Necrotizing enterocolitis (NEC) is the most common gastrointestinal disease of neonates and is associated with intestinal cell death, loss of barrier function, and progressive tissue damage often requiring surgical resection. Enterocyte necrosis is a putative characteristic of NEC and this cell death leads to the release of intracellular contents into their surroundings at the time of their demise. Cellular debris can trigger a feed-forward inflammatory loop in surrounding cells, potentially furthering tissue injury; which cellular components are responsible and whether this process contributes to NEC progression is not known. We conducted in vitro studies to evaluate the impact of mitochondrial exposures on immature intestinal cell survival and inflammatory responses in vitro. Isolated mitochondria from neonatal rat enterocytes (IEC-6) were lysed using a freeze/thaw cycle. IEC-6 cells were then treated with increasing concentrations of lysed mitochondria. Apoptotic and necrotic cell fractions were measured. A dose dependent increase in the number of apoptotic cells was found with increasing concentrations of lysed mitochondria at 72h. A statistically significant increase in necrotic cells was noted with 1X and 2X mitochondria when compared to control (p<0.05). These results demonstrate for the first time that exposure of cells to mitochondrial debris may result in increased immature intestinal epithelial cell death. Direct inflammatory stimulation of IEC-6 cells by LPS resulted in mitochondrial dysfunction by increasing proton leak from mitochondria. These studies support the view that mitochondrial from necrotic cells can elicit toxic effects on adjacent epithelial cells further promoting inflammatory and apoptotic processes and likely contributing to progressive intestinal cell loss. Further defining these pathways may provide novel strategies to intervene and improve NEC outcomes.
Necrotizing enterocolitis (NEC) is the most common gastrointestinal disease of neonates, afflicting 5-10% of infants in neonatal intensive care units, with fatality rates as high as 30%. The highest rate of death is among infants requiring surgery.1 Recent data from large, multicenter, neonatal network databases from the United States and Canada indicate a mean prevalence of about 7% in infants weighing between 501 to 1500 gram birth weight infants [1,2]. Although various risk factors have been described to play an important role in causing the pathogenesis of NEC including prematurity, hypoxia, formula feeding, bacterial infection, and intestinal ischemia [3], the exact mechanisms are not clearly defined. Moreover, the pathophysiology of classic necrotizing enterocolitis is incompletely understood. However, epidemiologic observations strongly suggest a multifactorial cause. The combination of a genetic predisposition, intestinal immaturity, and an imbalance in microvascular tone, accompanied by a strong likelihood of abnormal microbial colonization in the intestine and a highly immunoreactive intestinal mucosa, leads to a confluence of predisposing factors [1]. Previously, intestinal apoptosis has been identified as one of early events preceding overt inflammatory necrosis in a well-established rat model of NEC. In this model increased deoxyribonucleic acid (DNA) fragmentation and elevated caspase-3 activity preceded gross bowel morphological necrosis, signifying the role of intestinal apoptosis as a possible initiator of these events [4]. Predominant doctrine suggests that the induction of enterocyte apoptosis is necessary to allow for the turnover of the intestinal epithelium, which is required for maintenance of epithelial integrity. Enterocyte necrosis and the subsequent induction of the inflammatory response are generally associated with pathological barrier disruption [5].
Mitochondria appear to play a central role in the regulation of several cell death pathways [6,7]. Mitochondria are key players in both the intrinsic and extrinsic pathway of apoptosis and are released during necrosis. Recent studies have demonstrated that increased oxidative stress results in increased mitochondrial stress and thus cause neurotoxicity [8]. Mitochondria have been reported to play a role in necrosis through the production of reactive oxidative species following disruption of the association between the adenosine diphosphate (ADP)- adenosine triphosphate (ATP) translocase with cyclophilin D, or through release into the cytosol of caspase-independent cell death effectors such as apoptosis-inducing factor [9]. Mitochondria share several features with bacteria such as double membrane structure and circular genome. The innate immune system evolved to recognize conserved bacterial molecules that are largely foreign to host cells. Loss of cell membrane integrity and release of intracellular content grant necrotic cells the ability to induce an inflammatory response [10]. Recently, mitochondria have emerged as a source of damage-associated molecular patterns (DAMPs) and thus have role in DAMP-mediated immune stimulation [11-14]. It has been hypothesized that these sequestered molecules and proteins could trigger innate immune responses during a pathological insult such as necrotizing enterocolitis. DAMPs have been shown to activate pattern-recognition receptors on the innate immune system, which further propagate the inflammatory response leading to further cellular necrosis.5 Mitochondria themselves can act as DAMPs leading to a cascade of downstream signaling pathways and responding as effectors, primarily through the generation of reactive oxygen species (ROS), to facilitate the anti-microbial host-cell response [13].
Additionally, mitochondria are also likely to be adversely affected by increased inflammation and oxidants. Previous studies have demonstrated that oxidant injury and inflammation in intestinal epithelial cells is a potential mechanism of tissue injury that may contribute to the pathogenesis of the inflammatory bowel disease (IBD) [15]. More recent studies have demonstrated evidence of mitochondrial stress and alterations in mitochondrial function within the intestinal epithelium of patients with IBD and colitis [16]. It is widely known that commensal bacteria secrete molecules such as lipopolysaccharide (LPS), which can trigger Toll-like receptor (TLR) signaling pathways. Either the disruption of TLR signaling or increased inflammation via LPS may compromise the ability of the intestinal surface to protect and repair itself in the face of inflammatory or infectious insult [17]. Thus, although inflammation is thought to cause mitochondrial stress in intestinal cells, the direct effects of LPS on mitochondrial respiration are poorly understood. LPS induced mitochondrial dysfunction specifically in the intestinal epithelial cells maybe an important mediator in NEC pathophysiology. Mitochondria have also been recently identified as therapeutic targets in hepatocellular carcinoma as well as in an animal model of adjuvant arthritis [18,19]. Interestingly, the role of mitochondria as possible DAMP effectors in necrotizing enterocolitis is not studied till date. Here, we investigated the impact of mitochondrial exposures on immature intestinal cell survival and inflammatory responses in intestinal epithelial cells in vitro using our well-established model [20,21].
Cell culture
Neonatal rat intestinal epithelial cells were obtained from American Type Culture Collection (IEC-6, ATCC, Rockville, MD) and were maintained in high glucose (4.5 g/L) complete Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), 4 mM L-alanyl-L-glutamine, 3.47 µg/mL insulin, and penicillin/streptomycin in a humidified atmosphere of 5% CO2 and 95% air at 37°C. All studies were conducted using cells less than passage 20. Cells were passaged and seeded at a density of 250,000 cells/cm2 in complete media in collagen coated 75 cm3 flasks (Costar, Corning).
Mitochondrial isolation
Mitochondria were isolated from confluent IEC-6 cells using a Mitochondria Isolation Kit for Mammalian Cells (Thermo Scientific; Rockford, IL) as per manufacturer’s instructions. Following isolation, a portion of the isolated fraction was immediately fixed in paraformaldehyde/glutaraldehyde and post-fixed in osmium tetroxide overnight. The pellet was then dehydrated through a graded series of ethanol, and embedded in resin. Sections were examined on the electron microscope and images were captured at 15,000x magnification (Figure 1A). A portion of the isolated mitochondrial fraction was lysed with three freeze/thaw cycles at -20°C to provide a broken-mitochondrial preparation which was used for cell exposure studies (Figure 1B).
Isolated cell exposures to mitochondrial preparation
Cells were treated with increasing concentrations of isolated, lysed mitochondria or combination of lysed mitochondria and lipopolysaccharide (200µg/ml) for 72h. Lysed mitochondria were used at 1x, 2x, 5x concentrations (1x is equivalent to the amount of mitochondria isolated from a number of cells roughly equivalent to the number of cells being treated).
Immunofluorescence and image analysis
Following exposure, cells were stained with Annexin V/Propidium iodide FITC Apoptosis/Necrosis Detection Kit (R&D Systems; Minneapolis, MN) per package protocol and counterstained with DAPI. Cells were imaged at 10x with brightfield, FITC, Texas Red and DAPI filters. Four images were taken of each visual field to assess cell morphology, apoptosis (FITC), necrosis (TxRed), and cell nuclei (DAPI). The images were analyzed for % positive apoptosis (FITC staining) and % positive necrosis (TxRed staining) with a custom built image analysis subroutine (Figure 2).
Mitochondrial function measurement in IEC-6 cells
Rat intestinal epithelial cells (IEC-6) were plated in Seahorse cell culture plates at 80,000cells/well. Cells were allowed to attach for 24h followed by a 24h treatment with 0, 25, 50 or 75µg/ml of LPS in serum free media, previously demonstrated to be sub-lethal doses in this cell line. Following the LPS treatment, the cells were washed with mitochondrial media and cellular mitochondrial function was assessed using Seahorse XFe24 analyzer (Seahorse Biosciences, MA). Mitochondrial oxygen consumption rate (OCR) was measured after serial addition of uncouplers/inhibitors and OCR values were used to measure mitochondrial respiratory responses.
Data analysis
All data presented represent observations per group and represented as mean ± SE. A simultaneous comparison of groups was handled by one-way analyses of variance (ANOVA), with Student-Newman-Keuls post-hoc analysis. Statistical evaluations were performed using GraphPad Prism v 5.0 (GraphPad Software, La Jolla, CA). In all cases, significance was defined as p<0.05.
Mitochondrial structure in the isolated fractions from cells
Shown in Figure 1 are electron micrographs of the mitochondrial fraction isolated from confluent rat neonatal enterocytes. Figure 1A shows intact mitochondria, of varying size due to their removal/isolation from intact cellular structures. Figure 1B shows the mitochondrial fraction following repeat freeze-thaw cycles (see methods), wherein all structure has been destroyed. This isolated and disrupted mitochondrial preparation was used in subsequent experiments.
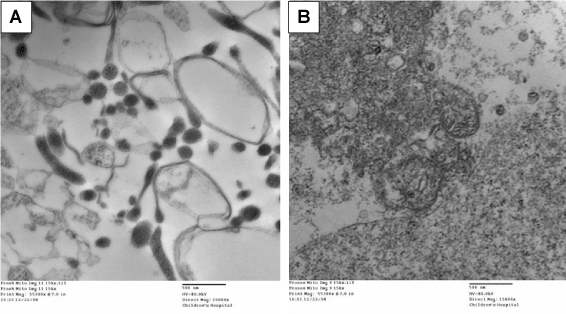
Figure 1. Representative electron microscopy images of isolated mitochondrial fractions from rat neonatal enterocytes. Panel A demonstrates preserved mitochondrial structure. Panel B demonstrates the disrupted mitochondria due to freeze-thaw cycles.
Detection of apoptotic vs. necrotic cell death
Figure 2 shows representative images of confluent wells of enterocytes that were probed for the prevalence of apoptotic vs. necrotic cell prevalence. Using a digital image analysis subroutine total numbers of nuclei (labeled with DAPI) were counted in a field of view, and then numbers of apoptosis- positive cells (Annexin-V positive, FITC-labeled) and numbers of necrosis-positive cells (propidium iodide labeled) were counted. This method was found to be reliable and reproducible with intra-assay and intra-day variability less than 7%.
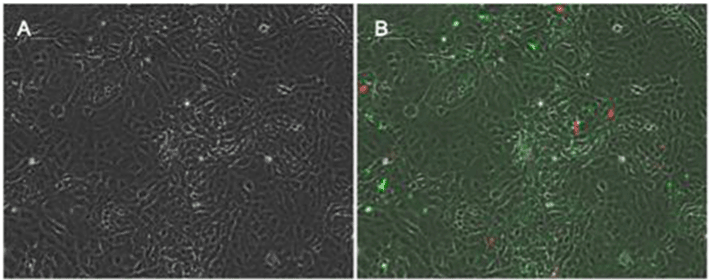
Figure 2. Representative images from confluent cells. Figure 2 A represents control cells under brightfield imaging conditions. Figure 2B demonstrates cells with FITC labeling imaged with TexasRed overlay to measure apoptotic vs. necrotic cell death in these cells.
Exposure to lysed mitochondria causes cell death
Shown in Figure 3 is the prevalence of apoptotic vs. necrotic neonatal enterocytes following exposures to lysed mitochondrial extract. Exposures were at 1x, 2x, or 5x of cell-source for mitochondria. At 72h there was a significant increase in apoptotic prevalence following exposure to the 5x concentration, whereas on significant effect on necrosis was observed.
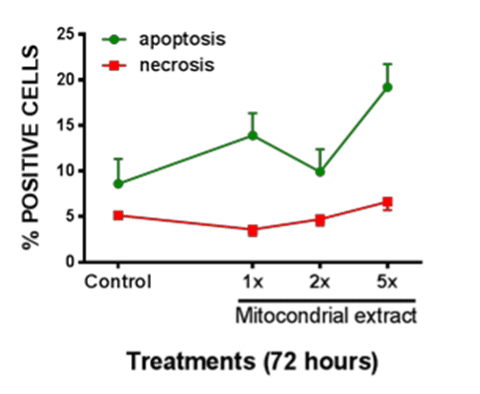
Figure 3. Apoptotic vs. Necrotic cell death in enterocytes was measured after exposure to various concentrations of lysed mitochondrial extract. Significant increase in apoptotic cell death was observed after 72h of exposure to lysed mitochondria. All data expressed as mean±SEM, * denotes p<0.05.
Combined exposure to lysed mitochondria and LPS enhances cell death
Figures 4 A and B show cellular effects of mitochondrial lysates in the absence or presence of LPS with respect to enterocyte apoptosis or necrosis. In these experiments lysed mitochondrial extract was investigated alone or in combination with LPS at 200 µg/ml. Significant differences were found at the combined exposure of 2x mitochondrial fraction and LPS for apoptosis prevalence. The prevalence of necrosis was also significantly enhanced at 1x and 2x mitochondrial extract when combined with LPS, whereas no effect was observed at the 5x mitochondrial plus LPS concentration in this setting.
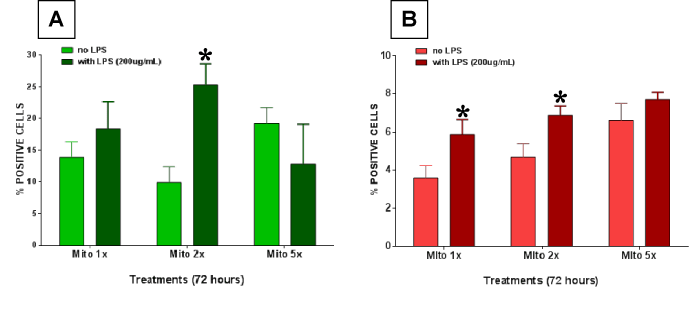
Figure 4. Enterocyte cell death was measured after treating the cells with lysed mitochondria with or without 200 µg/ml lippolysaccharide (LPS). (A) Apoptotic cell death and (B) necrotic cell death was measured. Significantly higher apoptotic cell death was observed in the combined exposure group. Necrosis was also significantly enhanced at 1x and 2x mitochondrial extract when combined with LPS. All data expressed as mean±SEM and * denotes p<0.05.
Lipopolysaccharide causes impaired mitochondrial respiration in rat intestinal epithelial cells
Baseline mitochondrial respiration was not significantly altered after LPS treatment. Additionally, LPS treatment did not alter ATP production or maximal respiration in mitochondria (data not shown). There was a significant increase in proton leak in mitochondria from cells treated with 50 and 75 µg/ml LPS as compared to untreated cells (Figure 5A). Basal respiratory control ratio (RCR) was significantly lower in 50 and 75 µg/ml LPS treated cells (p<0.05) (Figure 5B). Maximal RCR was significantly lower in all LPS treated cells (Figure 5C).
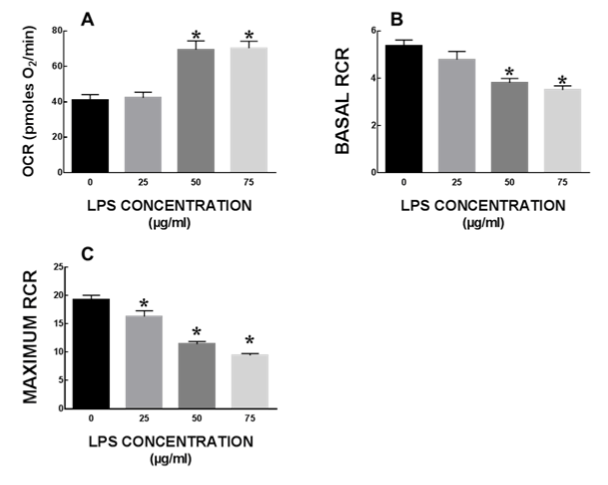
Figure 5. Mitochondrial respiration in IEC6 cells after LPS treatment. Mitochondrial function is severely impaired in intestinal epithelial cells at sub-lethal concentrations of LPS, mitochondrial function is severely impaired in intestinal epithelial cells. All data expressed as mean±SEM and * denotes p<0.05.
A crucial role of necrotic and apoptotic cell death in NEC is now well recognized. Our results demonstrate a concentration and time dependent apoptosis predominantly, but not necrosis in cells exposed to lysed mitochondria. Necrosis has long been described as a consequence of extreme physiochemical stress, such as heat, osmotic shock or mechanical stress, which kills cells quickly and directly. Therefore, this cell death has been described as uncontrolled and accidental necrosis characterized by loss of plasma membrane integrity and cellular collapse, though nuclei remain largely intact during this process [10,22,23]. Apoptosis refers to cell death in a programmed, well-coordinated manner that has well-defined morphological features followed by the quiescent removal of dying cells [5]. Activation of caspases, normally contained in cells as inactive precursor enzymes, plays a central role in apoptosis. Caspases can be activated through both intrinsic and extrinsic pathways. The precise role for enterocyte apoptosis versus necrosis in the maintenance of homeostasis remains incompletely understood. An earlier study demonstrated that apoptosis precedes gross morphologic changes in rat NEC model [4]. Abundant epithelial apoptosis has been observed in histologic specimens collected at the time of bowel resection in patients with NEC. Apoptosis of intestinal epithelium occurs during morphogenesis and also mediates the physiologic turnover of enterocytes [4]. These data indicate that there is substantial evidence of apoptosis in ‘necrotizing’ enterocolitis.
LPS is a known endotoxin present in the outer membrane of gram negative bacteria. Previous studies have demonstrated that LPS administration in association with systemic stress induces NEC in animals. Clinically, serum levels of LPS have shown to be increased in patients with both NEC and inflammatory bowel disease [21]. Additionally, LPS levels in stools of infants and mice that develop NEC have shown to be significantly higher as compared to their healthy counterparts [21]. An increase in the circulating concentration of LPS may exert deleterious effects on the intestinal epithelial monolayer, characterized by a reduction in barrier integrity, while concomitantly activating the subepithelial leukocytes, leading to the pro-inflammatory cytokine release that characterizes NEC. Signaling of LPS within mammalian cells is mediated by the membrane-bound receptor TLR-4, the pivotal role in LPS responsiveness of which was confirmed through the demonstration that mice bearing a single mutation in the TLR-4 gene are unresponsive to LPS [22]. Several previous studies have shown that TLR-4 is increased in the intestinal mucosa of mice, rats, and humans with NEC compared with controls and that activation of enterocyte TLR-4 leads to an increase in death of the cells that line the intestine through the process of apoptosis [21].
Our data shows there could be some possible interaction between LPS and mitochondria exposure on intestinal epithelial cells. At the highest mitochondrial concentration (5x), there was not a significant increase in apoptosis or necrosis. Additionally, in our preliminary studies the prevalence of apoptosis was minimally evident at 24h, but detectable at 72h for the LPS treatment, or the 5x mitochondrial exposure. These treatments demonstrated a 2- to 3-fold increase in prevalence of apoptotic cells at 72h, demonstrating the potential magnitude of longer term and chronic exposures of such agents to neonatal enterocytes. It is possible that a synergistic effect between LPS and mitochondrial lysates leads to increased apoptosis or a binding interaction between LPS and mitochondrial lysates rendering them useless in mounting an inflammatory response. LPS is known to impair hepatic mitochondrial function and cause mitochondrial DNA (mtDNA) damage [24,25]. Reduced basal and Maximal RCR suggest that mitochondria are uncoupled in cells that are treated with LPS. Moreover, increased proton leak from mitochondria may play a significant role in mitochondrial dysfunction and increased ROS production by mitochondria. Mitochondria play crucial roles both in cell survival and death, and mitochondrial damage may initiate apoptosis, necrosis, or both. mtDNA encodes crucial polypeptides of the electron transport chain (ETC) and mtDNA integrity is critical for maintenance of mitochondrial structure and function. LPS is known to impair mitochondria function via TLR4 mediated signalling leading to increased oxidants and impaired mitochondrial respiration [26]. TLR4 signaling is also directly known to contribute to mitochondrial ROS production in other settings [27]. Thus LPS and mitochondria may have synergistic effects in causing cellular death and thus may play an important role in pathogenesis of NEC.
2021 Copyright OAT. All rights reserv
Cellular necrosis elicits robust responses characterized by pro-inflammatory cytokine production and leukocyte recruitment [20], and are triggered via sensing of DAMPs. DAMPs are endogenous molecules that are usually sequestered within intracellular compartments of healthy cells but are released during pathological stress. This mechanism serves as an alert to indicate cell and tissue dysfunction, leading to containment of the injury and promotion of tissue repair [28]. Recently, mitochondrial constituents have been implicated as DAMPs, triggering responses to necrosis and cellular stress [13]. Mitochondria share several features with bacteria such as double membrane structure and circular genome. The innate immune system evolved to recognize conserved bacterial molecules that are largely foreign to host cells. Due to the close similarities between mitochondrial and bacterial constituents, it is highly likely that mitochondria might contain sequestered DAMPs that could trigger innate immune responses during pathophysiological stress and cellular injury such as NEC.
One of the limitations of this study is that it is purely ‘in-vitro’ and does not test in-vivo importance of these mediators. Further studies using our established rodent model of NEC will be undertaken to further elucidate the mechanistic aspects of the role of mitochondria in NEC pathophysiology. Our results presented here demonstrate that lysed mitochondria indeed have direct apoptotic effects on IEC-6 cells in this setting. Moreover we have demonstrated an apparent synergistic effect of mitochondrial fraction and LPS exposure on apoptosis in cells. Thus LPS mediated mitochondrial damage and resultant role of mitochondria as DAMPs may play a crucial role in the pathogenesis of NEC. Thus further studies are clearly warranted to investigate the role of mitochondria as DAMPs in-vivo and their interactions with LPS in NEC.
Necrotizing enterocolitis is a major morbidity in neonates with important prevalence and major consequences. It is common, progressive, and life-threatening. The mechanisms involved are likely related to innate immunity and related inflammation which ultimately converge on epithelial cell death. Mitochondria are possible participants or contributors and play an important role in apoptosis and necrosis. DAMPs and related processes are emerging as important aspects of necrosis in disease such as NEC. Our study shows evidence of mitochondrial effects on enterocytes in vitro with some apparent interactions with LPS. Further studies are warranted to study the interaction of mitochondrial lysates and LPS on intestinal epithelium.
We would like to acknowledge Danice Johnson MD for her effort on this project.
- Neu J, Walker WA (2011) Necrotizing enterocolitis. N Engl J Med 364: 255-264. [Crossref]
- Horbar JD, Carpenter JH, Badger GJ, Kenny MJ, Soll RF, et al. (2012) Mortality and neonatal morbidity among infants 501 to 1500 grams from 2000 to 2009. Pediatrics 129: 1019-1026. [Crossref]
- Zamora R, Grishin A, Wong C, Boyle P, Wang J, et al. (2005) High-mobility group box 1 protein is an inflammatory mediator in necrotizing enterocolitis: protective effect of the macrophage deactivator semapimod. Am J Physiol Gastrointest Liver Physiol 289: G643-G652. [Crossref]
- Jilling T, Lu J, Jackson M, Caplan MS (2004) Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatr Res 55: 622-629. [Crossref]
- Siggers RH, Hackam DJ (2011) The role of innate immune-stimulated epithelial apoptosis during gastrointestinal inflammatory diseases. Cell Mol Life Sci 68: 3623-3634. [Crossref]
- Arnoult D, Carneiro L, Tattoli I, Girardin SE (2009) The role of mitochondria in cellular defense against microbial infection. Semin Immunol 21: 223-232. [Crossref]
- Baines CP (2010) Role of the mitochondrion in programmed necrosis. Front Physiol 1: 156. [Crossref]
- Shin EJ, Dang DK, Tran HQ, Nam Y, Jeong JH, et al. (2016) PKCdelta knockout mice are protected from para-methoxymethamphetamine-induced mitochondrial stress and associated neurotoxicity in the striatum of mice. Neurochem Int 2016.
- Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev 15: 2922-2933. [Crossref]
- Kaczmarek A, Vandenabeele P, Krysko DV (2013) Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38: 209-223. [Crossref]
- Arnoult D, Soares F, Tattoli I, Girardin SE (2011) Mitochondria in innate immunity. EMBO Rep 12: 901-910. [Crossref]
- Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, et al. (2011) Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 32: 157-164. [Crossref]
- West AP, Shadel GS, Ghosh S (2011) Mitochondria in innate immune responses. Nat Rev Immunol 11: 389-402. [Crossref]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, et al. (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104-107.
- McKenzie SJ, Baker MS, Buffinton GD, Doe WF (1996) Evidence of oxidant-induced injury to epithelial cells during inflammatory bowel disease. J Clin Invest 98: 136-141. [Crossref]
- Novak EA, Mollen KP (2015) Mitochondrial dysfunction in inflammatory bowel disease. Front Cell Dev Biol 3: 62. [Crossref]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118: 229-241. [Crossref]
- Mehmood T, Maryam A, Zhang H, Li Y, Khan M, et al. (2016) Deoxyelephantopin induces apoptosis in HepG2 cells via oxidative stress, NF-kappaB inhibition and mitochondrial dysfunction. Biofactors.
- Zhang J, Song X, Cao W, Lu J, Wang X, et al. (2016) Autophagy and mitochondrial dysfunction in adjuvant-arthritis rats treatment with resveratrol. Sci Rep 6: 32928. [Crossref]
- Alcamo AM, Schanbacher BL, Huang H, Nankervis CA, Bauer JA, et al. (2012) Cellular strain amplifies LPS-induced stress signaling in immature enterocytes: potential implications for preterm infant NCPAP. Pediatr Res 72: 256-261. [Crossref]
- Richter JM, Schanbacher BL, Huang H, Xue J, Bauer JA, et al. (2012) LPS-binding protein enables intestinal epithelial restitution despite LPS exposure. J Pediatr Gastroenterol Nutr 54: 639-644. [Crossref]
- Krysko DV, Vanden Berghe T, D'Herde K, Vandenabeele P (2008) Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods 44: 205-221. [Crossref]
- Krysko DV, Vanden Berghe T, Parthoens E, D'Herde K, Vandenabeele P (2008) Methods for distinguishing apoptotic from necrotic cells and measuring their clearance. Methods Enzymol 442: 307-341. [Crossref]
- Choumar A, Tarhuni A, Lettéron P, Reyl-Desmars F, Dauhoo N, et al. (2011) Lipopolysaccharide-induced mitochondrial DNA depletion. Antioxid Redox Signal 15: 2837-2854. [Crossref]
- Suliman HB, Carraway MS, Piantadosi CA (2003) Postlipopolysaccharide oxidative damage of mitochondrial DNA. Am J Respir Crit Care Med 167: 570-579. [Crossref]
- Emre Y, Hurtaud C, Nübel T, Criscuolo F, Ricquier D, et al. (2007) Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem J 402: 271-278. [Crossref]
- Vogel RO, Janssen RJ, van den Brand MA, Dieteren CE, Verkaart S, et al. (2007) Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev 21: 615-624. [Crossref]
- Rock KL, Kono H (2008) The inflammatory response to cell death. Annu Rev Pathol 3: 99-126. [Crossref]