Lipoprotein lipase (LPL) is a member of a lipase family known to hydrolyze triglyceride molecules in plasma lipoproteins. LPL is predominantly expressed in adipose and muscle tissues, but is also highly expressed in the brain, where its specific roles are as yet unknown. Previously, we found that LPL is a novel Aβ-binding protein that plays a role in Aβ clearance and degradation in vitro. In this study, we generated LPL/APP-double-transgenic (Tg) mice and determined the effects of LPL overexpression on Aβ deposition and memory function in vivo. The LPL expression level was significantly higher in the brains of LPL/APP-double-Tg mice than in the brains of APP-Tg mice. However, unexpectedly, the levels of Aβ1-40 and Aβ1-42 in the brains of LPL/APP-double-Tg mice were similar to those of APP-Tg mice. It is also the case for the levels of Aβ deposition demonstrated by immunohistochemical analysis in the brains of these mice. In contrast, the passive avoidance test showed that memory impairment found in APP-Tg mice was improved in LPL/APP-double-Tg mice, whereas the novel objection recognition test showed no significant difference between these two groups. Our findings indicate that LPL does not seem to play a critical role in the brain to remove extracellular Aβ, but may have an effect to attenuate memory impairment in APP-Tg mice.
Key words
Lipoprotein lipase, Aβ burden, Alzheimer’s disease, memory impairment
Introduction
Lipoprotein lipase (LPL) catalyzes the hydrolysis of triacylglycerol (TG) and mediates the cellular uptake of lipoproteins by functioning as a “bridging molecule” between lipoproteins and sulfated glycosaminoglycans (GAGs) or lipoprotein receptors in blood vessels [1,2]. LPL is expressed in numerous organs and is also highly expressed in the brain [3,4]. Although the catabolic activity of LPL on TG is observed in the brain [5], the finding that apolipoprotein CII (apoCII), an essential cofactor for LPL, is not expressed in the brain [6,7] suggests that LPL has a novel non enzymatic function in the brain. It has been shown that LPL is accumulated in senile plaques (SPs) of Alzheimer’s disease (AD) brains [8]. In addition, it has been shown that single-nucleotide polymorphisms (SNPs) in the coding region of the LPL gene are associated with the incidence of AD and the severity of AD pathological features such as neurofibrillary tangles and SPs [9]. These results suggest that LPL may have a physiological role in the brain, whose alteration is associated with the pathogenesis of AD.
The formation and deposition of SPs in the brain is one of the pathological hallmarks of AD, and the levels of SP deposition and Aβ increase with AD progression [10]. Ab is a physiological peptide whose main species are 40 and 42 amino acids in length, and Ab1-42 is the predominant species in SPs [11]. Ab levels are determined by the balance between its production and degradation/clearance, and an attenuated Ab degradation/clearance has been suggested to be the cause of Ab accumulation [12]. Astrocytes and microglia directly take up and degrade Ab1-42 [13,14]. Ab degradation occurs in late endosomal-lysosomal compartments [15,16]. These lines of evidence led us to perform experiments to explore the role of LPL in Ab metabolism in AD model mice. Our previous study has shown that LPL is a novel Aβ binding protein, and LPL-bound Aβ is internalized, taken up by astrocytes in a CAG-dependent manner, transported to lysosomes, and finally degraded [17]. In this study, we performed experiments to determine whether the overexpression of LPL attenuates the progression of Aβ deposition and memory impairment in LPL/APP-double-transgenic (Tg) mice.
Materials and methods
Antibodies
A polyclonal anti-actin antibody was purchased from Sigma (St. Louis, MO). Monoclonal anti-Ab antibody (82E1) was purchased from IBL (Gunma, Japan). An anti-LPL antibody was purchased from Abcam Inc. (Cambridge, MA). The monoclonal antibodies, 22C11, which recognizes amino acids 66-81 of the N terminus of APP, ADAM10, specific for α-secretase, and PS1, specific for γ-secretase, were purchased from Millipore (USA). Other monoclonal antibody BACE1, specific for β-secretase was purchased from R&D (USA).
Generation of LPL-Tg mice
To construct a human LPL (hLPL) transgene, a human LPL Gateway Entry vector (pENTR221-hLPL) was purchased from DNAFORM (Yokohama, Japan). An hLPL fragment was cloned into a pBS-CAG-DEST vector (Pheonix Bio, Utsunomia, Japan) via a LR reaction to construct a pBS-CAG-hLPL vector, and the sequence was verified. The hLPL transgene was under the transcriptional control of a chicken actin promoter. Transgenic mice were generated by the pronuclear injection of C57BL/6J mice. The integration of hLPL cDNA in newborn mice was determined by Southern blot and PCR analysis of DNAs. Animal experiments were approved by the committee of animal experimentation of Nagoya City University Medical School.
Southern blot analysis
For Southern blotting of hLPL transgenic mice, 5 μg of tail DNA isolated using a standard protocol was digested with EcoRI, fractionated by agarose gel electrophoresis. Separated products were blotted on nylon membranes. DNA corresponding to the hLPL transgene was detected with a radiolabeled 1.6 kb fragment amplified by PCR from the pBS-CAG-hLPL vector as described above.
Generation of LPL/APP double-Tg mice
A J20 mouse (PDGFB-APPswInd20Lms/2J, J20) was purchased from Jackson’s lab (Sacramento, CA) and was used as APP-Tg mouse. The J20 mouse expresses a mutant form of the amyloid precursor protein bearing the K670/M671L and V717F mutations under the control of a PDGF-β promoter. To generate a strain of LPL/APP-double-Tg mice, LPL-Tg mice were crossed with APP-Tg mice. These mice were grown and analyzed at 15 months of age.
PCR genotyping of mice
Genomic DNAs were isolated from mouse tails and digested with 180 μl of 50 mM NaOH for 10 min at 95°C. Twenty microliters of 1 M Tris-HCl (pH 8.0) was added to the digest, followed by centrifugation at 12,000 rpm for 10 min. The resulting supernatant (1 μl) containing DNA was used for PCR using specific primers; 5’-TCTCCTGATGATGCAGATTTTGT-3’ and 5’-GTCCACATCTCCAA-GTCCTCTCT-3’. PCR was carried out using KOD Plus (TOYOBO, Osaka, Japan) for 35 cycles at 94°C (30 sec), 62°C (1 min), 72°C (1 min).
Novel object recognition test
The Novel object recognition test consisted of three sessions: habituation, training, and retention. Each mouse was individually habituated to a box (40 cm x 40 cm square and 40 cm high) by allowing it to explore the box without objects for 3 min for 3 days (habituation session). During the training session, two objects were placed in the back corner of the box. A mouse was then placed midway at the front of the box and the total time it spent exploring the two objects was recorded for 3 min. During the retention session, a mouse was placed back in the same box 24 h after the training session, in which one of the familiar objects used during the training session was replaced with a novel object. The animals were then allowed to explore each object and the time it spent exploring was recorded. Throughout the experiments, the objects used were counterbalanced in terms of their physical complexity and emotional neutrality. Preference index, that is, the ratio of the amount of time spent exploring any one of the two objects (training session) or the novel object (retention session) to the total amount of time spent exploring both objects, was used to measure cognitive function.
Passive avoidance test
To evaluate learning and memory abilities, a step-through passive avoidance test was used as described previously [18]. The apparatus (Passive avoidance system, Muromachi Kikai Co., Ltd., Tokyo, Japan) consisted of a light compartment (90 × 115 mm square and 150 mm high) and a dark compartment (140 × 175 mm square and 150 mm high) with a steel rod grid floor connected to a shock generator. For the acquisition trial, each mouse was placed in the light compartment and allowed to explore freely. The mouse was allowed to move to the dark compartment. The latency time until the mouse completely entered the dark compartment was measured. The door separating the light and dark compartments was closed as soon as the mouse entered the dark compartment. The mouse upon entering the dark compartment completely received a foot shock of 0.3 mA for 3 sec through a steel rod grid. After the acquisition trial, the mouse was returned to its home cage. Twenty-four hours later, the mouse was placed again in the apparatus. When the mice stayed in the light compartment, it is considered that it retained the memory of or remembered the aversive stimulus. When the mouse entered the dark compartment, it is considered that it had an impaired memory of the aversive stimulus. The maximum cut-off latency time was set at 300 sec.
Aβ ELISA
The frozen mouse cortex and hippocampus were first homogenized in 20 volumes of Tris-buffered saline (TBS; 10 mM Tris and 150 mM NaCl, pH 7.5) containing a protease inhibitor cocktail (Protease Inhibitor Cocktail, Roche, Mannheim, Germany). The homogenates were then centrifuged at 100,000 rpm for 20 min at 4 °C, as described elsewhere [19] with some modifications. Pellets were resuspended and further homogenized in 10 volumes of 6 M guanidine with the same protease inhibitors, followed by centrifugation at 100,000 rpm for 20 min. The supernatants were used for insoluble Aβ determination. Aβ1-40 and Aβ1-42 were assayed using ELISA kits (Wako Pure Chemical Industries, Osaka, Japan).
Immunohistochemical analysis
Paraffin-embedded brain sections of mice were deparaffinized in xylene and rehydrated in a graded ethanol series. Endogenous peroxidase was quenched using 3% H2O2 and was blocked with 5% goat serum in 0.25% Triton-X/PBS. Sections were incubated with an anti-Aβ antibody (82E1, IBL, Gunma, Japan) for 1 h in a humid chamber at room temperature followed by a biotin-labeled secondary antibody (Vector Lab, Burlingame, CA) and the visualized using a Vector ABC kit (Vector Lab.) and 3’3 diaminobenzidine (DAB).
Western blot analysis
The proteins separated by SDS-PAGE were electrophoretically transferred onto a polyvinylidene difluoride membrane (Millipore, USA). Nonspecific binding was blocked with 5% skim milk in phosphate-buffered saline containing 0.05% Tween 20. The blots were then incubated with primary antibodies overnight at 4°C. For the detection of proteins reactive with primary monoclonal or polyclonal antibodies, appropriate peroxidase-conjugated secondary antibodies were used in conjunction with SuperSignal Chemiluminesence to obtain images, and analyzed with the LAS4000 Mini Bio-Imaging Analyzer system (GE Healthcare).
Statistical analyses
The collected data were analyzed using GraphPad Prison software (San Diego, CA). The unpaired two-tail Student’s t-test was used for comparison. For novel object recognition test (Figure 2B), the difference between groups was analyzed by two-way ANOVA followed by Bonferroni multiple comparison post-hoc test. Results were considered significant when p < 0.05.
Results
To clarify the role of LPL in Aβ deposition and memory function in vivo, we generated LPL transgenic mice. The constructs used to generate transgenic mice are shown in Figure 1A. Ninety-eight lines of offspring were generated by pronuclear microinjection, and the presence of the transgene was confirmed by Southern blot (Figure 1B) and PCR analysis of the tail DNA using specific primers (Figure 1C). Two founder lines were established by mating founders with C57BL/6J wild-type (Wt) mice.
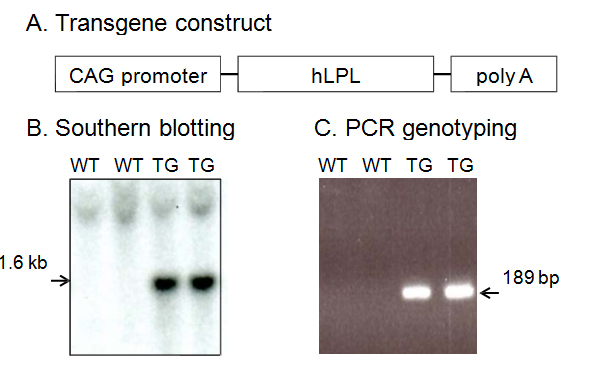
Figure 1. Generation of hLPL transgenic mice. A, Schematic map of the hLPL transgene construct containing the chicken actin (CAG) promoter followed by human LPL cDNA and the poly (A) sequence. B, Verification of germline transmission in the genomic DNAs from wild-type (Wt) and transgenic (Tg) mice by Southern blot analysis. C, Genomic PCR analysis of tail DNAs from Wt and Tg mice using specific primers to determine the genotypes of mice.
Next, to obtain double transgenic mice (LPL-/APP-double Tg mice), the LPL-Tg mice were crossed with the APP-Tg mice. The effects of LPL overexpression on the learning and memory functions in APP-Tg mice were examined. The passive avoidance test was performed to examine the memory functions in LPL-/APP-double Tg and LPL-Wt/APP-Tg mice. There was no significant difference between the acquisition trial time and the retention trial time in the LPL-Wt/APP-Tg mice. However, in the LPL-/APP-double Tg mice, the latency time in the retention trial was significantly longer than the acquisition time (Figure 2A). We also evaluated visual recognition memory by the novel-object recognition test. During the training session, there were no significant differences in exploratory preference between the two objects and total exploratory time between the groups (Figure 2Bb), suggesting that both groups of mice have similar levels of motivation, curiosity, and interest in exploring novel objects. In the retention session, the levels of exploratory preference for the novel objects were not significantly different between the LPL-/APP-double-Tg and LPL-Wt/APP-Tg mice (Figure 2Bb), indicating that LPL overexpression has no effect on the impaired visual recognition memory of APP-Tg mice.
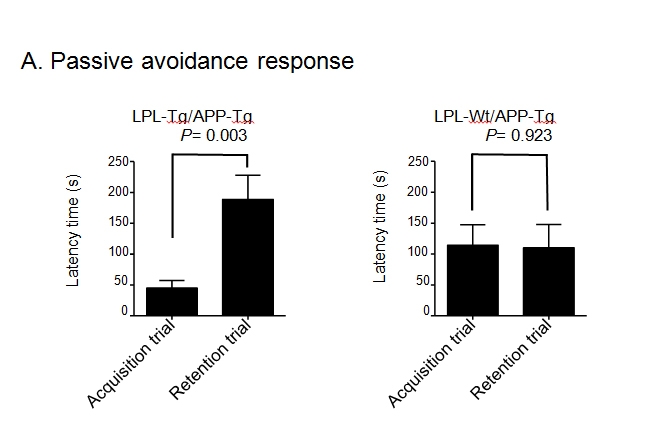
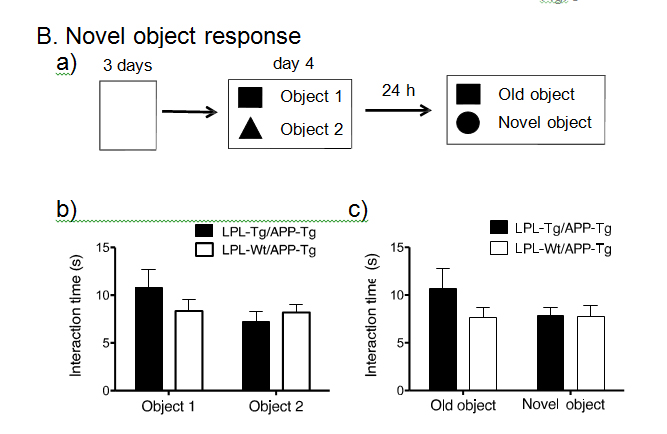
Figure 2. Examination of performances of LPL-/APP-double-Tg and LPL-Wt/APP-Tg mice in novel-object recognition test and step-through passive avoidance tests.
(A) Effect of LPL transgene on passive avoidance response in APP-Tg mice. The retention trial was carried out 24 hr after the acquisition trial. The time during which the mouse stayed in the light room was recorded as latency time. There was significant difference between the latency times in the acquisition trial and retention trial in LPL-Tg/APP-Tg mice (P=0.003), whereas, there was no significant difference between these trials in APP-Tg mice (P=0.928). (B) LPL-/APP-double-Tg and LPL-Wt/APP-Tg mice were subjected to the novel object response test. a), Diagram of the experimental design for the novel object recognition test. b) and c), There were no significant difference in the total interaction time between the two groups for both object recognition. Eleven mice were examined in each group.
Aβ deposition in the hippocampus and cortex was determined by ELISA. The Aβ1-40 and Aβ1-42 levels in the hippocampus samples from LPL-/APP-double-Tg mice were similar to those from LPL-Wt/APP-Tg mice (Figsures 3A and B). Similarly, the Aβ1-40 and Aβ1-42 levels in the cortical samples from LPL-/APP-double-Tg mice were similar to those from LPL-Wt/APP-Tg mice (Figures 3A and B).
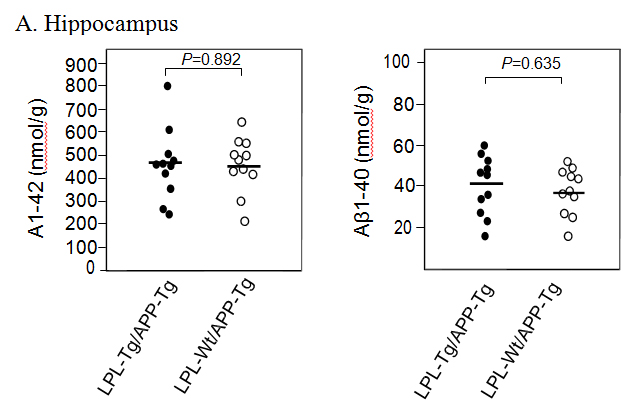
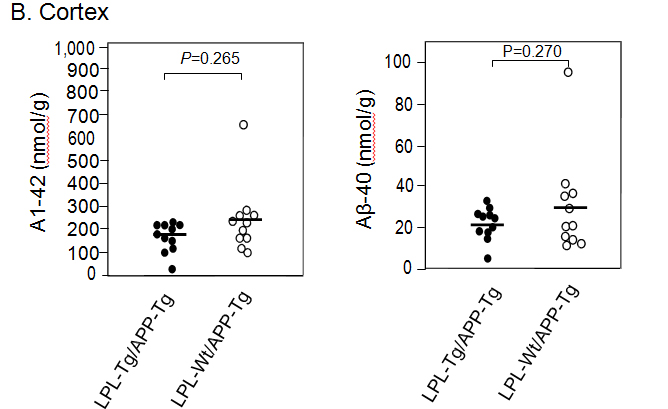
Figure 3. Ab levels in the brains of LPL-/APP-double-Tg and LPL-Wt/APP-Tg mouse. Hippocampi (A) and cortices (B) of the brains isolated from LPL-/APP-double-Tg and LPL-Wt/APP-Tg mice were homogenized, and the levels of guanidine-soluble Aβ1-40 and Aβ1-42 were determined using ELISA kits, as described in Materials and Methods. The data are the plots of Aβ levels of each sample and the bars indicate their average. There is no significant difference between the two groups analyzed by the unpaired Student’s t-test.
The levels of LPL, APP, ADAM10, BACE1, and PS1 were determined by Western blot analysis. Representative data are shown in Figure 4. The LPL level was higher in the LPL-/APP-double-Tg mouse brain than in the LPL-Wt/APP-Tg mouse brain (p<0.003). The levels of ADAM10, PS1, APP and BACE1 were not significantly different between these two groups.
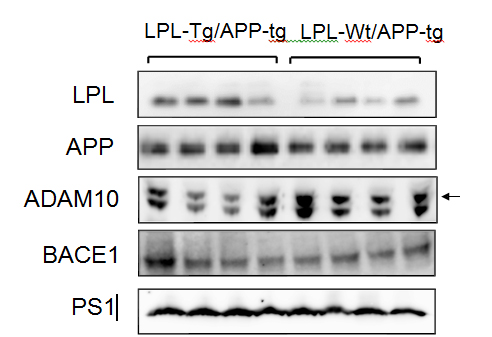
Figure 4. Expressions of LPL, APP, ADAM10, BACE1, and PS1 in LPL-/APP-double-Tg and LPL-Wt/APP-Tg mice. The cortices isolated from LPL-/APP-double-Tg and LPL-Wt/APP-Tg mice were homogenized and the homogenates were examined by Western blot analysis using antibodies specific for LPL, APP, ADAM10, BACE1, and PS1.
We next analyzed Aβ deposition by immunohistochemical analysis. Brain slices were processed for immunohistochemical analysis using the anti-Aβ antibody 82E1. Figures 5A and 5B show the representative photos of immunostained brain sections. Aβ deposition was observed mainly in the hippocampal regions in both types of mouse brains and few depositions were found in the cortical region. Figure 5C shows the results of quantitative analysis, showing that there is no significant difference in the ratio of deposited Aβ-area to the total hippocampal area between the two groups.
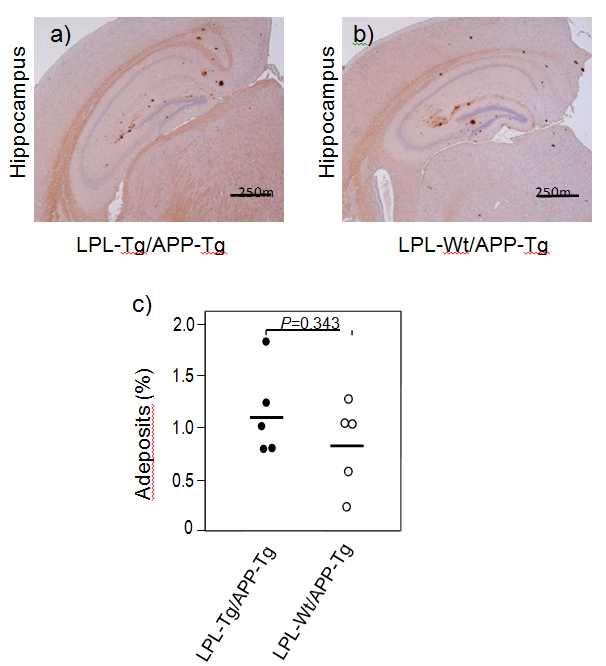
Figure 5. Ab deposition in LPL-/APP-double-Tg and LPL-Wt/APP-Tg mice. Sagittal brain sections of 15-month-old LPL-Tg/APP-Tg and LPL-Wt/APP-Tg mice were stained with the antibody 82E1 specific for Aβ. Representative images of the hippocampi from LPL-/APP-double-Tg (a) and LPL-Wt/APP-Tg (b) mice are shown. (c) Determinations of area of Aβ burden demonstrated by 82E1 staining in the hippocampus. There is no significant difference between the two groups.
Discussion
2021 Copyright OAT. All rights reserv
Previous studies have shown the expressions of mRNA and protein of LPL in the brain are found in several mammalian species [3,4,20]. However, the main lipoprotein fraction in the brain is HDL, which contains negligible or no triacylglycerol (TGs), and the brain lacks an essential cofactor, apoCII. These findings suggest that LPL in the brain has a different function from that in the systemic circulation, in which, with the cofactor apoCII, it catalyzes the hydrolysis of TGs [21]. In our previous study, we found the novel function of LPL, serving as an Ab-binding molecule; that is, exogenous LPL binds to Ab and promotes cellular binding and uptake of Ab in astrocytes [17]. The internalized Ab is degraded mainly via a lysosomal pathway. These findings suggest that an elevated LPL expression level enhances the uptake and removal of extracellular Ab by cells including glia, and that modulation of the LPL level may be a therapeutic target for AD treatment. Interestingly, a recent study has shown that Aβ stimulates LPL expression, which in turn induces Aβ phagocytosis in microglia, and silencing of LPL reduces microglial phagocytosis of Aβ [22]. These lines of evidence led us to perform this study to confirm whether LPL overexpression can reduce Aβ burden and ameliorate memory impairment in APP-Tg mice. The LPL overexpression in APP-Tg mice ameliorates memory impairment found in APP-Tg mice demonstrated by the passive avoidance test; however, the novel object recognition test showed there was no significant difference between these two groups. In addition, unexpectedly, we found that LPL overexpression in APP-Tg mice has no effect on brain Aβ deposition in APP-Tg mice.
There are several molecules involved in Aβ degradation and clearance [23], including neprilysin [24], endothelin-converting enzyme [25], insulin-degrading enzyme (IDE) [26,27], and angiotensin converting enzyme (ACE) [28-30]. One possibility may be that these proteases including endogenous LPL degrade Aβ in vivo and their effect is saturated in terms of Aβ degradation and/or clearance. Thus, overexpression of LPL has little effect on brain Aβ level, although our previous study has shown that LPL strongly enhances the cellular uptake of Ab, leading to increased degradation of Ab in astrocytes [17].
Previous studies have shown that SNPs in the coding region of the LPL gene are associated with AD development [31] and the severity of AD pathophysiological features [9]; however, the molecular mechanisms underlying this association remain unknown. It is possible that an altered function, probably not enhanced but impaired LPL function, would result in impaired Ab clearance and subsequent accumulation of Ab, accelerating AD development. Because LPL regulates the uptake and transport of vitamin E to the brain, the deficiency of which results in enhanced Ab accumulation and presynaptic defects accompanied by impaired learning and memory functions in vivo [32,33]. From these results, it is possible that a reduced LPL function in terms of regulation of vitamin E transport and also degradation of Aβ may enhance Ab accumulation and impair synaptic function, leading to the acceleration of AD development. Further study will be needed to address this issue using LPL-deficient mice.
In this study, we demonstrated that the overexpression of LPL has no effect on Aβ deposition, but a little effect to ameliorate memory impairment develops in APP-Tg mice. Although, LPL is an Aβ-binding protein and markedly enhances Aβ clearance from the extracellular spaces to internalize into astrocytes in vitro; these results may suggest that overexpression of LPL does not seem to play a critical role in additional Aβ clearance in vivo. For the effect of LPL overexpression on memory impairment, it is not unclear why memory impairment was ameliorated in LPL-Tg/APP-Tg mice whose brain Aβ deposition levels remained unchanged. There may be a possibility that LPL overexpression may has an effect on Aβ oligomer levels, which should be addressed in the next study.
This work was supported by JSPS KAKENHI Grant Number 15K15712 (to M.M.). The authors have no conflicts of interest to declare.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Grant-in-Aid for Scientific Research (B) and Challenging Exploratory Research.