A variety of proteins with potential for medical applications contain disulfide-bridges. In this review, the author describes investigations of such proteins, particularly those concerned with clarification of structure – function relationships and molecular engineering, focusing on three classes of targets, namely human lysozyme, plant lectins composed of hevein-type domains and extracellular domains of human Fas ligand and Fas receptor. With each class of proteins, biological functions relevant to therapeutic applications, structural features, development of sample preparation methods and the experimental results for the clarification of structure – function relationships, including those of protein engineering studies on the basis of their three-dimensional structures, is summarized in the light of our current knowledge. The studies were performed by elucidating details in structural and functional consequences after introducing ligand complex formations, site-directed mutagenesis and site-specific chemical modifications, which employed various biochemical and biophysical analyses in combination with newly developed recombinant expression and chemical synthesis methods. The main findings from the studies include basic principles concerning the structure – function relationships in the enzymatic catalysis by endoglycosidases and carbohydrate recognition by lectins as well as biotechnological advances in the use of extracellular domains of death ligands and death receptors. The experimental results reviewed here will contribute to the future development of novel disulfide-bridged proteins with therapeutic usefulness targeting unmet medical needs.
amaranthus caudatus anti-microbial peptide 2 (Ac-amp2), human Fas ligand, human Fas receptor, human lysozyme, Uritica dioica agglutinin (UDA), wheat-germ agglutinin (WGA)
aa: Amino Acid; Ac-amp2: Antimicrobial Peptides from Amaranthus caudatus seeds 2; ALPS: Autoimmune Lymphoproliferative Syndrome; CD: Cluster of Differentiation; CRD: Cysteine Rich Domain; hDcR3: Human Decoy Receptor 3; Ee-CBP: Antimicrobial Peptide from Euonymus europaeus L. bark; ECD: Extracellular Domain; ER: Endoplasmic Reticulum; Gal: galactose; GalNAc: N-acetylgalactosamine; GlcNAc: N-acetylglucosamine; GVHD: Graft-Versus-Host Disease; ITC: Isothermal Titration Calorimetry; Man: Mannose; MHC: Major Histocompatibility Complex; NMR: Nuclear Magnetic Resonance; NeuNAc: N-acetylneuraminicacid; PDB ID: Protein Data Bank Identification Code; PL-D2: Pokeweed Lectin-D isoform 2; NF-κB: Nuclear Factor-kappa B; PWM: Pokeweed Mitogen from Phytolacca americana bark; RA: Rheumatoid Arthritis; TNF: Tumor Necrosis Factor; WGA: Wheat Germ Agglutinin; UDA: Urtica dioica Agglutinin; WAMP: Antifungal Peptide from Triticum kiharae seeds
A wide variety of medically important proteins including but not limited to antibodies, enzymes, cell-surface binding proteins, cytokines and their cognate receptors, anti-microbial proteins, hormones, toxins and the proteins involved in either redox transformation or nutrition transportation, contain disulfide-bridges. These proteins are generally found at cell-surface or secreted into extracellular body fluids such as blood. The surrounding biochemical environments outside the cells are often not conducive to the survival of protein molecules due to the presence of many inactivating factors including proteases. Hence, the relevant proteins have to be stable enough to endure such circumstances. Disulfide-bridges are covalent bonds for binding two different parts of main peptide chain(s) in a protein molecule; therefore they give a protein molecule substantial structural stability [1]. The stabilizing effect of disulfide-bridges is mostly of an entropic nature and considered to be significantly larger than other non-covalent interactions, such as hydrogen bonding interactions and van der Waals interactions [2].
To date, many disulfide-bridged proteins including monoclonal antibodies, lysosomal enzymes, blood coagulation and fibrinolysis factors, serum proteins, insulin, growth hormones, interferons and interleukins have been already approved as practical protein medicines. However, there are still different classes of disulfide-proteins with a potential therapeutic activity, which may be applicable for fulfilling unmet medical needs. Among such proteins, the author focuses on three classes of disulfide-bridged proteins, namely human lysozyme, plant lectins composed of hevein-type domains and extracellular domains of human Fas ligand and human Fas receptor, in this review. First, therapeutic relevance is briefly summarized with each class of the proteins. Then, structural features emphasizing the three-dimensional aspects are explained. After that, efficient sample preparation methods for biochemical investigations achieved by either recombinant expression systems, affinity-based isolation from natural resources or chemical synthesis plus subsequent refolding, are described. Finally, representative topics concerning both structural analysis and functional characterization for elucidating the relationships between them and the biotechnological advances related to medical applications are presented.
A bacteriolytic enzyme with a potential for therapeutic applications
Human lysozyme was discovered in 1922 as a component of nasal fluid, which lysed a contaminated bacterium in the cultivation plate [3]. The susceptible bacterium was named Micrococcus lysodeikticus (presently Micrococcus luteus). The bacteriolytic enzymatic activity is initially considered to be effective only for Gram-positive bacteria lacking cell outer membrane such as Bacillus subtilis. However, subsequently it was found that this enzyme also killed Gram-negative bacteria [4,5]. In this case the synergetic action with other factors such as lactoferrin, which causes damage to the outer membrane for making the peptideglycan layer more accessible to lysozyme molecule, may take place [4]. Human lysozyme is contained in many human body fluids represented by airway surface liquid, tears and milk, and therefore considered to be essential as a major defense factor constituting the innate immunity of the human body [4,6]. Recently, serious problems in public health are emerging due to occurrence of drug-resistant bacterial pathogens. Human lysozyme is thought to be a safe anti-microbial agent, which can possibly substitute for low molecular weight drugs without showing unwanted allergic adverse effects, since it originates from the human body. Methicillin resistant Staphylococcus aureus, which is a major cause of worldwide nosocomical infections, is basically susceptible to lytic activity of lysozyme, though many strains showing different degree of resistance have been isolated [7]. Human lysozyme was also reexamined as an effective therapeutic drug for Pseudomonas aeruginosa lung infections to reduce bacterial burden and inflammation, which may be serious problems for patients suffering from opportunistic infection caused by significantly declined immune responses [8].
In some countries including Japan, a structurally homologous enzyme, chicken lysozyme has been used as an expectorant in medicine for patients suffering from colds, chronic paranasal sinus inflammation and so on. However, in this case, the effect of lysozyme would be mainly from its non-enzymatic antiphlogistic activity, since most common colds are caused not by bacteria but by viruses which are not susceptible to the lytic activity of lysozyme. It is questionable whether the reduction of the viscosity of sputum by the medication can be attributed to the enzymatic activity of lysozyme, since the chemical structure of the major mucopolysaccharides in sputum are not always identical to that of bacterial cell wall. Several reports on immune-potentiation in cancer treatments including the anti-metastatic activity of orally administered lysozyme have been published [9,10]. These non-enzymatic activities may be related to its highly cationic character under physiological pH condition [11]. However, the detailed mechanism at molecular level is still yet to be clarified. In connection to the diseases caused by the protein misfolding, eight kinds of mutant human lysozyme genes have been reported to be a pathogenic cause for hereditary non-neuropathic systemic amyloidosis to date [12].
A small globular monomeric protein with abundant secondary structures
Human lysozyme consists of 130 amino acid (aa) residues with molecular weight of approximately 14.7 kDa. It has four disulfide-bridges, which all connect its single main-chain to tightly fold up into a spheroid in the dimensions of 30 x 30 x 45 Å with well-extended α/β-type secondary structures (Figure 1A) [13]. The secondary structures comprise four α-helices, two 310-helices and one β-sheet region made from three anti-parallel chains. These structural features provide human lysozyme molecule a rather high thermodynamic stability showing denaturation temperature of 80.3°C at pH 4.5 [14]. A large excess number of basic amino acid residues (Arg, Lys, and His, 20 residues in total) over that of acidic amino acid residues (Glu and Asp, 11 residues in total) make the whole molecule very basic, which is shown by the isoelectric point of approximately 11.This molecule itself contains no carbohydrate chain, but recognizes either a carbohydrate homopolymer made of β-1,4 linked N-acetylglucosamine residues alone (chitin type substrate) or a β-1,4 linked alternating copolymer made of N-acetylglucosamine residues and N-acetylmuramicacid residues (bacterial cell-wall peptide glycan type substrate).On the surface of human lysozyme molecule, a deep active-site cleft, composed of six subsites designated A to F, exists for engulfing the above substrates, and cleaves the carbohydrate chains between the subsites D and E (Figures 1B and 1C).
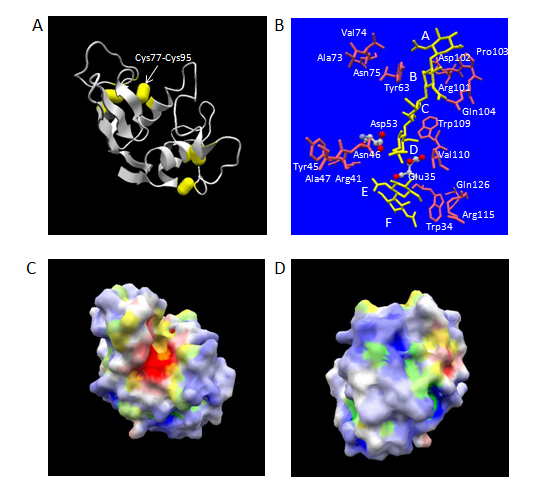
Figure 1. Three-dimensional structure of human lysozyme. The atomic coordinate data were obtained from PDB (ID: 1lzs, X-ray model). Panel A, overall structure. Secondary structures and disulfide-bonds are shown in ribbons and bales, respectively. Panel B, the structures of bound N-acetylchitohexaose substrate split into N-acetylchitotetraose and N-acetylchitobiose (in yellow) and aa residues of human lysozyme mentioned in the text [catalytic aa residues of ball-and-stick models in atom colors (red, oxygen; gray, carbon; blue, nitrogen) and other aa residues of stick only models in pink] are shown. Relevant aa residues’ names in human lysozyme are labelled. The labels, A, B, C, D, E and F, indicate the approximate positions of six subsites. Panels C and D, electrostatic potential distribution in molecular surface. Blue and green, positively charged regions; white and yellow, electrostatically neutral regions; red and orange, negatively charged regions. Blue, white and red, hydrophilic regions; green, yellow and orange, hydrophobic regions. Panel C, Front-side view exhibiting the catalytic cleft vertically in the same orientation as panels A and B; Panel D, back-side view by 180o rotation around the vertical axis.
Development of efficient recombinant production systems using several types of expression hosts
Initial attempt of intracellular overexpression of human lysozyme using an artificial human lysozyme gene designed with optimized codons for yeast in Escherichia coli resulted in the formation of insoluble and inactive inclusion bodies, though the enzymatic activity was partially regenerated by solubilization and dilution process [15]. Hence, a secretory expression system in Saccharomyces cerevisiae using chicken lysozyme secretion signal sequence was devised for successful production of soluble and fully active enzyme [16,17]. After the functional production in S. cerevisiae, other successful high-level expression systems of recombinant human lysozyme using various different hosts, including Aspergillus oryzae [18], Acremonium chrysogenum [19], Pichia pastoris [20,21], rice [22], transgenic mice [23,24] and transgenic cattle [25], followed. The highest level in the milk of tr/ansgenic mice reached 1.76 g/l, which is three times higher than human whey [24]. Recently, E. coli became an efficient producer of functionally active recombinant human lysozyme by co-expressing an enzyme inhibitor protein using an intracellular disulfide-bridges formation enhancing strain [26]. Another species of yeast, Kluyveromyces lactis was also used for an efficient secretory production of human lysozyme using a biofilm reactor [27].
The secretion level of human lysozyme depends on the secretion signal sequence in S. cerevisiae. Either chicken lysozyme signal peptide sequence and its derivatives or human serum albumin signal peptide sequence worked well, but the signal sequence derived from Aspergillus awamori glucoamylase failed to secrete human lysozyme [17]. A modified signal sequence containing ten times of Leu repeat sequence showed 1.8 times more effectiveness as compared with wild-type chicken lysozyme signal sequence [28]. Also, the deletion of a disulfide-bond between Cys77 and Cys95 (Figure 1A) gave an eight-fold efficient secretion in S. cerevisiae [29]. In P. pastoris, α-factor prepro sequence was proved to be much more efficient as compared to ch/icken lysozyme signal peptide sequence [20]. For research purposes of protein engineering studies of human lysozyme, S. cerevisiae systems have been used exclusively so far.
Extensive protein engineering studies concerning catalytic mechanism and enzymatic activity
After the determination of its detailed three-dimensional structure of chicken lysozyme as a first enzyme protein molecule by X-ray crystallography [30], chicken-type lysozymes including human lysozyme have always played the role of a model protein for engineering studies of many glycosidases, such as cellulases, amylases and so on, based on the available information in atomic details [31]. The enzymatic hydrolysis of substrates has primary significance as the fundamental medical applications on the basis of anti-bacterial activity of human lysozyme. In this review, the author focuses on protein engineering studies on the roles of aa residues of human lysozyme concerning its catalytic mechanism and the enzymatic activity. Other important aspects relevant to possible therapeutic applications at molecular level (e.g. amyloid formation) can be referred to elsewhere [32].
Precise positions of two catalytic carboxylate group containing residues (Glu35 and Asp53) are critical for the hydrolysis of substrates:
Human lysozyme is an endo-type β-glycosidase. β-glycosidases can be classified into two categories according to the stereo-chemistry of resulting terminal β-anomeric hydroxyl group derived from the scissile glycosidic bond in the cleaved product [33]. One is retaining enzymes and the other is inverting enzymes. Human lysozyme belongs to the former and its catalytic reaction is suggested to proceed via an oxocarbonium-ion type intermediate. In human lysozyme, the optimum pH of a chitin-type substrate hydrolysis was about 5.1, where Glu35 (pKa: 6.8) acts as a general acid, which is considered to first protonate the oxygen atom in the scissile bond. The bond cleavage is assisted by the electrostatic stabilization between the positive charge of resulting oxocarbonium-ion and the negative charge from the side-chain of Asp53 (pKa: 3.4) [34]. By analogy to the case of chicken lysozyme [35], the abnormal high pKa of Glu35 in human lysozyme can be considered partly due to the hydrophobic environment provided by a spatially adjacent residue, Trp109 (Figure 1B).
For the purpose of examining the importance of the precise side-chain positions of these carboxylate group containing residues in the hydrolysis of polysaccharide substrates, Glu35 and Asp53 residues were replaced with Asp and Glu, respectively [36-38]. Either replacement greatly reduced the hydrolytic activity against both M. luteus cell substrate and a soluble chitin-type substrate, ethylene glycol chitin. The extent of catalytic activity reduction was more significant in the replacement of Glu35 to Asp as compared to that of Asp53 to Glu.Human lysozyme with Glu53 mutation still showed 3.4% activity relative to wild-type enzyme against a chitin oligosaccharide substrate, p-nitrophenyl N-acetylchitopentaoside at 37°C, pH 5.0 in the reaction time of 30 min at initial concentration of 1.3 μM and 0.22 mM with regard to the enzyme and the substrate concentrations, respectively [39]. The extent of this enzymatic activity reduction was comparable to the case of a mutant chicken lysozyme, where the corresponding residue (Asp 52) was replaced to Asn, against M. luteus substrate (5.5 ± 2.5%) [40]. Interestingly, the relative activity of Glu53 mutant human lysozyme significantly decreased as the reaction time was prolonged (1.0 h, 1.0%; 2.0 h. 0.7%). Significant denaturation of the mutant enzyme was unlikely to occur within the reaction time, judging from the expected high thermostability under this pH condition [14]. Also, dissociation constants for either N-acetylchitotriose or N-acetylchitohexaose in solution were essentially kept between wild-type and Glu53 mutant enzymes [39]. Therefore, the above results might suggest that a half-covalent type intermediate was produced during the reaction in the case of Glu53 mutant, which would take much longer time to be cleaved as compared to the detachment of the intermediate simply stabilized by electrostatic interaction between the negative charge of Asp53 side chain and oxocarbonium ion intermediate of the substrate. The optimal pH of Glu53 mutant against M. luteus cell substrate was similar to the wild-type case, but the more reduced relative activity at alkaline side was observed as compared with wild-type enzyme [39]. On the other hand, Asp35 mutant exhibited approximately 0.003% relative Vmax/Km value to that of wild-type enzyme against M. luteus cell substrate at 30°C, pH 6.4.The optimal pH for this substrate was not significantly changed from that of wild-type enzyme, but the reduction of relative activity at both acidic and alkaline sides was more prominent in this case [38]. The specific activity of Gln35 mutant of a homologous enzyme, chicken lysozyme, was reported to be 0.1 ± 0.1 % relative to that of wild-type (Glu35) enzyme with the same substrate under the similar reaction conditions [40]. The detailed structures of both Asp35 [38] and Glu53 [41] mutants were determined by X-ray crystallography and no significant changes in the overall structures judging from the positional changes in equivalent α-carbon atoms were observed as compared to wild-type human lysozyme. Taken together, the above results evidently demonstrated that the precise positioning of two side-chain carboxylate groups of Glu35 and Asp53 was pivotal to the efficient catalysis by human lysozyme.
The van der Waals contact with the side-chain of Trp109 was found to be an important factor to determine the precise positioning of the side-chain of Glu35. Distinct movements ranging from 0.67 Å to 1.0 Å of equivalent α-carbon atoms in the region of the residues 110 to 118, 110 to 120, 104 to 115 and 100 to 111 were observed for Asp35 mutant, Ala35 mutant, Phe109 mutant and Ala109 mutant, respectively [38]. In all cases, root-mean square differences in equivalent α-carbon atoms of the entire molecules were less than the estimated coordinate errors (0.2 Å). The local movements introduced by the mutations of 109th residue resulted in a large decrease (13% and 16% of wild-type enzyme with regard to Phe109 and Ala109 mutants, respectively) in the Vmax value against M. luteus cell substrate, while maintaining the affinity for it [38]. This suggested that the van der Waals contact effectively contributed to fix the side-chain carboxylate group of Glu35 in wild-type enzyme to a maximally efficient position for protonation of the glycosidic oxygen atom. A similar reduction in the catalytic activity against both M. luteus cell and glycol chitin substrates without significant alteration of the affinity to N-acetylchitotriose ligand was reported for human lysozyme with Val110 to Pro mutation [42]. This residue is next to Trp109 and also locates at around subsite D (Figure 1B). It is reasonable to assume that the perturbation of the position of Glu35 carboxylate group introduced by the mutation is the cause of catalytic activity reduction in this case again, since no significant change in overall conformation was detected using circular dichroism spectra.
Aromatic side-chain group of Tyr63 is essential for the recognition of carbohydrate substrates at subsite B:
Human lysozyme has a deep cleft with six subsites designated A to F, each of which can accommodate either an N-acetylglucosamine residue or an N-acetylmuramicacid residue (subsites B, D and F only) (Figures 1B, 1D). To date, X-ray structures of the complexes with chitin-type oligosaccharides are available for human lysozyme. An X-ray structure of human lysozyme co-crystallized with N-acetylchitohexaose at pH 4.0 revealed that the hexasaccharide was cleaved into tertrasaccharide and disaccharide, which occupied subsites A to D and a position close to subsites E and F proposed on the basis of model building for chicken lysozyme, respectively [43]. The N-acetylchitotriose is thought to bind human lysozyme in two alternative modes, subsites A to C and subsites B to D by an NMR analysis. In either recognition modes, Tyr63 at subsite B is considered to play a central role for the substrate recognition [44].
In order to dissect the functional role of structural elements of Tyr 63 side-chain in the catalytic action of human lysozyme, Tyr63 was replaced with Phe, Trp, Leu or Ala [45]. X-ray crystallographic analysis showed that no appreciable change except for the difference in side chain structure of 63rd residue was introduced by the above mutations. While the lytic activity against M. luteus cell substrate was maintained in either mutation, relative activity toward non-charged chitin-type substrates greatly depended on whether the 63rd residue was an aromatic amino acid or not. This was also confirmed in the inhibition experiments of the lytic activity by N-acetylchitotriose. The effect of electrostatic interaction on the catalytic activity was further shown by the comparison of the relative activity to wild-type enzyme against non-charged soluble chitin-type substrate, ethyleneglycol chitin, and that against negatively charged soluble chitin-type substrate, carboxymethyl chitin [37]. Leu63 mutant showed 7 ± 4% relative activity against ethyleneglycol chitin, which was significantly less than the relative activity of 36 ± 5% against carboxymethyl chitin. The importance of the presence of an aromatic residue at 63rd position for chitin-type substrate was definitely demonstrated by the comparison of catalytic activity against p-nitrophenyl N-acetylchitopentaoside substrate [45]. The quantitative catalytic efficiency parameter, kcat/Km, under pH 5.0, 37°C of wild-type human lysozyme (Tyr63) was slightly altered from 250 ± 80 M-1s-1 to 130 ± 30 M-1s-1 and 70 ± 16 M-1s-1 with Phe63 mutant and Trp63 mutant, respectively, which corresponded to G values in catalysis of 0.4 ± 0.4 kcalmol-1 and 0.8 ± 0.3 kcalmol-1. In contrast, Leu63 mutant and Ala63 mutant exhibited the kcat/Km values of 4.3 ± 2.0 M-1s-1 and 6.4 ± 4.9 M-1s-1, respectively, which corresponded to the G values of 2.6 ± 0.6 kcalmol-1 and 2.5 ± 0.8 kcalmol-1. Further, the preference in cleavage pattern of p-nitrophenyl N-acetylchitopentaoside also significantly shifted from 4:1 to 3:2 by the loss of aromatic side-chain of 63rd residue, which indicated that a large decrease in relative total affinity of subsites A, B, C and D to that of subsites E and F against this substrate.
X-ray structural analysis of site-specifically conjugated derivatives of disaccharides:
In order to further examine the functional role of the interactions between the carbohydrate residue bound at subsite B and Tyr63 residue in the ligand recognition event in human lysozyme, human lysozyme derivatives site-specifically conjugated with 2’, 3’-epoxypropyl β-glycoside derivatives of three kinds of disaccharides, namely N-acetylchitobiose (GlcNAc-β1, 4-GlcNAc), N-acetyl lactosamine (Gal-β1, 4-GlcNAc) and β-1, 4 linked mannosyl N-acetylglucosamine (Man- β1, 4-GlcNAc), were synthesized. The detailed structures of these three kinds of site-specifically labeled human lysozymes were analyzed by X-ray crystallography [46,47]. All conjugations were conducted by the ester linkage formation with the side-chain carboxylate group of Asp53. The geometry of the glycosidic linkage in the N-acetylchitobiose moiety of the conjugated complex were essentially the same as observed for the unconjugated complex with β-1, 4 linked N-acetylglucosamine oligomers [46]. The binding mode of N-acetylglucosamine residue at subsite C was totally maintained, therefore the site-specific labeling reactions were considered to proceed via the recognition of N-acetylglucosamine residue at subsite C. The inactivation of lytic activity against M. luteus cells substrate occurred by the conjugation. The half lifetime of residual lytic activity of human lysozyme in the presence of 2’, 3’-epoxypropyl β-glycosides of Gal-β1, 4-GlcNAc and Man- β1,4-GlcNAc was prolonged by approximately twice as much compared with the corresponding derivative of GlcNAc-β1, 4-GlcNAc [47]. This clearly presented the weaker affinity of the former two derivatives than the latter one, which should be ascribed to the difference in the affinity at subsite B.
The comparison of three-dimensional structures revealed the origin of substrate recognition specificity at subsite B. The apolar face of N-acetylglucosamine residue at subsite B in N-acetylchitobiose conjugated human lysozyme showed a stacking geometry almost parallel to the phenolic side-chain group of Tyr63 (Figure 2A) making 5 possible CH-π interactions at subsite B [48]. In contrast, the parallelism between the galactose residue at subsite B and the phenolic side-chain group of Tyr63 in the N-acetyllactosamine conjugated human lysozyme was significantly worse (Figure 2B) and the number of possible CH-π interactions at subsite B decreased to 4 [48]. Cooperatively, the counterpart of possible direct hydrogen bond concerning Asp102 side-chain was altered from 6-OH group to axial 4-OH group of galactose residue [46]. Further, the three-dimensional structures of Leu63 mutant human lysozyme in conjugation with 2’, 3’-epoxypropyl β-glycoside of N-acetylchitobiose was also determined [49]. By the abolishment of CH-π interactions between the saccharide residue at subsite B and the side-chain group of 63rd residue due to the replacement of Tyr63 with Leu, the N-acetylglucosamine residue at subsite B in the Leu63 mutant conjugate significantly moved away from the side-chain of Leu63 as compared to the relative positions of the corresponding N-acetylglucosamine residue in the wild-type human lysozyme. The above results suggested that the cooperativity of CH-π interactions and conventional hydrogen bonding interactions are an origin of the substrate recognition specificity at subsite B in human lysozyme, and clearly demonstrated the direct contribution of aromatic side-chain group to the recognition process.
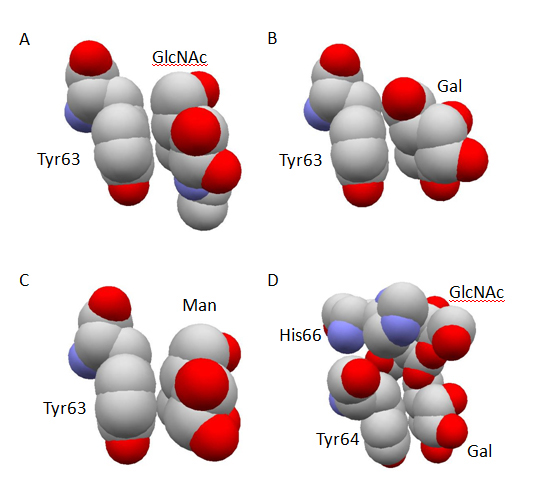
Figure 2. Stacking geometry between aromatic amino-acid residue and carbohydrate residue. The atomic coordinate data were obtained from PDB. Only relevant residues of space-filling models are shown in atom colors (red, oxygen; gray, carbon; blue, nitrogen) and labeled. Panel A, human lysozyme conjugated with 2, 3’-epoxypropyl β-glycoside of GlcNAc-β1, 4-GlcNAc (PDB ID: 1rey); Panel B, human lysozyme conjugated with 2, 3’-epoxypropyl β-glycoside of Gal-β 1, 4-GlcNAc (PDB ID: 1rez); Panel C, human lysozyme conjugated with 2, 3’-epoxypropyl β-glycoside of Man-β 1, 4-GlcNAc (PDB ID: 1rem); Panel D, glutaraldehyde-crosslinked WGA-3 complexed with GlcNAc-β 1, 6-Gal (PDB ID: 1k7t).
In spite of the reduced affinity suggested by the lower reaction rate in the conjugation with Man-β1, 4-GlcNAc as compared to that with GlcNAc-β1, 4-GlcNAc, the parallelism of the mannose residue with Tyr63 side-chain was maintained [47] (Figure 2C), and the total number of possible CH-π interactions at subsite B was not changed [48]. The original possible direct hydrogen bond between Gln104 side-chain and carbonyl oxygen atom of the N-acetylglucosamine residue at subsite B was lost, but Gln104 found axial 2-OH group of the mannose residue as the new counterpart of hydrogen bond. Therefore, the weaker affinity of Man-β1, 4-GlcNAc as compared with N-acetylchitobiose should be ascribed to factors other than the total number of possible CH-π interactions and direct hydrogen bonding interactions, which still remain to be clarified.
Importance of positively charged side-chain of Arg115 for the affinity around subsites E and F:
It has been suggested that the binding of substrate at subsites E and F of the catalytic cleft in chicken-type lysozyme had two distinct possibilities of recognition by conformational energy calculations [50]. One is the productive binding mode involving “right side” region of the cleft and the other is the non-productive binding mode involving “left side” region of the cleft. The latter binding modes are reported as the mode in charge of the initial binding of substrate in chicken lysozyme using site-directed mutagenesis studies [51]. In human lysozyme, the corresponding regions responsible for the former and the latter binding modes includes Arg115 and Trp34 residues, and Tyr45, Asn46 and Ala47 residues, respectively (Figure 1B). In the initial model building study of chicken lysozyme in complex with N-acetylchitohexaose, Arg114 in chicken lysozyme, which corresponds to Arg115 in human lysozyme, was considered to form double hydrogen bonds with the N-acetylglucosamine residue at subsite F [30].
For the purpose of surveying the role of Arg115 in human lysozyme, this residue was replaced with Lys, His, Gln or Glu, and the effect on their enzymatic functions was assessed by examining the catalytic activity against M. luteus cell and ethyleneglycol chitin substrates [52]. Neither the apparent Km value nor Vmax value against M. luteus cells were significantly affected by the replacement of Arg115 with Lys, which revealed the specific hydrogen bonding interactions between Arg115 and N-acetylglucosamine residue at subsite F were actually not valid in the hydrolysis of this substrate. The activities of the above mentioned mutant human lysozymes against ethyleneglycol chitin were mostly dependent on the charge state of 115th residue. In contrast to the full retention of activity (90 ± 2% ~ 105 ± 4%) under the condition that the side-chain of 115th residue remains positively charged state, the relative activity to wild-type enzyme became significantly lower (58 ± 5% ~ 78 ± 2%) under the condition that the net charge of 115th residue changed to neutral, and further decreased (49 ± 3% ~ 58 ± 3%) under the condition that the side-chain was negatively charged. These results clearly suggest that Arg115 of human lysozyme was important not for providing the hydrogen bonding interactions with the carbohydrate residue at subsite F, but for possessing a positive charge to maintain the local structure essential for the recognition of substrates at around subsite F.
The direct structural evidence about this hypothesis was obtained from X-ray structural analysis of His115 and the Glu115 mutants crystallized at pH 4.5 [53]. Under this pH condition, the His115 residue should be protonated to have a positive charge and the side-chain carboxylate group of Glu115 would be dissociated to have a negative charge. The main-chain structure of His115 mutant was identical to that of wild-type human lysozyme, whereas the main-chain position of Glu115 mutant presented a large difference from that of wild-type in the region of 100th to 130th residues constituting the “right side” of catalytic cleft at subsites E and F in human lysozyme. This structural deformation introduced by the Glu115 mutation was considered to be derived from the difference in the interaction mode of the side group of 115th residue and the spatially adjacent side-chain group of Trp34.The positively charged imidazolyl group of His115 mutant shared the same plane as the guanidinium group of Arg115 in wild-type human lysozyme so that they maintain a possible cation-π interaction. On the other hand, the carboxylate group of Glu115 moved away from the face of indole ring of Trp34 as if they avoid proximity of the negative charge to the π-electrons.
Chicken-type lysozymes including human lysozyme catalyze not only the hydrolysis of substrates but also transglycosylation reaction involving the reaction products of hydrolysis.The transglycosylation reaction was also used for the enzymatic synthesis of p-nitrophenyl 35-O-β-N-acetyl glucosaminyl-α-maltopentaoside by chicken lysozyme [54]. The transglycosylation reaction with human lysozyme was originally evidenced by the observation that the net amounts of N-acetylchitotriose and N-acetylchitobiose in the case of 3:2 split as well as those of N-acetylchitotetraose and N-acetylglucosamine in the case of 4:1 split were substantially different from each other during the experimental time-course measurements of the amounts of reaction products using a starting substrate, N-acetylchitopentaose [55]. This observation was further supported by virtual computer simulations supposing the occurrence of transglycosylation reaction. Subsites E and F in the catalytic cleft are responsible for the transglycosylation, since the transglycosylation reaction occurs when the oligosaccharide product binds to these subsites and reacts with the oxocarbonium intermediate as a substitute for a water molecule in the hydrolysis reaction. The effect of mutations at 115th residue on the transglycosylation reaction was demonstrated by the comparison of the reaction time courses of wild-type and 115th mutant human lysozymes for N-acetylchitopentaose and N-acetylchitohexaose substrates [52]. In either case, the profile of time courses concerning the amounts of oligosaccharide reaction products at pH 5.0 entirely depended on the charge state of 115th residue’s side-chain, which reflected the structural change mentioned above.
High structural flexibility around “right side” lobe of subsites A and B and “left side” lobe of subsite A in expressing the catalytic activity:
Among the potentially flexible regions consisting of coiled-loop structures in chicken lysozyme, the largest positional fluctuation of main-chain atoms was observed for the region consisting of the residues from 102 to 104 in the molecular dynamics simulations [56].This was supported by the comparison of two independent molecules contained in the unit cells of monoclinic crystal of chicken lysozyme, in which 100th to 104th residues exhibited significant structural variations between them [57]. Pro103 residue in human lysozyme has been reported to be unimportant for the thermodynamic stability by the replacement with Gly [14].
The region from 100th to 104th residues forms the “right side” lobe of subsites A and B in the catalytic cleft (Figure 1B), and Asp102 residue in human lysozyme is thought to contribute to the specific recognition of the N-acetylglucosamine residues occupied at subsites A and B by making the hydrogen bonding interactions with the carbohydrate moieties via its side-chain carboxylate group [43 ] . In order to examine the functional importance of structural integrity in this region, Asp102 in human lysozyme was replaced with Asn or Glu.In addition, a deletion mutant Pro103 was created. All these mutants showed a considerable residual activity compared with wild-type enzyme against M. luteus substrate. The catalytic efficiency parameter, Vmax/Km values, for Asn 102, Glu102 and Pro103 mutants were 76%, 40% and 55% of that of wild-type human lysozyme (M. Muraki, unpublished result). The variableness of the corresponding residue in chicken lysozyme (Asp101) was also implicated by the replacement of Asp101 with Gly in turkey lysozyme. In both enzymes, N-acetylchitotriose bound at subsites A, B and C in essentially the same manner, in spite of the difference in the side-chain group of 101st< residue [58]. The structural flexibility around subsite A in human lysozyme was also suggested by the insertion of several sequences ranging from 4 to 12 residues in length containing Arg-Gly-Ser sequence into the position between Val74 and Asn75 residues [59]. These residues are nearly located at the “left side” lobe of subsite A in the substrate binding cleft (Figure 1B) and were involved in a coiled-loop region (Figure 1A). All mutants containing the insertion sequences retained nearly identical relative lytic activity (85 ~ 109 %) against M. luteus cell substrate as compared to wild-type enzyme.
Increase and decrease of net molecular surface charge alters pH and ionic strength dependencies of lytic activity against M. luteus cell substrate:
As mentioned above, the electrostatic interactions play an essential role for the recognition of M. luteus cell substrate by human lysozyme. The importance of basic amino acid residues on the surface of lysozyme in the lytic activity against this substrate was originally pointed out by the acetylation of chicken lysozyme [60]. A computer-program assisted calculation of electrostatic potentials in whole human lysozyme molecule revealed the molecular surface of this protein was mostly positively charged except in the vicinity of catalytic residues (Figures 1C, 1D).
Among various chicken-type lysozymes of different species origin, a number of discrepancies in amino-acid residues have been identified in spite of the well-conserved molecular size. For example, there are 14, 41 and 52 aa residues replacement including deletions from 130 aa residues of human lysozyme with regard to baboon, cow and chicken lysozymes, respectively. Using the information on these differences in amino-acid residues and that on the three-dimensional structure, several mutant human lysozymes, which had either one or two units of increased and decrease in positive charge on the molecular surface, were created by site-directed mutagenesis to examine their effects on the lytic activity against M. luteus cell substrate [61]. Overall, the increase and decrease in the positive charge shifted the optimal ionic-strength under the optimal pH condition for wild-type enzyme to higher and lower directions, respectively. Two mutant enzymes with double amino-acid residues replacements were also created. One had an increased positive charge by two units due to the replacements of Val74 to Arg and Gln126 to Arg, and the other a decreased positive charge by two units due to the replacements of Arg 41 to Gln and Arg101 to Ser. Even with only two units change in positive charge, these mutants presented totally different pH- lytic activity profiles examined under several ionic-strength conditions from those of wild-type human lysozyme. The results indicated that the (Arg74+Arg126) mutant showed significantly higher lytic activity than wild-type enzyme under the conditions of higher pH and higher ionic-strength. In contrast, the (Gln41+Ser101) mutant became a better catalyst under the conditions of lower pH and lower ionic-strength as compared with wild-type human lysozyme. A similar observation was subsequently reported for the mutant possessing an N-terminal additional lysine residue [62]. These observations can be explained by the presence of optimal electrostatic interactions between the enzyme and M. luteus cell substrate for expressing maximal lytic activity.
Further, the co-existence of poly L-lysine hydrochloride considerably affect the lytic activity in a dose-responded manner depending on the net surface positive charge of wild-type and the mutant human lysozymes described above, for which no effect on the lytic activity was observed by addition of the same amount of monomeric L-lysine hydrochloride [61]. This suggested that the lytic activity can be controlled by the presence of polyelectrolytes, which also means that it would be possible to enhance the lytic activity in the presence of a polyelectrolyte by engineering the surface charge of human lysozyme. In fact, this possibility proved to be true in the case that a less positively charged mutant containing double mutations of Arg101 to Asp and Arg115 to His exhibited 3-fold, 7-fold and 43-fold increased half maximal inhibitory concentration values in the bacteriolytic activity against M. luteus cell substrate for three kinds of poly-anionic electrolytes, alginate, mucin and DNA, respectively [5]. From a medical point of view, it is noteworthy that this mutant retained 98.5% of relative activity against a clinically important gram-negative bacterium, P. aeruginosa. Alginate is a major mucopolysaccharide component consisting of biofilms, which is closely relevant to nosocomical infection caused by this pathogenic bacterium. A detailed three-dimensional structure of the above surface electrostatic-potential reshaped double mutant was determined by X-ray crystallography [63]. Electrostatic potential analysis based on the resolved structure showed a significantly expanded negative potential field restricted to in and around the substrate binding cleft.
Alteration of catalytic and ligand binding properties using site-specific chemical modifications:
Chemical modification can be a powerful method to alter functional properties of enzyme molecules.Site-specific intermolecular cross-linking was achieved by the introduction of a free sulfhydryl group on the surface of human lysozyme and the subsequent reaction with a compound containing two maleimide groups [64]. One of the two alternative residues, either Arg41 or Ala73, was replaced with Cys, then reacted with N, N’-bis (3-maleimide propionyl)-2-hydroxy-1, 3-propandiamine. Three kinds of site-specifically crosslinked human lysozyme dimers between two Cys41 mutants, one Cys41 mutant and one Cys73 mutant, and two Cys73 mutants were created.Among them, only Cys41-Cys41 dimer exhibited the altered enzymatic property from monomeric wild-type enzyme and other dimers. The Cys41-Cys41 dimer showed substantially reduced (38 ± 3% of wild-type) activity against a chitin-type oligomer substrate, p-nitrophenyl N-acetylchitopentaoside, while maintaining (87 ± 5% of wild type) the activity against a chitin-type high-polymer substrate, ethyleneglycol chitin. Consequently, the ratio of the relative activity of the Cys41-Cys41 dimer against the latter substrate over that against the former substrate became 2.3 ± 0.3. In contrast, the ratio remained at 1.0 ± 0.1 and 1.1 ± 0.1 with regard to the Cys41-Cys73 dimer and the Cys73-Cys73 dimer, respectively.Interestingly, the activity of the Cys41-Cys41 dimer against the oligomer substrate was enhanced as the ionic-strength of the reaction medium increased in spite of non-charged property of the substrate, whereas those of the other dimers were essentially constant [64]. The above results may be explained by the spatial proximity of two catalytic clefts of the monomer parts in the Cys41-Cys41 dimer. In the hydrolysis of p-nitrophenyl N-acetylchitopentaoside, the resulting products, such as N-acetylchitotriose and N-acetylchitotetraose, were no longer a good substrate, but were effective inhibitors, since these products still retain the similar affinity for the catalytic cleft to that of the original substrate. The difference in ionic-strength of reaction medium can alter the degree of spatial proximity of the two catalytic clefts in the dimer, which would affect the efficiency of the inhibition. In spite of the presence of two carbohydrate binding sites in the human lysozyme dimers, none of them showed hemagglutination activity toward blood red cells (M. Muraki, unpublished result). This observation was consistent with the result that only the oligomers bigger than the trimer of non-specifically cross-linked chicken lysozyme behaved as lectin-like molecules [65].
Another interesting result was obtained in the site-specific conjugation of wild-type and Glu102 mutant human lysozymes with 2’, 3’-epoxypropyl β-glycoside of N-acetyllactosamine [66,67]. Dual labeling of the active site cleft of human lysozyme occurred via the first ligand assisted recognition of the second ligand. In both cases, the first ligand part and the second part were covalently attached to the side-chain carboxylate group of Asp53 residue and that of Glu35 residue, respectively. The second ligand part locates in parallel to the first ligand part and many close atomic contacts mediated by hydrogen bonding network and van der Waals interactions were observed between the two parts, which were firmly supported by a number of possible hydrogen bonding and CH-π interactions between the first ligand part and the protein part.
Carbohydrate binding proteins with therapeutic potentials via recognition of various types of pathogenic cells and immunological cells
Lectin molecules composed of hevein domains were isolated from a variety of plant organs including germ (e.g. WGA from Triticum aestivum) [68], root (e.g. PWM from Phytolacca americana) [69], rhizome (e.g. UDA from Uritica dioica) [70], bark (e.g. Ee-CBP from Euonymus europaeus L.) [71], latex (e.g. Hevein from Hevea brasiliensis) [72] and seed (e.g. Ac-amp2 from Amaranthus caudatus) [73]. These protein molecules specifically recognize N-acetylglucosamine oligomer structures found in the surfaces of immunological cells and pathogenic microbes in common. In this review, the author focuses on studies concerning three kinds of representative molecules among them, namely WGA, UDA and Ac-amp2.
WGA was found as a component in wheat-germ lipase preparations, which selectively agglutinated malignant cells, in 1963 [74]. The cancer cell specificity displayed by WGA was further characterized to be derived from the cell-surface changes during the transformation of normal cells to malignant cells [75]. Then, the resistance of cancer cells against WGA was found to be closely related to the metastatic character of the tumor cell lines derived from melanoma [76] and sarcoma [77]. Due to this cell-discriminating feature based on the carbohydrate binding specificity, WGA has been subjected to the experiments aimed at many biomedical applications using nanoparticles. This type of application includes drug-targeting either to brain via intranasal administration [78] and photoluminescence imaging analysis of cancer cells [79]. WGA shows not only specific binding but also cytotoxic activity against various cancer cells including pancreatic cancer [80], colon cancer [81] and leukemia cells [82]. The cytotoxity was suggested to be caused by the induction of apoptosis associated with G2/M phase cell cycle arrest via a mitochondrial pathway independent of Fas receptor and caspase 3 [83,84]. Another WGA function related to therapeutic applications includes inhibitory effect to the binding of pathogenic IgG auto-antibodies against desmoglein 1 in Pemphigus Foliaceus patients [85].
WGA is a dietary molecule contained in wheat flour as a component of wheat germ and can significantly affect human health via influence on the immunological response of gastrointestinal epithelium. A biophysical study suggested that this highly stable protein molecule was partially unfolded at low pH in stomach but attained its fully active conformation again once it reaches the intestine [86]. This property makes WGA attractive as a carrier of peroral drug delivery, since it binds specifically to sugars expressed by gastrointestinal epithelial cells [87]. In this connection, stimulation of the synthesis of pro-inflammatory cytokines in human peripheral mononuclear cells by WGA at nanomolar concentration is also intriguing [88].
UDA was found as a plant lectin showing interferon γ -inducing activity toward human lymphocytes in 1984 [70]. UDA exists as a mixture of six isolectins, which all share the identical molecular size, carbohydrate-binding specificity and agglutination properties. All isolectins showed identical human interferon γ -inducing activity, which should lead to enhancement of the antiviral function of lymphocytes [89]. In fact, it was shown that this lectin molecule possesses strong antiviral activity against many serious pathogenic viruses including human immunodeficiency virus [90], severe acute respiratory syndrome associated corona virus [91] and dengue virus [92]. One of the most striking differences of UDA from other plant lectins is that this T-lymphocyte mitogenic lectin behaves as a superantigen specific to Vβ 8.3+ T-cell population by using MHC class I antigen and MHC class II antigen as its receptors [93]. It was demonstrated that the deletion of Vβ 8.3+ T-cells using UDA prevented the development of the systemic lupus erythematosus-like disease in an autoimmune model MRL lpr/lpr mice, which has a homozygous mutation concerning Fas receptor gene [94]. Interestingly, the binding of UDA toward MHC class II was shown to be completely inhibited by its specific carbohydrate ligand, N-acetylchitotriose, in a dose-dependent manner [95]. UDA has also been examined for a safe anti-prostatic hypertrophy agent contained in stinging nettle extracts as a main therapeutic component [96].
Ac-amp2 was first isolated in 1992 as an anti-microbial protein, which exhibited strong growth inhibition activity against plant pathogenic fungi such as Botrytis cinerea and Fusarium culmorum [73]. This chitin binding protein shares the structural homogeneity to hevein in the alignment of primary sequences assisted by the invariant positioning of its 6 Cys residues, and therefore considered to be a truncated hevein domain. This molecule also showed the inhibitory activity against some Gram-positive bacteria including Bacillus megaterium, but not against Gram-negative bacteria including E. coli. Because of its small molecular size, Ac-amp2 is often classified as an anti-microbial peptide [97]. This class of proteins is a promising candidate for novel potential therapeutics for treatment of immune-suppressed patients with systemic fungal infections, since anti-microbial peptides are produced by a wide range of organisms including human and play a significant role in protecting the host against pathogens [98]. Of note, Ac-amp2 did not show the cytotoxic effect against either human umbilical endothelial cells or human skin-muscle fibroblast cells at concentrations up to 500 μg/ml [73].
A tiny globular domain devoid of extended secondary structures: tandem repeat and truncation
A fundamental structure of hevein domain is a small globule composed of 43 aa residues containing four disulfide-bridges as is the case for human lysozyme. However, the folded structure of this domain cannot afford to contain extended secondary structures due to its globular shape and tiny size. The chitin binding lectins belonging to this group consist of single or multiple tandem repeat of hevein-type domains in their primary structures. So far, the three-dimensional structures of WGA, UDA and Ac-amp2 have been revealed using X-ray crystallography and NMR analysis. The crystal structures of all WGA isolectins (WGA-1, 2 and 3) have been determined by X-ray crystallography [99, 100].It revealed that WGA is a dimeric protein with dimensions of 40 x 40 x 70 Å consisting of two identical protomers of 171 aa residues polypeptide, each of which are composed of four hevein-type domains called domain A, B, C and D from its N-terminal end (Figure 3A). On the other hand, UDA is a monomeric protein consisting of two tandem repeat of hevein domains with a total number of 89 aa residues including a hinge region composed of 4 aa residues between the two domains (Figure 3B). The detailed crystal structures of two isolectins (UDA-I and VI as identified by mass spectrometry analysis) have been elucidated to date [101,102]. Although the main-chain structure and the four disulfide bridges of the two hevein-type domains in UDA can be well superimposed to each other, the overall spatial orientations of the two domains were shown to be totally different from those of the first and second domains of WGA [101]. Ac-amp2 is a C-terminal truncated version of hevein-type domain containing three disulfide bridges within the compact structure of 30 aa residues (Figure 3C) [73]. The three-dimensional solution structure delineated by NMR analysis displayed strong homology to the domain B of WGA concerning the main-chain structure [103].
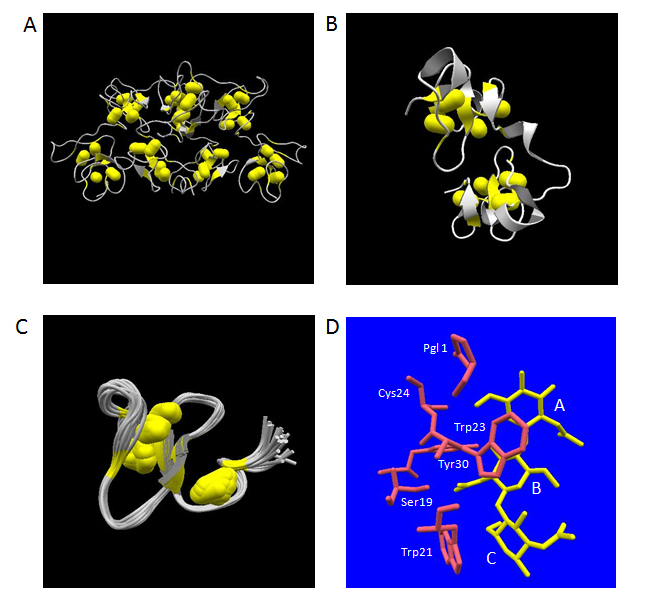
Figure 3. Three-dimensional structures of plant lectins composed of hevein-type domains. The atomic coordinate data were obtained from PDB. Secondary structures and disulfide-bonds are shown in ribbons and bales, respectively. Panel A, WGA-3 (PDB ID: 1wgt, X-ray model); Panel B, UDA-VI (PDB ID: 1ehh, X-ray model); Panel C, Ac-amp2 (PDB ID; 1mmc, 26 NMR models overlapped). Panel D, the structures of bound N-acetylchitotriose (in yellow) and aa residues mentioned in the text (in pink) are shown. Relevant aa residues’ names in UDA-VI are labelled. The labels, A, B and C, indicate the three N-acetylglucosamine residues starting from the non-reducing end.
Isolation from natural resources, recombinant production and chemical synthesis
To date, the samples for biochemical analysis of lectins composed of hevein-type domains have been most conveniently isolated from the plant tissues that contain the objective lectins. This is probably because the raw materials contained in plant origin are often readily available in large quantities unlike human proteins. A frequently used method to isolate the lectins from the crude aqueous extract of plant materials is affinity chromatography as exemplified by the use of ovomucoid sepharose for WGA [104] or chitin powder for UDA [70]. It was necessary to conduct further fractionation using either ion-exchange chromatography or reversed-phase chromatography for obtaining pure isolectins of WGA and UDA, since the affinity purified materials were a mixture of the isolectins.In the case of Ac-amp2, it was possible to obtain a pure sample directly using two steps of anion-exchange and cation-exchange chromatography without the necessity of affinity purification step by virtue of its highly basic character (pI > 10) [73].
A preparation method for recombinant WGA-2 was developed using a secretory expression in S. cerevisiae [105]. In this system, it was necessary to use a special strain KS58-2Ddel holding the ssl1 mutation, which causes a supersecretion phenotype of human lysozyme, for efficient (150~200 μg/l) secretion of functional WGA-2.This secretion amount was further increased 3 to 4 fold by the co-expression of protein disulfide isomerase gene derived from S. cerevisiae. In contrast, the use of the parent wild-type strain KK4 reduced the secretion amount to 1/20 value, even though the same chicken lysozyme-derived signal sequence was used. Another yeast secretion system consisted of a combination of S. cerevisiae α-factor prepro signal sequence and P. pastoris GS115 host strain, which was successful in the secretion of human lysozyme, failed to secrete functional WGA (M. Muraki, unpublished result). A highly homologous protein, barley lectin, showing 95% sequence identity to WGA was over-expressed in E. coli [106]. The intracellularly accumulated protein as an inclusion body was successfully renatured using a redox buffer system consisting of oxidized and reduced glutathione to give a final yield of 400~500 μg of the purified functional protein from 1g of E. coli cells. The obtained recombinant barley lectin was crystallized. The refolding strategy of inactive protein using the glutathione redox buffer system was also applicable for single hevein-type domain proteins, WIN2 [107] and Ac-amp2 [108]. In this case, the peptide possessing each corresponding primary sequence was chemically prepared using 9-fluorenylmethyloxycarbonyl-based peptide synthesis and then refolded to the native three-dimensional structure. The use of acetoamidomethyl protection group for Cys combined with the deprotection by mercury (II) acetate and the purification by reversed-phase column chromatography were essential for the preparation of the functional product. Another possible method for the preparation of a single hevein-type domain lectin was reported for WAMP-1a, which used the expression of fusion protein with thioredoxin in E. coli [109]. The hevein-type domain containing five disulfide-bridges was successfully purified by cyanogen bromide cleavage and subsequent fractionation using a reversed-phase chromatography to give the functional product in a final yield of ~8 mg per liter of the bacterial culture.
Crystallography and solution studies concerning the carbohydrate ligand recognition mechanism
After the pioneering study on the X-ray crystallographic structural analysis of WGA in complex with N-acetylchitobiose, N-acetylchitotetraose and N-acetylneuraminicacid [110], structural elucidations of the carbohydrate ligand recognition mechanism about the lectin molecules composed of hevein-type domains have been conducted mainly using X-ray crystallography and NMR analysis.
Among this type of lectins, the most extensive studies have been performed with regard to WGA isolectins.The complex structures of N-acetylneuraminyl lactose (NeuNAc-α2, 3-Gal-β1, 4-Glc) with WGA-1 and WGA-2 (at 2.2 Å resolution) [111], that of glycopeptides containing T5-sialotetrasaccharide (NeuNAc-α2, 3-Gal-β1, 3-GalNAc-α2, 6-NeuNAc) from glycophorin A of erythrocytes transmembrane with WGA-1 (at 2.0 Å resolution) [112] and that of GlcNAc-β1, 6-Gal, GlcNAc-β1, 4-GlcNAc and GlcNAc-β1, 6-Gal-β1, 4-Glc with glutaraldehyde-crosslinked WGA-3 (at 2.4, 2.2 and 2.2 Å resolution, respectively) [113] have been determined by X-ray crystallography. All the bound carbohydrate moieties were almost exclusively found in the contact region between domain B and domain C belonging to each different protomer composed of four hevein-type domains. Recently, all theoretically predicted eight carbohydrate binding sites in WGA were revealed to be simultaneously functional by the crystallographic analysis of WGA-3 complexed with a synthetic divalent GlcNAc ligand [114].
It is noteworthy that the non-reducing end of GlcNAc in N-acetylchito-oligosaccharides occupied essentially the same location with N-acetylneuraminicacid residues in an orientation where the carbohydrate moiety always made three specific polar hydrogen bonding interactions. Those were the hydrogen bond between the carbonyl-oxygen atom of its N-acetoamide group and the OH-group of Ser62 in WGA domain B, that between its 3-OH group and the OH-group of Tyr73 in WGA domain B and that between the amido-nitrogen atom of its N-acetoamide group and the carboxylate-group of Glu115 in WGA domain C [115]. By adapting this orientation, the counterpart of hydrogen bond involving the OH-group of Ser114 was altered from the 4-OH group of N-acetylglucosamine residue to the carboxylate-group of N-acetylneuraminicacid residue. Another important aspect in the recognition of the carbohydrate ligands by WGA was the presence of three aromatic aa residues (Tyr64, Tyr66 and Tyr73 for WGA-1, Tyr64, His66 and Tyr73 for WGA-1 and WGA-3) in the binding site. As exemplified by the recognition of saccharide moiety in the subsite B of human lysozyme (Figures 2A-2C), an aromatic side-chain group can make a face-to- face stacking contact via many CH-π interactions, which substantially contributes to determine the orientation of the carbohydrate-ligand conformation by adjusting the glycosidic dihedral angles to make the best fit between the protein part and the ligand part. This strategy was found in essentially all structures of the complexes, and evidently presented by comparison of the recognition mode in glutaraldehyde-crosslinked WGA-3 complexed with GlcNAc-β1, 4-GlcNAc to that in the same protein complexed with GlcNAc-β1, 6-Gal [113]. As compared to the GlcNAc-β1, 4-GlcNAc, which represents a dissacharide part structure of chitin, GlcNAc-β1, 6-Gal has an extra methylene group adjacent to the glycosidic oxygen between two six atoms-membered rings. Nevertheless, the latter ligand exhibited no less strong affinity than the former ligand in solution. The crystallographic analysis revealed that this was accomplished by adjusting the conformation of the bonds between the two six atoms-membered carbohydrate rings for best fitting to the side-chains of Tyr64 and His66, which were involved in the recognition process of the carbohydrate ligand via face to face stacking contacts (Figure 2D).
With respect to UDA, a monomeric lectin composed of two hevein-type domains, the complex structures with two kinds of carbohydrate ligands, N-acetylchitotriose [101,116] and N-acetylchitotetraose [116], have been studied by X-ray crystallography at 1.9 Å and 1.4 Å, respectively. In the tetraose complex, the position of the first to the third N-acetylglucosamine residues from the non-reducing end overlapped with those in the triose complex, leaving the fourth N-acetylglucosamine residue protruding outside of the molecule without significant close contacts with the protein part. In the crystal structures of the UDA isolectin VI – N-acetylchitotriose complex, the ligands were found between a couple of UDA monomers as a state sandwiched by the two hevein-type domains at the N-terminal and the C-terminal sides. The same situation was observed in the more recently determined crystal structure of PL-D2 from pokeweed, another monomeric lectin composed of two hevein-type domains, in complex with N-acetylchitotriose [117]. Also in UDA, a similar strategy to that of WGA was observed for the recognition of the N-acetylchitotriose part of the ligands (Figure 3D). In this case, the specific polar hydrogen bonding interactions concerning the first N-acetylglucosamine residue from the non-reducing end were made through the main-chain carbonyl oxygen of Cys24 and the side-chain of N-terminal pyroglutamicacid group. Other hydrogen bonding interactions were also observed between the second N-acetylglucosamine residue of the ligand and the side-chain OH-groups of Ser19 and Tyr30. The face-to-face stacking contacts via CH-π interactions with the second and the third N-acetylglucosamine residues were accomplished by the side-chains of Trp23 and Trp21, respectively.
On the other hand, structural information on the molecular recognition mechanism of single hevein-type domain lectins has been mostly investigated using NMR analyses, which include the nuclear Overhauser effect spectroscopy experiments on hevein, pseudohevein and Ac-amp2 in complex with N-acetylchitotriose [118,119]. The results obtained from these experiments confirmed the important roles of three conserved aromatic aa residues and one serine residue in the ligand-binding site, which were revealed by the crystallographic experiments with WGA and UDA. The structurally corresponding residues to Tyr64, His66/Tyr66, Tyr73 and Ser62 in WGA were Trp21 (UDA)/Phe18 (Ac-amp2), Trp23 (UDA)/Tyr20 (Ac-amp2), Tyr30 (UDA)/Tyr27 (Ac-amp2) and Ser19 (UDA)/Ser16 (Ac-amp2), respectively. Therefore, the conclusions for the carbohydrate ligand recognition mechanism obtained in the X-ray structural determinations of the complexes were proved to be also applicable to the interactions in solution.
Isothermal titration calorimetry (ITC) experiments as well as NMR titration experiments are valuable methodologies for evaluating the strength and quantitative thermodynamic parameters of lectins – carbohydrate ligands interactions. The association constants for N-acetylchito-oligosaccharides determined using ITC experiments commonly increased as the length of the ligands became longer with regard to WGA [120], UDA [121] and hevein [122], which was also confirmed by NMR titration experiments [122]. However, ITC experiments under similar solvent conditions showed that the binding enthalpy of N-acetylchitobiose toward UDA was substantially smaller than WGA, and much closer to that of hevein.Further, the binding enthalpy values of N-acetylchito-oligosaccharides toward WGA were always more than twice those of toward UDA, whereas the binding free energy values were essentially identical to each other [121]. These results suggested that UDA behaves as a monomeric binder to the carbohydrate ligands in solution, despite the sandwiched structure observed in the crystal structures of UDA – N-acetylchitotriose/N-acetylchitotetraose complexes.N-acetyllactosamino-oligosaccharides containing the sequence of either GlcNAc-β1, 6-Gal or GlcNAc-β1, 6-GalNAc are known to exhibit no less weaker binding strength toward a WGA-immobilized column than N-acetylchito-oligosaccharides [123]. In fact, ITC experiments demonstrated the greater association constants for GlcNAc-β1, 6-Gal as compared with GlcNAc-β1, 4-GlcNAc [113]. Concerning the oligosaccharide ligands with a terminal sialic acid residue,NMR titration experiments showed that α-2,6-linked N-acetylneuraminyl lactose and the corresponding lactosamine binds several times more strongly to WGA than α-2,3-linked N-acetylneuraminyl lactose in solution, though detailed mechanism has not yet been clarified [124]. Apart from ITC and NMR titration experiments, two mutants of WGA-2 concerning the Tyr73 and Phe116, which both locate at the contact region between domain B and domain C of two protomers, have been characterized with respect to the roles of these residues in the binding of N-acetylchitotriose using equilibrium dialysis experiments cooperated by X-ray analyses of the three-dimensional structures without ligands [125]. This study showed the possibility of enhancing the affinity toward the saccharide ligand by introducing a new hydrogen bond into the protomers’ contact site using site-directed mutagenesis.
In order to assess the functional importance of two aromatic aa residues in Ac-amp2 responsible for stacking with apolar faces of carbohydrate residues in the recognition process of the ligands, several kinds of mutants including the alteration to non-natural aa residues were created and their affinity toward chitin column was evaluated [108]. Among them, some mutations of Phe18 to the residues possessing larger aromatic ring side-chains, such as Trp, β-(1-naphthyl)alanine and β-(2-naphthyl)alanine brought about the affinity enhancement toward chitin. In contrast, a great loss of the affinity occurred by the mutations to β-cyclohexylalanine and alanine residues, which resulted in the dearomatization and the loss of benzene ring in Phe18 side-chain, respectively. The enhanced binding strength presented by Trp18 and β-(2-naphthyl)alanine mutants as compared with wild-type Ac-amp2 were further quantitatively confirmed by NMR titration experiments using N-acetylchitotriose as the ligand [126]. The above results demonstrated that the ligand affinity in plant lectins composed of hevein-type domains could be artificially controlled by tuning the CH-π interactions between the stacking aromatic side-chains and the carbohydrate ligands.
Carbohydrate residues are generally considered to be hydrophilic as a whole molecule due to the presence of many hydroxyl groups. However, carbohydrate residues possess an apolar face comprised of axial CH-groups [127]. Recently, the attractive forces governed by CH-π interactions has been well recognized as playing an important role for the formation of functional structure and molecular recognition processes in a variety of biological macromolecules including recognition of carbohydrate molecules by proteins [128]. The net energy per single CH-π interaction is considered to be much less than an electrostatic interaction or a conventional hydrogen bonding interaction. However, it is effective even in a polar environment such as in water and may cooperate with each other by themselves and also with conventional polar hydrogen bonding interactions [129]. Carbohydrate ligands including N-acetylchito-oligosaccharides for plant lectins composed of hevein-type domains as well as chitin-type polysaccharide substrates for human lysozyme have no net electrostatic charges, which made the relative contribution of CH-π interactions within total binding energy become significant and therefore highlighted in the recognition event.
Close relation to many serious diseases in the human body
The encoding genes for human Fas receptor and human Fas ligand was identified in 1991 [130] and 1994 [131], respectively. Several cell-death inducing signaling systems conducted by the proteins called death ligands and death receptors cause apoptosis in the human body. Among them, human Fas ligand – human Fas receptor system represents one of the most important systems, which has a serious relation to the maintenance of human health by eliminating harmful cells to the body [132]. The apoptotic signaling process using this system is triggered by the specific binding of the extracellular domain of Fas ligand (hFasLECD) to that of Fas receptor (hFasRECD) existing on the surface of target cells. Physiologically, this apoptotic process is considered to be directly involved in the implementation of the removal of emerging cancerous cells, virus-infected cells and auto-reactive immune cells by natural killer cells and activated T-cells. Therefore, the uncontrolled state of this system can shortly lead to the onset of many serious diseases caused from the abnormal immune systems, which includes various types of cancers and autoimmune diseases such as rheumatoid arthritis (RA) in the cases of dysfunction, and graft-versus-host diseases (GVHD), fulminant hepatitis and spinal cord injury in the cases of excessive function. The defect of normal lymphocyte apoptosis caused by the mutations in human Fas ligand and Fas receptor genes is known to result in the disorders called autoimmune lymphoproliferative syndrome (ALPS) [133]. It is also noteworthy that hFasLECD exhibited the inhibitory effects on the angiogenesis both to the synovial tissue cells from the joints of RA patients in vitro and to the in vivo exposed Matrigels embedded in mice by inducing apoptosis of synoviocytes [134].
The binding of Fas ligand to Fas receptor also brings about non-apoptotic processes under the conditions that the normal apoptosis is inhibited for some reasons. For example, either in the caspase 8 deficient cells or in the case that the function of caspase 8 is inhibited in the cells, the NF-κB mediated cell activation via receptor interacting protein 1 was thought to take place by the Fas ligand binding to Fas receptor [135]. Fas ligand – Fas receptor signaling was also reported to induce the NF-κB activation mediated production of an inflammatory cytokine, interleukin 8 [136]. Continuous inflammation of tissues via constitutive NF-κB activation can lead to the onset of cancers, which is exemplified by the occurrence of hepatoma originated from chronic hepatitis [137]. From a clinical point of view, another important aspect of the Fas ligand – Fas receptor system is the possibility of usage as diagnostic biomarkers of diseases.Soluble Fas ligand (sFasL) and soluble Fas receptor (sFasR) in human serum have been extensively investigated as potentially useful biomarkers for a number of diseases including malignant tumors [138-140], cardiovascular diseases [141], viral infections [142-144], autoimmune diseases [145], central nervous system diseases [146], inflammatory diseases [147] and diabetic complications [148]. The ratio of sFasL to sFasR was also proposed to be a promising prognostic and predictive factor in response to chemotherapy [149,150]. It is important to note that sFasL levels of sera were significantly increased in patients suffering from early phase Stevens-Johnson syndrome and toxic epidermal necrosis, which will proceed to severe cutaneous adverse reactions by drugs such as skin detachment without treatment [151].
A trimeric protein with extended-sheet structure and single disulfide-bridge (hFasLECD) and a monomeric protein with multiple short -sheet structures and extensive disulfide-bridges (hFasRECD)
Human Fas ligand is a Type-II membrane protein, which belongs to tumor necrosis factor (TNF) ligand superfamily [152]. Its extracellular domain (ECD) locating at C-terminal side of hFasL protein consisted of 179 aa residues (aa 103 – 281) and contains a single disulfide-bridge between Cys202 and Cys233 (Figure 4). This ECD exists as a homo-trimeric state without the help of the N-terminal side intracellular and transmembrane domains.Recently determined three-dimensional structure of hFasLECD – human decoy receptor 3 (hDcR3) complex revealed that the protomer in the assembled trimer of hFasLECD has an extended β-sheet structure composed of two β-strands, each made of five antiparallel β-strands (Figure 4). More than 40 % of residues are involved in the formation of the β-sheet structure. The overall structure of trimeric hFasLECD resembles a bell-shaped truncated pyramid with a height of approximately 60 Å. An hFasLECD monomer contains three possible N-glycosylation sites at Asn184, Asn250 and Asn260.
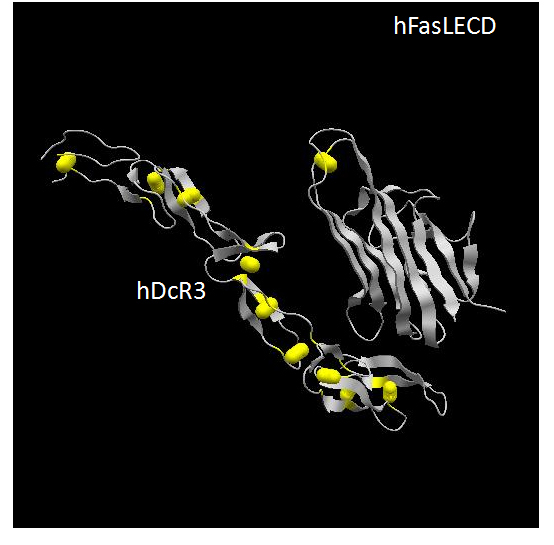
Figure 4. Three-dimensional structure of hFasLECD – hDcR3 complex. The atomic coordinate data were obtained from PDB (ID: 4msv, X-ray model).Only single protomer part of hFasLECD and one bound DcR3 monomer within the complex composed of one trimeric hFasLECD molecule and three monomeric DcR3 molecules are presented. Secondary structures and disulfide-bonds are shown in ribbons and bales, respectively.
Human Fas receptor is a Type-I membrane protein, which belongs to TNF receptor superfamily [152]. In contrast to hFasLECD, the hFasRECD composed of 157 aa residues locates at the N-terminal side of hFasR (aa 1 – 157) and contains three compact Cys-rich domains (CRD) called CRD1, CRD2 and CRD3, each of which consists of about 40 aa residues.These CRDs connected in tandem are considered to form the overall three-dimensional structure of an elongated-shape as observed in DcR3 (Figure 4). The total number of disulfide-bridges in hFasRECD is nine. The three-dimensional structure of hFasRECD in complex with a Fab domain of an anti-hFasRECD antibody has been clarified by X-ray crystallography [153], though information on a number of residues at N- and C-terminal sides is still missing in the structure. Due to compact size of the CRDs, the multiple β-sheet regions assigned within hFasRECD structure are rather small and composed of 2 to 4 aa residues. An hFasRECD monomer contains two possible N-glycosylation sites at Asn102 and Asn120. The requirement of the N-terminal part and the CRD1 region as a pre-association domain for the implementation of apoptosis, which relates to the onset of ALPS, has been reported [154].
The three-dimensional structure of hFasLECD – hFasRECD complex has not been experimentally delineated yet. However, by analogy to the already determined three-dimensional structures of other homologous complexes, such as human lymphotoxin α(LTα) – human TNF receptor 1 complex [155], human tumor necrosis factor (TNFα) – human TNF receptor 2 complex [156], human TNF related apoptosis inducing ligand (TRAIL) ECD – human death receptor 5 (DR5) complex [157] and hFasLECD – hDcR3 complex (PDB ID: 4msv), it is reasonable to assume that three monomeric hFasRECDs bind to the three inter-protomer interfaces formed between adjacent monomers in a trimeric hFasLECD to result in 3 to 3 receptor – ligand assembly.
Efficient recombinant production using yeast cell systems and insect cell systems
hFasLECD and hFasRECD are not available abundantly from natural human resources such as blood, therefore it is important to develop efficient recombinant production systems for obtaining these proteins in large amounts to perform biochemical studies. So far, the recombinant production systems for these proteins have been developed using bacterial cells, yeast cells, insect cells and mammalian cells. Among them, the systems using mammalian cells including human embryo kidney and monkey COS-1 cells were employed in the analysis which requires relatively small amount of biologically functional proteins [158,159]. For the purpose of biochemical analysis requiring larger amount of samples, several other heterologous systems using the cell systems of lower animals and single cellular organisms have been devised for the recombinant production of hFasLECD and hFasRECD.
With respect to hFasLECD, the most efficient heterologous system developed so far for the functional production of mg level amount protein available was the secretory expression system using a methylotrophic yeast, P. pastoris [160]. The secretion level was found to be significantly increased by the truncation of N-terminal region (aa 103-138) [161] and also the addition of DYKDDDDK-(Gly)5 tag sequence at the N-terminus [162]. The hFasRECD binding activity of these recombinant products was confirmed by receptor-mediated co-immunoprecipitation and size-exclusion chromatography analysis. Recently, an improved production method using a disposable culture-bag has been devised for this system [163]. As compared with other human death-ligand ECD such as TNFα, LTα or TRAIL ECD, hFasLECD is hard to efficiently produce in E. coli [160]. This situation may be related to the critical role of N-glycosylation, especially at Asn260 site, for the secretion in yeast. Asn-linked glycans are known to be involved in the folding process of glycoproteins in endoplasmic reticulum (ER) of eukaryotic cells including yeasts via calnexin/calreticulin cycle [164]. In P. pastoris, the secretion of triple mutant of hFasLECD lacking all possible N-glycosylation sites (Asn184, Asn250 and Asn260) was completely repressed, in spite of the efficient secretion of double N-glycosylation site deletion mutant concerning Asn184 and Asn250 [162]. This suggested that the presence of nascent N-glycan holding a terminal glucose residue at Asn260 site was critical for the normal folding of hFasLECD, which avoided translocation into the channel for ER-associated degradation pathway [165]. This hypothesis is supported by the experimental result that the secretion of the double N-glycosylation site deletion mutant concerning Asn184 and Asn250 was totally blocked using a glycoengineered P. pastoris strain called SuperMan5, which can only attach an impaired Man5 oligosaccharide at the N-glycosylation site (M. Muraki. unpublished result). In this connection, the strong direct binding of hFasLECD to calreticulin, a chaperon lectin molecule in ER, in human system was also reported [166]. Another unique production method of hFasLECD was reported with the system using an amoeba host, Dictyostelium discoideum, which secretes biologically active soluble protein at a less efficient level as compared with P. pastoris [167]. Importantly, a recombinant hFasLECD produced in an E. coli system was used as the source for the crystallization experiments in the recently published X-ray structure of hFasLECD – hDcR3 complex (PDB ID: 4msv), which suggested that the non-glycosylated hFasLECD protein was efficiently refolded to a biologically functional form.
On the other hand, the most efficient heterologous production system for the recombinant hFasRECD reported to date employs silkworm larvae as the expression host [160]. Inspired by the successful secretory production of recombinant mouse FasRECD either with a hexahistidine tag or with a human IgG1-Fc domain tag at the C-terminus in Trichoplusia ni cell line [168], a secretion system for the fusion protein of hFasRECD to human IgG1-Fc domain was developed using the larvae of Bombyx mori [169]. As compared with the system for the same fusion protein in another insect cell line, Spodoptera frugiperda, the B. mori larvae system presented much higher expression level per unit volume of the secretion product. This production system was further improved so that an efficient isolation of hFasRECD fragment became possible by incorporating a protease cleavage site sequence, which was specific for thrombin digestion [170]. Although the production yield was found to be lower than the case with wild-type hFasRECD, the double alteration mutant to Gln concerning two N-glycosylation sites at Asn102 and Asn120 was also secreted into the body fluid at a significant level in the B. mori larvae system [163]. This suggested that N-glycosylation had an increasing effect on the level of production, but not always a prerequisite for the secretion of hFasRECD in insect cell systems in this case. In contrast, secretory production of wild-type hFasRECD in P. pastoris employing exactly the same expression unit construct used for the successful secretory production of hFasLECD [162], which had only a substitution of hFasLECD gene with hFasRECD gene, failed to work even in the presence of the two N-glycosylation sites (M. Muraki, unpublished results). This is probably because the much higher density of disulfide-bridges within smaller divided domains containing less extended secondary structures prevented hFasRECD molecule from efficient folding in the ER compartment of P. pastoris, which constitutes a critical process for the correct disulfide-bridges formation.
Great potential in medical applications as protein therapeutics
The basic integrity in biological functions concerning cell-death inducing activity of wild-type recombinant hFasLECD produced in P. pastoris was originally shown using the mice sensitized with Propionibacterium acnes [171]. The recombinant hFasLECD derivatives secreted from P. pastoris were examined by a receptor-mediated co-immunoprecipitation method using the purified recombinant hFasRECD-Fc fusion protein produced in the body fluid of recombinant B. mori larvae, and proved to be functionally active [161-163]. The functional activity of hFasRECD-Fc fusion protein and isolated hFasRECD fragment from the fusion protein produced in B. mori larvae was examined by size-exclusion chromatography analysis of the mixtures with the N-terminal truncated tag-free derivative of hFasLECD produced in P. pastoris [170]. This revealed that the isolated hFasRECD interacted less strongly with the hFasLECD as compared to the divalent hFasRECD-Fc derivative, showing the contribution of antibody-like avidity in the latter case.
At present, no direct experimental information on the recognition mechanism between hFasLECD and hFasRECD at three-dimensional level has been obtained by X-ray crystallographic analysis yet.Site-directed mutagenesis studies aiming at the identification of critical aa residues for hFasLECD – hFasRECD interaction have been conducted based on the artificial molecular models generated from homologous death ligand ECD’s and death receptor ECD’s structures as a template using knowledge-based computer algorisms [158]. Hence, the available information is still rather limited, and the effect of mutagenesis has been mainly assessed using cell-death inducing activity assay and enzyme-linked immunosorbent assay (ELISA). Concerning hFasLECD, two aa residues, Pro206 and Tyr218, located at the binding interface with hFasRECD were replaced to three kinds of different aa residues (Arg, Asp or Phe) independently [158]. The Arg206, Asp206, Phe206 and Arg218 mutants greatly reduced the activities in both assays mentioned above, whereas the Asp218 and Phe218 mutants showed a rather moderate reduction of the activities. The role of each structurally separated domain part in hFasLECD responsible for the self-association into trimeric structure and that of N-glycosylation in the receptor binding was investigated using a series of deletion mutants from its C-terminal [159]. With regard to hFasRECD, fifteen kinds of aa residues (two in CRD1, ten in CRD2 and three in CRD3) were replaced with Ser independently [172,173]. Among them, two mutations concerning Arg86 and Arg87 completely abolished the binding activity to hFasLECD. Four mutations concerning Phe42, Lys73, Lys78 and Glu98 showed little or marginal effect and the others had intermediate reduction of binding activity except for the Ser118 mutant, which was not expressed at the level of detection by ELISA. Also, the roles of each CRD in hFasRECD for the ligand binding were examined using a CRD1 deletion mutant and a series of swapping mutants with the corresponding CRDs in human TNF receptor 1 (TNFR 1) ECD [174].This revealed that no single CRD or pair of CRDs is sufficient for hFasLECD binding as well as that the specificity toward hFasLECD was mainly determined by the CRD2 and the CRD3.
In spite of the limited information on the intra-domain structure – function relationships, hFasLECD and hFasRECD have been thought to possess great potential for the treatment of diseases as protein therapeutics, since the apoptosis induction via the FasR-mediated extrinsic pathway has strong clinical relevance to serious diseases in the human body. Consequently, many engineered derivatives of hFasLECD and hFasRECD have been developed to improve their potential therapeutic functions searching for novel effective biopharmaceuticals targeted to the diseases, which are considered to originate from immunological disorders related to hFasL – hFasR signaling system.
A soluble trimeric hFasLECD molecule, as well as membrane bound full-length form of hFasL, shows specific binding to the membrane bound form of hFasR existing on the surface of targeted cells, however the binding of single trimeric hFasLECD molecule alone is not usually able to trigger efficient apoptosis on malignant cells. This was considered to be derived from the lack of multiple hFasRs’ assembly on the surface of targeted cells, which made the clustering of hFasR in the lipid rafts of cell membrane a promising target for cancer therapy [175]. It was revealed that the association of two adjacent trimeric hFasLECD molecules was the minimum requirement for the efficient formation of death-inducing signaling complex [176]. For the purpose of making this association possible, many artificial methods to combine more than two trimeric hFasLECD molecules have been devised. Among the developed engineered molecules, the most promising agent judging from the experimental results against malignant cells was a hexameric cluster agonist called MegaFasL, which consisted of two hFasLECD trimers combined by the collagen domain of human adiponectin [176]. This agonist molecule presently referred to as APO010 by the manufacturer has been successfully examined for its cell-death inducing activity against many malignant cells including several types of leukemia [177,178], ovarian carcinoma [179], gastrointestinal stromal tumors [180] and glioma [181], and also glioblastoma stem-like cells [182] as potential therapeutic agents aimed at in vivo and ex vivo usage.According to Clinical Trial.gov (ID: NCT00437736), a phase I dose finding study of APO010 in patients with solid tumors has been already completed.Other reported methods to combine more than two hFasLECD trimers include the exploitation of human IgG1-Fc domain [176], self-association module of human leukemia inhibitory factor receptor gp190 [183], fibritin trimerization domain from bacteriophage T4 [184] and an isoleucine zipper motif for self-oligomerization [185].
Another important means for the engineering of hFasLECD molecule was the addition of targeting specificity by the fusion of binding fragments specific to the cell-surface antigens. The cell-surface antigens targeted by scFv antibodies included a fibroblast activation protein [186,187], CD20 on B-cell derived malignant cell lines [188], CD7 on leukemic T-cells [189] and auto-reactive T-cells in RA and juvenile idiopathic arthritis [190] and human tumor-associated antigens, TAG-72 and TAL6, which were expressed on the surface of Jurkat-Ras cells and HeLa cells [191]. Cytotoxic T-lymphocyte-associated protein 4 protein on fibroblast-like synoviocyte cells, which can induce T-cell anergy by blocking CD28 costimulatory signal, was also examined as a fusion partner to hFasLECD for the treatment of RA [192]. One derivative was designed as a prodrug by further fusing with hFasRECD, which was selectively activated on the cancer cells expressing matrix metalloprotease 2 [187]. The production of the above genetically-fused proteins with complicated structures generally required the usage of mammalian cells, represented by HEK293 cells or CHO-K1 cells, as the expression host in order to assure the functional integrity of both effector components in the recombinant fusion proteins. Recently, a novel recombinant derivative of hFasLECD with an N-terminal tag containing single reactive Cys residue per protomer, which may be applicable for the enhancement of its therapeutic functions by the conjugation with either other proteins or low molecular-weight compounds by site-specific chemical modifications, was efficiently produced in P. pastoris, a more convenient host for handling in productive expression [163]. This recombinant hFasLECD product containing an N-terminal DYKDDDDK-tag sequence was site-specifically conjugated either with N-ethylmaleimide or with polyethylenglycol derivatives containing either single or double terminal maleimide group(s). Both conjugated products retained specific receptor binding activity to glycosylated and non-glycosylated hFasRECD-Fc produced in B. mori, and the former product was confirmed to show significant cytotoxic activity against a colorectal cancer cell line, HT-29, by crosslinking with an anti-DYKDDDDDK antibody [163].
Since the initial observation on the prevention of cytotoxic T lymphocytes (CTL)-induced fulminant hepatitis by mouse FasRECD-human IgG1 Fc domain chimera protein in mice [193], effective engineered hFasRECD molecules have been investigated for suppressing harmful biological functions caused by hFasL.So far, two clinically important strategies, which can contribute to the treatment of serious diseases, have been devised. One is the abrogation of excessive apoptosis induction caused by hFasL in CTL [194] and the other is inhibition of unwanted non-apoptotic signaling processes, proliferation and migration, mediated by hFasR on the surface of malignant tumor cells displaying resistance to normal apoptosis [195]. Both strategies have been proved to be efficiently attained by the divalent fusion protein of hFasRECD connected to human IgG1 Fc domain (hFasRECD-Fc), which was named APG101 by the manufacturer in clinical trials. The former strategy was demonstrated to be effective for the treatment of GVHD and myelodysplastic syndrome (MDS) and the latter for that of glioblastoma multiforme (GBM), and has already entered clinical trials of phase I with MDS (ID: NCT01736436) and phase II with GBM (ID: NCT 01071837), respectively. Other methods to combine multiple hFasRECD using C-terminal domain of osteoprotegerin, cartilage matrix protein and cartilage oligomeric matrix protein (COMP) as the fusion partners were also reported [196]. Among them, the hFasRECD-COMP fusion protein containing pentamerized hFasRECDs showed at least 20-fold stronger activity than hFasRECD-Fc in blocking the cytotoxic activity of antibody-crosslinked hFasLECD against four kinds of malignant tumor cell-lines in vitro.
It is essential to clarify the action mechanism and origin of ligand recognition specificity for the development of novel engineered proteins with a potential for biomedical applications. Extracellular disulfide-bridged proteins with therapeutic potentials represent attractive and promising candidates as biopharmaceuticals due to their enhanced structural stability compared to intracellular proteins. To date, many protein biopharmaceuticals with disulfide-bridges are available in the market. Among them, the most clinically and commercially successful class of therapeutic proteins is monoclonal antibodies, and their molecular design for novel applications is still evolving by integrating the merits of low-molecular weight compounds [197]. However, there is a need to search for different classes of disulfide-bridged proteins other than already launched proteins as commercially available drugs to fulfill the unmet-medical needs, which cannot be attained by low-molecular weight drugs alone. In this paper, the author reviewed the therapeutic relevance and structure – function relationships revealed by employing various biochemical and biophysical analyses in combination with newly developed recombinant expression and chemical synthesis methods concerning three different classes of disulfide-bridged proteins, which possess such potential for medical applications.
Despite the merits of high substrate/ligand recognition specificity of proteins, some difficulties generated by obstructive factors also exist in the case of disulfide-bridged proteins described here. For example, Streptococcus pneumoniae, a virulent bacterium causing many serious infectious diseases including bacteremia and meningitis, is basically susceptible to lytic activity by lysozyme. However, an enzyme named peptideglycan N-acetylglucosamine deacetylase A encoded in this bacterium’s genome inhibits the susceptibility to lysozyme by producing deacetylated peptideglycans, which are no more efficiently recognized by this enzyme [198]. Plant lectins composed of hevein-domains are of non-human origin. Therefore, the direct administrations of this class of proteins to human blood systems may be hampered by their immunogenic properties in the human systems, in spite of the substantial therapeutic potentials for treatment of human diseases, especially as an antiviral agent for AIDS treatment [199]. As for human Fas ligand – Fas receptor system, it was suggested that the glycoproteins of filoviruses including Ebolavirus and Marburgvirus, which are known to cause severe hemorrhagic fever in humans, interfere with the Fas-receptor mediated apoptosis via steric shielding as an immune evasion mechanism [200]. In the joint of RA patients, an abundant expression of hDcR3, a decoy receptor to hFasL, was observed, which works as an efficient competitive inhibitor for hFasL mediated apoptosis against hyperplastic synovial fibroblasts [201]. Also, the aberrant sialylation on cell-surface has been pointed out to be closely related to the evasion mechanism of malignant tumors by impairing FasL – FasR pathway mediated apoptosis [202]. Further investigations based on advanced engineering technology, taking these inhibitory mechanisms into account, will be necessary for maximizing their therapeutic potentials.
Finally, development of an efficient production system is a prerequisite for the successful biochemical and pharmaceutical characterization of proteins. From the viewpoint of sample preparation for investigations, another important issue to be overcome is glycosylation problems. Most disulfide-bridged glycoproteins are produced as secretory proteins in the biosynthesis, and Asp-linked N-glycosylation are considered to assist productive protein folding in the ER compartment of secretory pathway [164]. As exemplified in this paper, many useful methods for the recombinant production of disulfide-bridged therapeutic proteins have been developed using heterologous expression systems such as yeasts and insects. However, practical manufacturing of glycosylated pharmaceutical proteins for human use dominantly depends on mammalian cell-culture systems derived from Chinese hamster’s ovary and human embryonic kidney under the existing circumstance [203]. This situation can be ascribed to the differences in glycosylation forms between mammals and other lower organisms to a large extent. Future development of either the production methods of glycosylated proteins with humanized carbohydrate chains using non-mammalian systems including in vitro methods or the faster and more inexpensive production methods using mammalian cell systems will greatly accelerate both basic and applied research on disulfide-bridged proteins with a potential for medical applications.
- Dombkowski AA, Sultana KZ, Craig DB (2014) Protein disulfide engineering. FEBS Lett 588: 206-212. [Crossref]
- Nick Pace C, Scholtz JM, Grimsley GR (2014) Forces stabilizing proteins. FEBS Lett 588: 2177-2184. [Crossref]
- Fleming A, Allison VD (1922) Observations on a Bacteriolytic Substance ("Lysozyme") found in Secretions and Tissues. Br J Exp Pathol 3: 252-260. [Crossref]
- Travis SM, Singh PK, Welsh MJ (2001) Antimicrobial peptides and proteins in the innate defense of the airway surface. Curr Opin Immunol 13: 89-95. [Crossref]
- Scanlon TC, Teneback CC, Gill A, Bement JL, Weiner JA, et al. (2010) Enhanced antimicrobial activity of engineered human lysozyme. ACS Chem Biol 5: 809-818. [Crossref]
- Wang G (2014) Human antimicrobial peptides and proteins. Pharmaceuticals (Basel) 7: 545-594. [Crossref]
- Figueiredo TA, Ludovice AM, Sobral RG (2014) Contribution of peptidoglycan amidation to beta-lactam and lysozyme resistance in different genetic lineages of Staphylococcus aureus. Microb Drug Resist 20: 238-249. [Crossref]
- Griswold KE, Bement JL, Teneback CC, Scanlon TC, Wargo MJ, Leclair LW (2014) Bioengineered lysozyme in combination therapies for Pseudomonas aeruginosa lung infections. Bioengineered 5: 143-147. [Crossref]
- Sava G, Ceschia V, Pacor S, Zabucchi G (1991) Observations on the antimetastatic action of lysozyme in mice bearing Lewis lung carcinoma. Anticancer Res 11: 1109-1113. [Crossref]
- Sava G (1996) Pharmacological aspects and therapeutic applications of lysozymes. EXS 75: 433-449. [Crossref]
- Pellegrini A, Thomas U, von Fellenberg R, Wild P (1992) Bactericidal activities of lysozyme and aprotinin against gram-negative and gram-positive bacteria related to their basic character. J Appl Bacteriol 72: 180-187. [Crossref]
- Jean E, Ebbo M, Valleix S, Benarous L, Heyries L, et al. (2014) A new family with hereditary lysozyme amyloidosis with gastritis and inflammatory bowel disease as prevailing symptoms. BMC Gastroenterol 14: 159. [Crossref]
- Artymiuk PJ, Blake CC (1981) Refinement of human lysozyme at 1.5 A resolution analysis of non-bonded and hydrogen-bond interactions. J Mol Biol 152: 737-762. [Crossref]
- Herning T, Yutani K, Inaka K, Kuroki R, Matsushima M, et al. (1992) Role of proline residues in human lysozyme stability: a scanning calorimetric study combined with X-ray structure analysis of proline mutants. Biochemistry 31: 7077-7085. [Crossref]
- Muraki M, Jigami Y, Tanaka H, Harada N, Kishimoto F, et al. (1986) Expression of synthetic human lysozyme gene in Escherichia coli. Agric Biol Chem 50: 713-723.
- Jigami Y, Muraki M, Harada N, Tanaka H (1986) Expression of synthetic human-lysozyme gene in Saccharomyces cerevisiae: use of a synthetic chicken-lysozyme signal sequence for secretion and processing. Gene 43: 273-279. [Crossref]
- Yoshimura K, Toibana A, Kikuchi K, Kobayashi M, Hayakawa T, et al. (1987) Differences between Saccharomyces cerevisiae and Bacillus subtilis in secretion of human lysozyme. Biochem Biophys Res Commun 145: 712-718. [Crossref]
- Tsuchiya K, Tada S, Gomi K, Kitamoto K, Kumagai C, et al. (1992) High level expression of the synthetic human lysozyme gene in Aspergillus oryzae. Appl Microbiol Biotechnol 38: 109-114. [Crossref]
- Morita S, Kuriyama M, Nakatsu M, Suzuki M, Kitano K (1995) Secretion of active human lysozyme by Acremonium chrysogenum using a Fusarium alkaline protease promoter system. J Biotechnol 42: 1-8. [Crossref]
- Oka C, Tanaka M, Muraki M, Harata K, Suzuki K, et al. (1999) Human lysozyme secretion increased by alpha-factor pro-sequence in Pichia pastoris. Biosci Biotechnol Biochem 63: 1977-1983. [Crossref]
- Xiong R, Chen J (2008) Secreted expression of human lysozyme in the yeast Pichia pastoris under the direction of the signal peptide from human serum albumin. Biotechnol Appl Biochem 51: 129-134. [Corssref]
2021 Copyright OAT. All rights reserv
- Lönnerdal B (2002) Expression of human milk proteins in plants. J Am Coll Nutr 21: 218S-221S. [Crossref]
- Yu Z, Meng Q, Yu H, Fan B, Yu S, et al. (2006) Expression and bioactivity of recombinant human lysozyme in the milk of transgenic mice. J Dairy Sci 89: 2911-2918. [Crossref]
- Liu S, Li X, Lu D, Shang S, Wang M, et al. (2012) High-level expression of bioactive recombinant human lysozyme in the milk of transgenic mice using a modified human lactoferrin BAC. Transgenic Res 21: 407-414. [Crossref]
- Yang B, Wang J, Tang B, Liu Y, Guo C, et al. (2011) Characterization of bioactive recombinant human lysozyme expressed in milk of cloned transgenic cattle. PLoS One 6: e17593. [Crossref]
- Lamppa JW, Tanyos SA, Griswold KE (2013) Engineering Escherichia coli for soluble expression and single step purification of active human lysozyme. J Biotechnol 164: 1-8. [Crossref]
- Ercan D, Demirci A (2014) Enhanced human lysozyme production in biofilm reactor by Kluyveromyces lactisK7. Biochem Eng J 92: 2-8.
- Yamamoto Y, Taniyama Y, Kikuchi M, Ikehara M (1987) Engineering of the hydrophobic segment of the signal sequence for efficient secretion of human lysozyme by Saccharomyces cerevisiae. Biochem Biophys Res Commun 149: 431-436. [Crossref]
- Taniyama Y, Yamamoto Y, Nakao M, Kikuchi M, Ikehara M (1988) Role of disulfide bonds in folding and secretion of human lysozyme in Saccharomyces cerevisiae. Biochem Biophys Res Commun 152: 962-967. [Crossref]
- Blake CC, Johnson LN, Mair GA, North AC, Phillips DC, et al. (1967) Crystallographic studies of the activity of hen egg-white lysozyme. Proc R Soc Lond B Biol Sci 167: 378-388. [Crossref]
- Strynadka NC, James MN (1996) Lysozyme: a model enzyme in protein crystallography. EXS 75: 185-222. [Crossref]
- Swaminathan R, Ravi VK, Kumar S, Kumar MV, Chandra N (2011) Lysozyme: a model protein for amyloid research. Adv Protein Chem Struct Biol 84: 63-111. [Crossref]
- Speciale G, Thompson AJ, Davies GJ, Williams SJ (2014) Dissecting conformational contributions to glycosidase catalysis and inhibition. Curr Opin Struct Biol 28C: 1-13. [Crossref]
- Kuramitsu S, Ikeda K, Hamaguchi K, Fujio H, Amano T (1974) Ionization constants of Glu 35 and Asp 52 in hen, turkey, and human lysozymes. J Biochem 76: 671-683. [Crossref]
- Inoue M, Yamada H, Yasukochi T, Kuroki R, Miki T, et al. (1992) Multiple role of hydrophobicity of tryptophan-108 in chicken lysozyme: structural stability, saccharide binding ability, and abnormal pKa of glutamic acid-35. Biochemistry 31: 5545-5553. [Crossref]
- Muraki M, Jigami Y, Morikawa M, Tanaka H (1987) Engineering of the active site of human lysozyme: conversion of aspartic acid 53 to glutamic acid and tyrosine 63 to tryptophan or phenylalanine. Biochim Biophys Acta 911: 376-380. [Crossref]
- Muraki M, Morikawa M, Jigami Y, Tanaka H (1987) The roles of conserved aromatic amino-acid residues in the active site of human lysozyme: a site-specific mutagenesis study. Biochim Biophys Acta 916: 66-75. [Crossref]
- Muraki M, Goda S, Nagahora H, Harata K (1997) Importance of van der Waals contact between Glu 35 and Trp 109 to the catalytic action of human lysozyme. Protein Sci 6: 473-476. [Crossref]
- Muraki M, Harata K, Hayashi Y, Machida M, Jigami Y (1991) The importance of precise positioning of negatively charged carboxylate in the catalytic action of human lysozyme. Biochim Biophys Acta 1079: 229-237. [Crossref]
- Malcolm BA, Rosenberg S, Corey MJ, Allen JS, de Baetselier A, et al. (1989) Site-directed mutagenesis of the catalytic residues Asp-52 and Glu-35 of chicken egg white lysozyme. Proc Natl Acad Sci U S A 86: 133-137. [Crossref]
- Harata K, Muraki M, Hayashi Y, Jigami Y (1992) X-ray structure of Glu 53 human lysozyme. Protein Sci 1: 1447-1453. [Crossref]
- Kikuchi M, Yamamoto Y, Taniyama Y, Ishimaru K, Yoshikawa W, et al. (1988) Secretion in yeast of human lysozymes with different specific activities created by replacing valine-110 with proline by site-directed mutagenesis. Proc Natl Acad Sci U S A 85: 9411-9415. [Crossref]
- Song H, Inaka K, Maenaka K, Matsushima M (1994) Structural changes of active site cleft and different saccharide binding modes in human lysozyme co-crystallized with hexa-N-acetyl-chitohexaose at pH 4.0. J Mol Biol 244: 522-540. [Crossref]
- Mine S, Ueda T, Hashimoto Y, Imoto T (2000) Analysis of the internal motion of free and ligand-bound human lysozyme by use of 15N NMR relaxation measurement: a comparison with those of hen lysozyme. Protein Sci 9: 1669-1684. [Crossref]
- Muraki M, Harata K, Jigami Y (1992) Dissection of the functional role of structural elements of tyrosine-63 in the catalytic action of human lysozyme. Biochemistry 31: 9212-9219. [Crossref]
- Muraki M, Harata K, Sugita N, Sato K (1996) Origin of carbohydrate recognition specificity of human lysozyme revealed by affinity labeling. Biochemistry 35: 13562-13567. [Crossref]
- Muraki M, Harata K, Sugita N, Sato Ki (1998) X-ray structure of human lysozyme labelled with 2',3'-epoxypropyl beta-glycoside of man-beta1,4-GlcNAc. Structural change and recognition specificity at subsite B. Acta Crystallogr D Biol Crystallogr 54: 834-843. [Crossref]
- Muraki M (2002) The importance of CH/pi interactions to the function of carbohydrate binding proteins. Protein Pept Lett 9: 195-209. [Crossref]
- Muraki M, Harata K, Sugita N, Sato KI (2000) Protein-carbohydrate interactions in human lysozyme probed by combining site-directed mutagenesis and affinity labeling. Biochemistry 39: 292-299. [Crossref]
- Pincus MR, Scherage HA (1979) Conformational energy calculations of enzyme-substrate and enzyme-inhibitor complexes of lysozyme. 2. calculation of the structures of complexes with a flexible enzyme. Macromolecules 12: 633-644.
- Inoue M, Yamada H, Yasukochi T, Miki T, Horiuchi T, et al. (1992) Left-sided substrate binding of lysozyme: evidence for the involvement of asparagine-46 in the initial binding of substrate to chicken lysozyme. Biochemistry 31: 10322-10330. [Crossref]
- Muraki M, Morikawa M, Jigami Y, Tanaka H (1989) A structural requirement in the subsite F of lysozyme. The role of arginine 115 in human lysozyme revealed by site-directed mutagenesis. Eur J Biochem 179: 573-579. [Crossref]
- Harata K, Muraki M, Jigami Y (1993) Role of Arg115 in the catalytic action of human lysozyme. X-ray structure of His115 and Glu115 mutants. J Mol Biol 233: 524-535. [Crossref]
- Matsui H, Kawagishi H, Usui T (1990) Enzymatic synthesis of p-nitrophenyl 3(5)-O-beta-N-acetylglucosaminyl-alpha-maltopentaoside by lysozyme; a novel substrate for human amylase assay. Biochim Biophys Acta 1035: 90-96. [Crossref]
- Fukamizo T, Torikata T, Kuhara S, Hayashi K (1982) Human lysozyme-catalyzed reaction of chitooligosaccharides. J Biochem 92: 709-716. [Crossref]
- Post CB, Brooks BR, Karplus M, Dobson CM, Artymiuk PJ, et al. (1986) Molecular dynamics simulations of native and substrate-bound lysozyme. A study of the average structures and atomic fluctuations. J Mol Biol 190: 455-479. [Crossref]
- Harata K (1994) X-ray structure of a monoclinic form of hen egg-white lysozyme crystallized at 313 K. Comparison of two independent molecules. Acta Crystallogr D Biol Crystallogr 50: 250-257. [Crossref]
- Harata K, Muraki M (1997) X-ray structure of turkey-egg lysozyme complex with tri-N-acetylchitotriose. Lack of binding ability at subsite A. Acta Crystallogr D Biol Crystallogr 53: 650-657. [Crossref]
- Yamada T, Matsushima M, Inaka K, Ohkubo T, Uyeda A, et al. (1993) Structural and functional analyses of the Arg-Gly-Asp sequence introduced into human lysozyme. J Biol Chem 268: 10588-10592.
- Yamasaki N, Hayashi K, Masaru F (1968) Acetylation of lysozyme, part II. mechanism of lysis by lysozyme. Agr Biol Chem 32: 64-68.
- Muraki M, Morikawa M, Jigami Y, Tanaka H (1988) Engineering of human lysozyme as a polyelectrolyte by the alteration of molecular surface charge. Protein Eng 2: 49-54. [Crossref]
- Tsuchiya Y, Morioka K, Yoshida K, Shirai J, Kokuho T, et al. (2007) Effect of N-terminal mutation of human lysozyme on enzymatic activity. Nucleic Acids Symp Ser (Oxf) : 465-466. [Crossref]
- Gill A, Scanlon TC, Osipovitch DC, Madden DR, Griswold KE (2011) Crystal structure of a charge engineered human lysozyme having enhanced bactericidal activity. PLoS One 6: e16788. [Crossref]
- Muraki M, Jigami Y, Harata K (1994) Alteration of the substrate specificity of human lysozyme by site-specific intermolecular cross-linking. FEBS Lett 355: 271-274. [Crossref]
- Mega T, Hase S (1994) Conversion of egg-white lysozyme to a lectin-like protein with agglutinating activity analogous to wheat germ agglutinin. Biochim Biophys Acta 1200: 331-333. [Crossref]
- Muraki M, Harata K, Sugita N, Sato K (1999) Dual affinity labeling of the active site of human lysozyme with an N-acetyllactosamine derivative: first ligand assisted recognition of the second ligand. Biochemistry 38: 540-548. [Crossref]
- Muraki M, Harata K (2003) X-ray structural analysis of the ligand-recognition mechanism in the dual-affinity labeling of c-type lysozyme with 2',3'-epoxypropyl beta-glycoside of N-acetyllactosamine. J Mol Recognit 16: 72-82. [Crossref]
- Allen AK, Neuberger A, Sharon N (1973) The purification, composition and specificity of wheat-germ agglutinin. Biochem J 131: 155-162. [Crossref]
- Börjeson J, Reisfeld R, Chessin LN, Welsh PD, Douglas SD (1966) Studies on human peripheral blood lymphocytes in vitro. I. Biological and physicochemical properties of the pokeweed mitogen. J Exp Med 124: 859-872. [Crossref]
- Peumans WJ, De Ley M, Broekaert WF (1984) An unusual lectin from stinging nettle (Urtica dioica) rhizomes. FEBS Lett 177: 99-103.
- Van den Bergh KP, Proost P, Van Damme J, Coosemans J, Van Damme EJ, et al. (2002) Five disulfide bridges stabilize a hevein-type antimicrobial peptide from the bark of spindle tree (Euonymus europaeus L.). FEBS Lett 530: 181-185. [Crossref]
- Van Parijs J, Broekaert WF, Goldstein IJ, Peumans WJ (1991) Hevein: an antifungal protein from rubber-tree (Hevea brasiliensis) latex. Planta 183: 258-264. [Crossref]
- Broekaert WF, Marien W, Terras FR, De Bolle MF, Proost P, et al. (1992) Antimicrobial peptides from Amaranthus caudatus seeds with sequence homology to the cysteine/glycine-rich domain of chitin-binding proteins. Biochemistry 31: 4308-4314.
- Aub JC, Tieslau C, Lankester A (1963) Reactions of Normal and Tumor Cell Surfaces to Enzymes. I. Wheat-Germ Lipase and Associated Mucopolysaccharides. Proc Natl Acad Sci USA 50: 613-619. [Crossref]
- Burger MM (1973) Surface changes in transformed cells detected by lectins. Fed Proc 32: 91-101. [Crossref]
- Tao TW, Burger MM (1977) Non-metastasising variants selected from metastasising melanoma cells. Nature 270: 437-438. [Crossref]
- Dennis JW, Carver JP, Schachter, H (1984) Asparagine-linked oligosaccharides in murine tumor cells: comparison of a WGA-resistant (WGAr) nonmetastatic mutant and a related WGA-sensitive (WGAs) metastatic line. J Cell Biol 99: 1034-1044.
- Liu Q, Shen Y, Chen J, Gao X, Feng C, et al. (2012) Nose-to-brain transport pathways of wheat germ agglutinin conjugated PEG-PLA nanoparticles. Pharm Res 29: 546-558. [Crossref]
- Yuan H, Khoury CG, Hwang H, Wilson CM, Grant GA, et al. (2012) Gold nanostars: surfactant-free synthesis, 3D modelling, and two-photon photoluminescence imaging. Nanotechnology 23: 075102. [Crossref]
- Schwarz RE, Wojciechowicz DC, Picon AI, Schwarz MA, Paty PB (1999) Wheatgerm agglutinin-mediated toxicity in pancreatic cancer cells. Br J Cancer 80: 1754-1762. [Crossref]
- Preobrazhenska EV, Stoika RS (2002) Effects of TGF-beta1, fluorouracil and cytotoxic lectins on HT-29 and SW-480 human colon cancer cells. Exper Oncol 24: 188-193.
- Ohba H, Bakalova R, Muraki M (2003) Cytoagglutination and cytotoxicity of Wheat Germ Agglutinin isolectins against normal lymphocytes and cultured leukemic cell lines - relationship between structure and biological activity. Biochim Biophys Acta 1619: 144-150. [Crossref]
- Miyoshi N, Koyama Y, Katsuno Y, Hayakawa S, Mita T, et al. (2001) Apoptosis induction associated with cell cycle dysregulation by rice bran agglutinin. J Biochem 130: 799-805. [Crossref]
- Gastman B, Wang K, Han J, Zhu ZY, Huang X, et al. (2004) A novel apoptotic pathway as defined by lectin cellular initiation. Biochem Biophys Res Commun 316: 263-271. [Crossref]
- Ortiz-Urda S, Elbe-Burger A, Smolle J, Marquart Y, Chudnovsky Y, et al. (2003) The plant lectin wheat germ agglutinin inhibits the binding of pemphigus foliaceus autoantibodies to desmoglein 1 in a majority of patients and prevents pathomechanisms of pemphigus foliaceus in vitro and in vivo. J Immunol 171: 6244-6250. [Crossref]
- Portillo-Téllez Mdel C, Bello M, Salcedo G, Gutiérrez G, Gómez-Vidales V, et al. (2011) Folding and homodimerization of wheat germ agglutinin. Biophys J 101: 1423-1431. [Crossref]
- Dalla Pellegrina C, Rizzi C, Mosconi S, Zoccatelli G, Peruffo A, et al. (2005) Plant lectins as carriers for oral drugs: is wheat germ agglutinin a suitable candidate? Toxicol Appl Pharmacol 207: 170-178. [Crossref]
- Dalla Pellegrina C, Perbellini O, Scupoli MT, Tomelleri C, Zanetti C, et al. (2009) Effects of wheat germ agglutinin on human gastrointestinal epithelium: insights from an experimental model of immune/epithelial cell interaction. Toxicol Appl Pharmacol 237: 146-153. [Crossref]
- Van Damme EJ, Broekaert WF, Peumans WJ (1988) The Urtica dioica Agglutinin Is a Complex Mixture of Isolectins. Plant Physiol 86: 598-601. [Crossref]
- Balzarini J, Van Laethem K, Hatse S, Froeyen M, Peumans W, et al. (2005) Carbohydrate-binding agents cause deletions of highly conserved glycosylation sites in HIV GP120: a new therapeutic concept to hit the achilles heel of HIV. J Biol Chem 280: 41005-41014. [Crossref]
- Kumaki Y, Wandersee MK, Smith AJ, Zhou Y, Simmons G, et al. (2011) Inhibition of severe acute respiratory syndrome coronavirus replication in a lethal SARS-CoV BALB/c mouse model by stinging nettle lectin, Urtica dioica agglutinin. Antiviral Res 90: 22-32. [Crossref]
- Alen MM, Kaptein SJ, De Burghgraeve T, Balzarini J, Neyts J, et al. (2009) Antiviral activity of carbohydrate-binding agents and the role of DC-SIGN in dengue virus infection. Virology 387: 67-75. [Crossref]
- Rovira P, Buckle M, Abastado JP, Peumans WJ, Truffa-Bachi P (1999) Major histocompatibility class I molecules present Urtica dioica agglutinin, a superantigen of vegetal origin, to T lymphocytes. Eur J Immunol 29: 1571-1580. [Crossref]
- Musette P, Galelli A, Chabre H, Callard P, Peumans W, Truffa-Bachi P, Kourilsky P, Gachelin G (1996) Urtica dioica agglutinin, a V beta 8.3-specific superantigen, prevents the development of the systemic lupus erythematosus-like pathology of MRL lpr/lpr mice. Eur J Immunol 26: 1707-1711. [Crossref]
- Galelli A, Truffa-Bachi P (1993) Urtica dioica agglutinin. A superantigenic lectin from stinging nettle rhizome. J Immunol 151: 1821-1831. [Crossref]
- Chrubasik JE, Roufogalis BD, Wagner H, Chrubasik S (2007) A comprehensive review on the stinging nettle effect and efficacy profiles. Part II: urticae radix. Phytomedicine 14: 568-579. [Crossref]
- van der Weerden NL, Bleackley MR, Anderson MA (2013) Properties and mechanisms of action of naturally occurring antifungal peptides. Cell Mol Life Sci 70: 3545-3570. [Crossref]
- van 't Hof W, Veerman EC, Helmerhorst EJ, Amerongen AV (2001) Antimicrobial peptides: properties and applicability. Biol Chem 382: 597-619. [Crossref]
- Wright CS (1989) Comparison of the refined crystal structures of two wheat germ isolectins. J Mol Biol 209: 475-487. [Crossref]
- Harata K, Nagahora H, Jigami Y (1995) X-ray structure of wheat germ agglutinin isolectin 3. Acta Crystallogr D Biol Crystallogr 51: 1013-1019. [Crossref]
- Harata K, Muraki M (2000) Crystal structures of Urtica dioica agglutinin and its complex with tri-N-acetylchitotriose. J Mol Biol 297: 673-681. [Crossref]
- Harata K, Schubert WD, Muraki M (2001) Structure of Urtica dioica agglutinin isolectin I: dimer formation mediated by two zinc ions bound at the sugar-binding site. Acta Crystallogr D Biol Crystallogr 57: 1513-1517. [Crossref]
- Martins JC, Maes D, Loris R, Pepermans HA, Wyns L, et al. (1996) H NMR study of the solution structure of Ac-AMP2, a sugar binding antimicrobial protein isolated from Amaranthus caudatus. J Mol Biol 258: 322-333. [Crossref]
- LeVine D, Kaplan MJ, Greenaway PJ (1972) The purification and characterization of wheat-germ agglutinin. Biochem J 129: 847-856. [Crossref]
- Nagahora H, Ishikawa K, Niwa Y, Muraki M, Jigami Y (1992) Expression and secretion of wheat germ agglutinin by Saccharomyces cerevisiae. Eur J Biochem 210: 989-997. [Crossref]
- Schroeder MR1, Raikhel NV (1992) Isolation and characterization of pro-barley lectin expressed in Escherichia coli. Protein Expr Purif 3: 508-511. [Crossref]
- Muraki M, Morii H, Harata K (1998) Chemical synthesis of chitin binding domain of potato WIN2 protein. Protein Pept Lett 5: 193-198.
- Muraki M, Morii H, Harata K (2000) Chemically prepared hevein domains: effect of C-terminal truncation and the mutagenesis of aromatic residues on the affinity for chitin. Protein Eng 13: 385-389. [Crossref]
- Odintsova TI, Vassilevski AA, Slavokhotova AA, Musolyamov AK, Finkina EI, et al. (2009) A novel antifungal hevein-type peptide from Triticum kiharae seeds with a unique 10-cysteine motif. FEBS J 276: 4266-4275. [Crossref]
- Wright CS (1980) Crystallographic elucidation of the saccharide binding mode in wheat germ agglutinin and its biological significance. J Mol Biol 141: 267-291. [Crossref]
- Wright CS (1990) 2.2 A resolution structure analysis of two refined N-acetylneuraminyl-lactose--wheat germ agglutinin isolectin complexes. J Mol Biol 215: 635-651. [Crossref]
- Wright CS, Jaeger J (1993) Crystallographic refinement and structure analysis of the complex of wheat germ agglutinin with a bivalent sialoglycopeptide from glycophorin A. J Mol Biol 232: 620-638. [Crossref]
- Muraki M, Ishimura M, Harata K (2002) Interactions of wheat-germ agglutinin with GlcNAc beta 1,6Gal sequence. Biochim Biophys Acta 1569: 10-20. [Crossref]
- Schwefel D, Maierhofer C, Beck JG, Seeberger S, Diederichs K, et al. (2010) Structural basis of multivalent binding to wheat germ agglutinin. J Am Chem Soc 132: 8704-8719. [Crossref]
- Wright CS, Kellogg GE (1996) Differences in hydropathic properties of ligand binding at four independent sites in wheat germ agglutinin-oligosaccharide crystal complexes. Protein Sci 5: 1466-1476. [Crossref]
- Saul FA, Rovira P, Boulot G, Damme EJ, Peumans WJ, et al. (2000) Crystal structure of Urtica dioica agglutinin, a superantigen presented by MHC molecules of class I and class II. Structure 8: 593-603. [Crossref]
- Hayashida M, Fujii T, Hamasu M, Ishiguro M, Hata Y (2003) Similarity between protein-protein and protein-carbohydrate interactions, revealed by two crystal structures of lectins from the roots of pokeweed. J Mol Biol 334: 551-565. [Crossref]
- Asensio JL, Siebert HC, von Der Lieth CW, Laynez J, Bruix M, et al. (2000) NMR investigations of protein-carbohydrate interactions: studies on the relevance of Trp/Tyr variations in lectin binding sites as deduced from titration microcalorimetry and NMR studies on hevein domains. Determination of the NMR structure of the complex between pseudohevein and N,N',N"-triacetylchitotriose. Proteins 40: 218-236. [Crossref]
- Verheyden P, Pletinckx J, Maes D, Pepermans HA, Wyns L, et al. (1995) 1H NMR study of the interaction of N,N',N"-triacetyl chitotriose with Ac-AMP2, a sugar binding antimicrobial protein isolated from Amaranthus caudatus. FEBS Lett 370: 245-249.
- Bains G, Lee RT, Lee YC, Freire E (1992) Microcalorimetric study of wheat germ agglutinin binding to N-acetylglucosamine and its oligomers. Biochemistry 31: 12624-12628. [Crossref]
- Lee RT, Gabius HJ, Lee YC (1998) Thermodynamic parameters of the interaction of Urtica dioica agglutinin with N-acetylglucosamine and its oligomers. Glycoconj J 15: 649-655. [Crossref]
- Asensio JL, Cañada FJ, Siebert HC, Laynez J, Poveda A, et al. (2000) Structural basis for chitin recognition by defense proteins: GlcNAc residues are bound in a multivalent fashion by extended binding sites in hevein domains. Chem Biol 7: 529-543. [Crossref]
- Renkonen O, Penttilä L, Niemelä R, Vainio A, Leppänen A, et al. (1991) N-acetyllactosaminooligosaccharides that contain the beta-D-GlcpNAc-(1----6)-D-Gal or beta-D-GlcpNAc-(1----6)-D-GalNAc sequences reveal reduction-sensitive affinities for wheat germ agglutinin. Carbohydr Res 213: 169-183. [Crossref]
- Kronis KA, Carver JP (1982) Specificity of isolectins of wheat germ agglutinin for sialyloligosaccharides: a 360-MHz proton nuclear magnetic resonance binding study. Biochemistry 21: 3050-3057. [Crossref]
- Nagahora H, Harata K, Muraki M, Jigami Y (1995) Site-directed mutagenesis and sugar-binding properties of the wheat germ agglutinin mutants Tyr73Phe and Phe116Tyr. Eur J Biochem 233: 27-34. [Crossref]
- Chavez M, Andreu C, Vidal P, Aboitiz N, Freire F, et al. (2005) On the importance of carbohydrate-aromatic interactions for the molecular recognition of oligosaccharides by proteins: NMR studies of the structure and binding affinity of AcAMP2-like peptides with non-natural naphthyl and fluoroaromatic residues. Chemistry 11: 7060-7074. [Crossref]
- Asensio JL, Ardá A, Cañada FJ, Jiménez-Barbero J (2013) Carbohydrate-aromatic interactions. Acc Chem Res 46: 946-954. [Crossref]
- Nishio M, Umezawa Y, Fantini J, Weiss MS, Chakrabarti P (2014) CH-π hydrogen bonds in biological macromolecules. Phys Chem Chem Phys 16: 12648-12683. [Crossref]
- Nishio M (2011) The CH-π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Phys Chem Chem Phys 13: 13873-13900.
- Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, et al. (1991) The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66: 233-243. [Crossref]
- Takahashi T, Tanaka M, Inazawa J, Abe T, Suda T, et al. (1994) Human Fas ligand: gene structure, chromosomal location and species specificity. Int Immunol 6: 1567-1574. [Crossref]
- Nagata S (1997) Apoptosis by death factor. Cell 88: 355-365. [Crossref]
- Nagata S (1998) Human autoimmune lymphoproliferative syndrome, a defect in the apoptosis-inducing Fas receptor: a lesson from the mouse model. J Hum Genet 43: 2-8. [Crossref]
- Kim WU, Kwok SK, Hong KH, Yoo SA, Kong JS, et al. (2007) Soluble Fas ligand inhibits angiogenesis in rheumatoid arthritis. Arthritis Res Ther 9: R42. [Crossref]
- Kataoka T, Budd RC, Holler N, Thome M, Martinon F, et al. (2000) The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol 10: 640-648. [Crossref]
- Imamura R, Konaka K, Matsumoto N, Hasegawa M, Fukui M, et al. (2004) Fas ligand induces cell-autonomous NF-kappaB activation and interleukin-8 production by a mechanism distinct from that of tumor necrosis factor-alpha. J Biol Chem 279: 46415-46423. [Crossref]
- He G, Karin M (2011) NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res 21: 159-168. [Crossref]
- Holdenrieder S, Stieber P (2010) Circulating apoptotic markers in the management of non-small cell lung cancer. Cancer Biomark 6: 197-210. [Crossref]
- Boroumand-Noughabi S, Sima HR, Ghaffarzadehgan K, Jafarzadeh M, Raziee HR, et al. (2010) Soluble Fas might serve as a diagnostic tool for gastric adenocarcinoma. BMC Cancer 10: 275. [Crossref]
- Soni S, Rath G, Deval R, Salhan S, Mishra AK, et al. (2011) Prognostic significance of soluble fas and soluble fas ligand in serum of patients with complete hydatidiform moles. Am J Reprod Immunol 66: 230-236. [Crossref]
- Cardinal H, Brophy JM, Bogaty P, Joseph L, Hébert MJ, et al. (2010) Usefulness of soluble fas levels for improving diagnostic accuracy and prognosis for acute coronary syndromes. Am J Cardiol 105: 797-803. [Crossref]
- Bahr GM, Capron A, Dewulf J, Nagata S, Tanaka M, et al. (1997) Elevated serum level of Fas ligand correlates with the asymptomatic stage of human immunodeficiency virus infection. Blood 90: 896-898.
- Hosaka N, Oyaizu N, Than S, Pahwa S (2000) Correlation of loss of CD4 T cells with plasma levels of both soluble form Fas (CD95) Fas ligand (FasL) in HIV-infected infants. Clin Immunol 95: 20-25. [Crossref]
- Raghuraman S, Abraham P, Daniel HD, Ramakrishna BS, Sridharan G (2005) Characterization of soluble FAS, FAS ligand and tumour necrosis factor-alpha in patients with chronic HCV infection. J Clin Virol 34: 63-70. [Crossref]
- Sahin M, Aydintug O, Tunc SE, Tutkak H, NaziroÄŸlu M (2007) Serum soluble Fas levels in patients with autoimmune rheumatic diseases. Clin Biochem 40: 6-10. [Crossref]
- Towfighi A, Skolasky RL, St Hillaire C, Conant K, McArthur JC (2004) CSF soluble Fas correlates with the severity of HIV-associated dementia. Neurology 62: 654-656. [Crossref]
- Endo S, Inoue Y, Fujino Y, Yamada Y, Sato N, et al. (2000) Soluble Fas and soluble Fas L levels in patients with acute pancreatitis. Res Commun Mol Pathol Pharmacol 108: 179-186. [Crossref]
- Protopsaltis J, Kokkoris S, Nikolopoulos G, Spyropoulou P, Katsaros T, et al. (2007) Correlation between increased serum sFas levels and microalbuminuria in type 1 diabetic patients. Med Princ Pract 16: 222-225. [Crossref]
- Reimer T, Koczan D, Muller H, Friese K, Thiesen HJ, et al. (2002) Tumour Fas ligand:Fas ratio greater than 1 is an independent marker of relative resistance to tamoxifen therapy in hormone receptor positive breast cancer. Breast Cancer Res 4: R9.
- Nadal C, Maurel J, Gallego R, Castells A, Longaron R, et al. (2005) FAS/FAS ligand ratio: a marker of oxaliplatin-based intrinsic and acquired resistance in advanced colorectal cancer. Clin Cancer Res 11: 4770-4774. [Crossref]
- Murata J, Abe R, Shimizu H (2008) Increased soluble Fas ligand levels in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis preceding skin detachment. J Allergy Clin Immunol 122: 992-1000. [Crossref]
- Bodmer JL, Schneider P, Tschopp J (2002) The molecular architecture of the TNF superfamily. Trends Biochem Sci 27: 19-26. [Crossref]
- Chodorge M, Züger S, Stirnimann C, Briand C, Jermutus L, et al. (2012) A series of Fas receptor agonist antibodies that demonstrate an inverse correlation between affinity and potency. Cell Death Differ 19: 1187-1195. [Crossref]
- Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, et al. (2000) Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 288: 2354-2357. [Crossref]
- Banner DW, D'Arcy A, Janes W, Gentz R, Schoenfeld HJ, et al. (1993) Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell 73: 431-445. [Crossref]
- Mukai Y, Nakamura T, Yoshikawa M, Yoshioka Y, Tsunoda S, et al. (2010) Solution of the structure of the TNF-TNFR2 complex. Sci Signal 3: ra83. [Crossref]
- Hymowitz SG, Christinger HW, Fuh G, Ultsch M, O'Connell M, et al. (1999) Triggering cell death: the crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol Cell 4: 563-571. [Crossref]
- Schneider P, Bodmer JL, Holler N, Mattmann C, Scuderi P, et al. (1997) Characterization of Fas (Apo-1, CD95)-Fas ligand interaction. J Biol Chem 272: 18827-18833. [Crossref]
- Orlinick JR, Elkon KB, Chao MV (1997) Separate domains of the human fas ligand dictate self-association and receptor binding. J Biol Chem 272: 32221-32229. [Crossref]
- Muraki M (2012) Heterologous production of death ligands' and death receptors' extracellular domains: structural features and efficient systems. Protein Pept Lett 19: 867-879. [Crossref]
- Muraki M (2008) Improved secretion of human Fas ligand extracellular domain by N-terminal part truncation in Pichia pastoris and preparation of the N-linked carbohydrate chain trimmed derivative. Protein Expr Purif 60: 205-213. [Crossref]
- Muraki M (2006) Secretory expression of synthetic human Fas ligand extracellular domain gene in Pichia pastoris: influences of tag addition and N-glycosylation site deletion, and development of a purification method. Protein Expr Purif 50: 137-146. [Crossref]
- Muraki M (2014) Improved production of recombinant human Fas ligand extracellular domain in Pichia pastoris: yield enhancement using disposable culture-bag and its application to site-specific chemical modifications. BMC Biotechnol 14: 19.
- Helenius A1, Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73: 1019-1049. [Crossref]
- Banerjee S, Vishwanath P, Cui J, Kelleher DJ, Gilmore R, et al. (2007) The evolution of N-glycan-dependent endoplasmic reticulum quality control factors for glycoprotein folding and degradation. Proc Natl Acad Sci U S A. [Crossref]
- Duus K, Pagh RT, Holmskov U, Højrup P, Skov S, et al. (2007) Interaction of calreticulin with CD40 ligand, TRAIL and Fas ligand. Scand J Immunol 66: 501-507. [Crossref]
- Zhen L, Zhinan X, Shanling Z, Keju J, Yinghua L (2012) Production, purification and cytotoxity of soluble human Fas ligand expressed by Escherichia coli and Dictyostelium discoideum. Biochem. Eng. J. 62: 86-91.
- Mahiou J, Abastado JP, Cabanie L, Godeau F (1998) Soluble FasR ligand-binding domain: high-yield production of active fusion and non-fusion recombinant proteins using the baculovirus/insect cell system. Biochem J 330: 1051-1058. [Crossref]
- Muraki M, Honda S (2010) Efficient production of human Fas receptor extracellular domain-human IgG1 heavy chain Fc domain fusion protein using baculovirus/silkworm expression system. Protein Expr Purif 73: 209-216. [Crossref]
- Muraki M, Honda S (2011) Improved isolation and purification of functional human Fas receptor extracellular domain using baculovirus - silkworm expression system. Protein Expr Purif 80: 102-109. [Crossref]
- Tanaka M, Suda T, Yatomi T, Nakamura N, Nagata S (1997) Lethal effect of recombinant human Fas ligand in mice pretreated with Propionibacterium acnes. J Immunol 158: 2303-2309. [Crossref]
- Starling GC, Bajorath J, Emswiler J, Ledbetter JA, Aruffo A, et al. (1997) Identification of amino acid residues important for ligand binding to Fas. J Exp Med 185: 1487-1492. [Crossref]
- Starling GC, Kiener PA, Aruffo A, Bajorath J (1998) Analysis of the ligand binding site in Fas (CD95) by site-directed mutagenesis and comparison with TNFR and CD40. Biochemistry 37: 3723-3726. [Crossref]
- Orlinick JR, Vaishnaw A, Elkon KB, Chao MV (1997) Requirement of cysteine-rich repeats of the Fas receptor for binding by the Fas ligand. J Biol Chem 272: 28889-28894. [Crossref]
- Mollinedo F, Gajate C (2006) Fas/CD95 death receptor and lipid rafts: new targets for apoptosis-directed cancer therapy. Drug Resist Updat 9: 51-73. [Crossref]
- Holler N, Tardivel A, Kovacsovics-Bankowski M, Hertig S, Gaide O, et al. (2003) Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol Cell Biol 23: 1428-1440. [Crossref]
- Greaney P, Nahimana A, Lagopoulos L, Etter AL, Aubry D, et al. (2006) A Fas agonist induces high levels of apoptosis in haematological malignancies. Leuk Res 30: 415-426. [Crossref]
- Nahimana A, Aubry D, Lagopoulos L, Greaney P, Attinger A, et al. (2011) A novel potent Fas agonist for selective depletion of tumor cells in hematopoietic transplants. Blood Cancer J 1: e47. [Crossref]
- Etter AL, Bassi I, Germain S, Delaloye JF, Tschopp J, et al. (2007) The combination of chemotherapy and intraperitoneal MegaFas Ligand improves treatment of ovarian carcinoma. Gynecol Oncol 107: 14-21. [Crossref]
- Rikhof B, van der Graaf WT, Meijer C, Le PT, Meersma GJ, et al. (2008) Abundant Fas expression by gastrointestinal stromal tumours may serve as a therapeutic target for MegaFasL. Br J Cancer 99: 1600-1606. [Crossref]
- Eisele G, Roth P, Hasenbach K, Aulwurm S, Wolpert F, et al. (2011) APO010, a synthetic hexameric CD95 ligand, induces human glioma cell death in vitro and in vivo. Neuro Oncol 13: 155-164. [Crossref]
- Eisele G, Wolpert F, Decrey G, Weller M (2013) APO010, a synthetic hexameric CD95 ligand, induces death of human glioblastoma stem-like cells. Anticancer Res 33: 3563-3571. [Crossref]
- Daburon S, Devaud C, Costet P, Morello A, Garrigue-Antar L, et al. (2013) Functional characterization of a chimeric soluble Fas ligand polymer with in vivo anti-tumor activity. PLoS One 8: e54000. [Crossref]
- Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, et al. (2008) Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 13: 235-248. [Crossref]
- Shiraishi T, Suzuyama K, Okamoto H, Mineta T, Tabuchi K, et al. (2004) Increased cytotoxicity of soluble Fas ligand by fusing isoleucine zipper motif. Biochem Biophys Res Commun 322: 197-202. [Crossref]
- Samel D, Muller D, Gerspach J, Assohou-Luty C, Sass G, et al. (2003) Generation of a FasL-based proapoptotic fusion protein devoid of systemic toxicity due to cell-surface antigen-restricted Activation. J Biol Chem 278: 32077-32082. [Crossref]
- Watermann I, Gerspach J, Lehne M, Seufert J, Schneider B, et al. (2007) Activation of CD95L fusion protein prodrugs by tumor-associated proteases. Cell Death Differ 14: 765-774. [Crossref]
- Bremer E, ten Cate B, Samplonius DF, Mueller N, Wajant H, et al. (2008) Superior activity of fusion protein scFvRit:sFasL over cotreatment with rituximab and Fas agonists. Cancer Res 68: 597-604. [Crossref]
- Bremer E, ten Cate B, Samplonius DF, de Leij LF, Helfrich W (2006) CD7-restricted activation of Fas-mediated apoptosis: a novel therapeutic approach for acute T-cell leukemia. Blood 107: 2863-2870. [Crossref]
- Bremer E, Abdulahad WH, de Bruyn M, Samplonius DF, Kallenberg CG, et al. (2011) Selective elimination of pathogenic synovial fluid T-cells from rheumatoid arthritis and juvenile idiopathic arthritis by targeted activation of Fas-apoptotic signaling. Immunol Lett 138: 161-168. [Crossref]
- Chan DV, Sharma R, Ju CY, Roffler SR, Ju ST (2013) A recombinant scFv-FasLext as a targeting cytotoxic agent against human Jurkat-Ras cancer. J Biomed Sci 20: 16. [Crossref]
- Zhang W, Wang B, Wang F, Zhang J, Yu J (2012) CTLA4-FasL fusion product suppresses proliferation of fibroblast-like synoviocytes and progression of adjuvant-induced arthritis in rats. Mol Immunol 50: 150-159. [Crossref]
- Kondo T, Suda T, Fukuyama H, Adachi M, Nagata S (1997) Essential roles of the Fas ligand in the development of hepatitis. Nat Med 3: 409-413. [Crossref]
- Hartmann N, Messmann JJ, Leithäuser F, Weiswange M, Kluge M, et al. (2013) Recombinant CD95-Fc (APG101) prevents graft-versus-host disease in mice without disabling antitumor cytotoxicity and T-cell functions. Blood 121: 556-565. [Crossref]
- Tuettenberg J, Seiz M, Debatin KM, Hollburg W, von Staden M, et al. (2012) Pharmacokinetics, pharmacodynamics, safety and tolerability of APG101, a CD95-Fc fusion protein, in healthy volunteers and two glioma patients. Int Immunopharmacol 13: 93-100. [Crossref]
- Holler N, Kataoka T, Bodmer JL, Romero P, Romero J, et al. (2000) Development of improved soluble inhibitors of FasL and CD40L based on oligomerized receptors. J Immunol Methods 237: 159-173. [Crossref]
- Rader C (2014) Chemically programmed antibodies. Trends Biotechnol 32: 186-197. [Crossref]
- Vollmer W, Tomasz A (2000) The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J Biol Chem 275: 20496-20501. [Crossref]
- François KO, Balzarini J (2012) Potential of carbohydrate-binding agents as therapeutics against enveloped viruses. Med Res Rev 32: 349-387. [Crossref]
- Noyori O, Nakayama E, Maruyama J, Yoshida R, Takada A (2013) Suppression of Fas-mediated apoptosis via steric shielding by filovirus glycoproteins. Biochem Biophys Res Commun 441: 994-998. [Crossref]
- Hayashi S, Miura Y, Nishiyama T, Mitani M, Tateishi K, et al. (2007) Decoy receptor 3 expressed in rheumatoid synovial fibroblasts protects the cells against Fas-induced apoptosis. Arthritis Rheum 56: 1067-1075. [Crossref]
- Büll C, Stoel MA, den Brok MH, Adema GJ (2014) Sialic acids sweeten a tumor's life. Cancer Res 74: 3199-3204. [Crossref]
- Zhu J (2012) Mammalian cell protein expression for biopharmaceutical production. Biotechnol Adv 30: 1158-1170. [Crossref]