoietic malignancies demonstrate loss-of-function mutations of BAX. Blood 91: 2991-2997.[Crossref]
- 104. Lee JW, Soung YH, Kim SY, Nam SW, Kim CJ, et al. (2004) Inactivating mutations of proapoptotic Bad gene in human colon cancers. Carcinogenesis 25: 1371-1376.[Crossref]
- 105. Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, et al. (2010) A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis 1: e40.[Crossref]
- 106. Smith AJ, Karpova Y, D'Agostino R Jr, Willingham M, Kulik G (2009) Expression of the Bcl-2 protein BAD promotes prostate cancer growth. PLoS One 4: e6224.[Crossref]
- 107. Holler N, Zaru R, Micheau O, Thome M, Attinger A, et al. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489-495.[Crossref]
- 108. Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, et al. (2003) A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. JBiolChem 278: 51613-51621.[Crossref]
- 109. Temkin V, Huang Q, Liu H, Osada H, Pope RM (2006) Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol Cell Biol 26: 2215-2225.[Crossref]
- 110. Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, et al. (1998) The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. BiochimicaetBiophysicaActa 1366: 177-196.
- 111. Choi K, Kim J, Kim GW, Choi C (2009) Oxidative stress-induced necrotic cell death via mitochondira-dependent burst of reactive oxygen species. CurrNeurovasc Res 6: 213-222.[Crossref]
- 112. Nieminen AL, Gores GJ, Wray BE, Tanaka Y, Herman B, et al. (1988) Calcium dependence of bleb formation and cell death in hepatocytes. Cell Calcium 9: 237-246.[Crossref]
- 113. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, et al. (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652-658.[Crossref]
- 114. Piao L, Li Y, Kim SJ, Byun HS, Huang SM, et al. (2009) Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP production and necrotic cell death. Cancer Res 69: 3397-3404.[Crossref]
- 115. Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC (2011) Targeted delivery of a cisplatinprodrug for safer and more effective prostate cancer therapy in vivo. ProcNatlAcadSci USA 108: 1850-1855.[Crossref]
- 116. Swaminathan SK, Roger E, Toti U, Niu L, Ohlfest JR, et al. (2013) CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J Control Release 171: 280-287.[Crossref]
- 117. Garcia AG, Nedev H, Bijian K, Su J, Alaoui-Jamali MA, et al. (2013) Reduced in vivo lung metastasis of a breast cancer cell line after treatment with Herceptin mAb conjugated to chemotherapeutic drugs. Oncogene 32: 2527-2533.[Crossref]
- 118. Spugnini EP, Biroccio A, De Mori R, Scarsella M, D'Angelo C, et al. (2011) Electroporation increases antitumoral efficacy of the bcl-2 antisense G3139 and chemotherapy in a human melanoma xenograft. JTransl Med 9: 125. [Crossref]
- 119. Dalafave DS, Prisco G (2010) Inhibition of Antiapoptotic BCL-XL, BCL-2, and MCL-1 Proteins by Small Molecule Mimetics. Cancer Inform 9: 169-177.[Crossref]
- 120. Li N, Zhang CX, Wang XX, Zhang L, Ma X, et al. (2013) Development of targeting lonidamine liposomes that circumvent drug-resistant cancer by acting on mitochondrial signaling pathways. Biomaterials 34: 3366-3380. [Crossref]
- 121. Calvino E, Estañ MC, Simón GP, Sancho P, Boyano-AdánezMdel C, et al. (2011) Increased apoptotic efficacy of lonidamine plus arsenic trioxide combination in human leukemia cells. Reactive oxygen species generation and defensive protein kinase (MEK/ERK, Akt/mTOR) modulation. BiochemPharmacol 82: 1619-1629. [Crossref]
- 122. Chen B, Xu M, Zhang H, Wang JX, Zheng P, et al. (2013) Cisplatin-induced non-apoptotic death of pancreatic cancer cells requires mitochondrial cyclophilin-D-p53 signaling. BiochemBiophys Res Commun 437: 526-531. [Crossref]
- 123. Carlson EA, Rao VK, Yan SS (2013) From a cell's viewpoint: targeting mitochondria in Alzheimer's disease. Drug Discovery Today: Therapeutic Strategies 10: 2.