A coincidence of Muller cell sheen dystrophy (MCSD) and congenital color vision deficiency in a 36 years old man is presented in this case report. Best corrected visual acuity (BCVA) was 1.0 in both eyes. He failed to read Ishihara color plates (0/13 in both eyes). Dilated fundus examination showed bilateral wrinkled glistening inner retinal surface at the posterior pole. In the macular images of optical coherence tomography, internal limiting membrane layer had an undulating view. Maternal ocular examination and color vision test revealed no pathology. His father was not alive, and he had no pedigree. To our knowledge this is the first case in the literature, diagnosed with MCSD associated with congenital color vision deficiency.
Muller cell sheen dystrophy, familial internal limiting membrane dystrophy, dyschromatopsy
Familial internal limiting membrane (ILM) dystrophy was first described by Polk and coworkers [1]. They reported a dominantly inherited retinal dystrophy characterized by glistening inner retinal surface with a visual loss in later life. Although Polk defined the disorder as “familial ILM dystrophy”, it was defined as “Muller cell sheen dystrophy” in the later reports, owing to suspects that the primary affected part was Muller cells [2].
Trichromatic color vision deficiency is a relatively frequent trichromatic disorder in which visual acuity (VA) is spared and photophobia and nystagmus are absent. It differs from rod monchromatism and blue cone monochromatism with its milder symptoms [3,4]
In the presenting case report, clinical features of a Muller cell sheen dystrophy associated with incomplete achromatopsia is presented.
A 36 years old male with no complaint applied to our clinic for driver license approval examination. He had no specific disease in his past medical history. His best corrected visual acuity (BCVA) was 1.0 in both eyes. He failed to read Ishihara color plates (0/13 in both eyes). Intraocular pressure was 14 in right, and 15 mm.Hg in left eye. Anterior segment had a normal view in slit lamp examination. Dilated fundus examination showed bilateral wrinkled glistening inner retinal surface at the posterior pole (Figure 1). In the macular images of optical coherence tomography (OCT) (Zeiss HD-OCT 5000, Carl Zeiss Meditec, Jena, Germany), internal limiting membrane (ILM) layer had an undulating view (Figure 2-3). ERG revealed no pathology (Figure 4). Blood count and other systemic investigations revealed no abnormal finding. Maternal ocular examination and color vision test revealed no pathology. His father was not alive, and he had no pedigree.
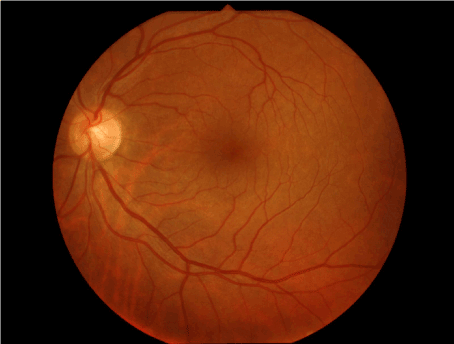
Figure 1. Wrinkled and glistening inner retinal surface of macular periphery.
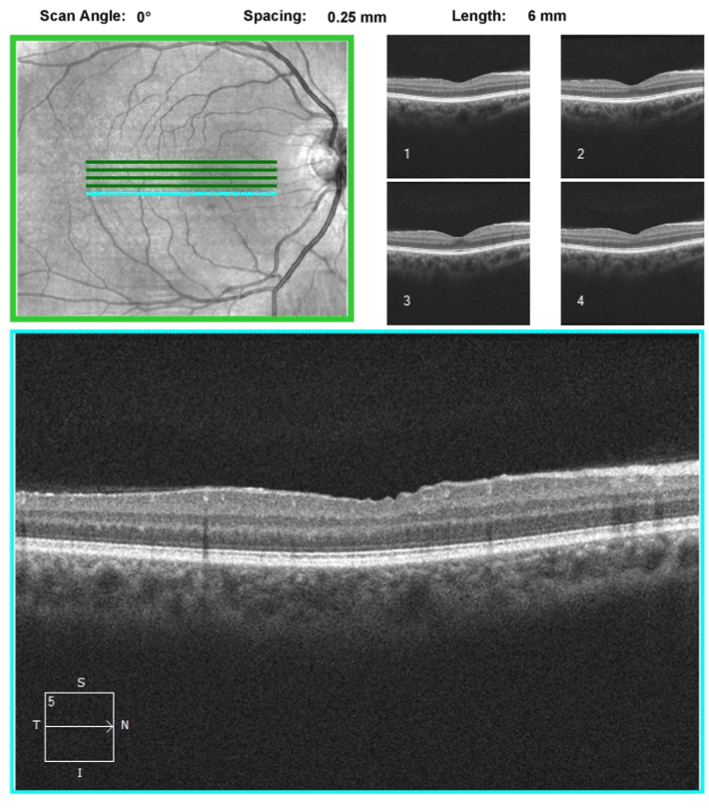
Figure 2. Cross sectional OCT view of wrinkled ILM.
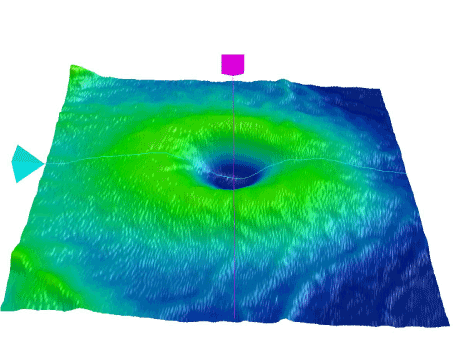
Figure 3. OCT surface view of undulating inner retinal surface of macular periphery.
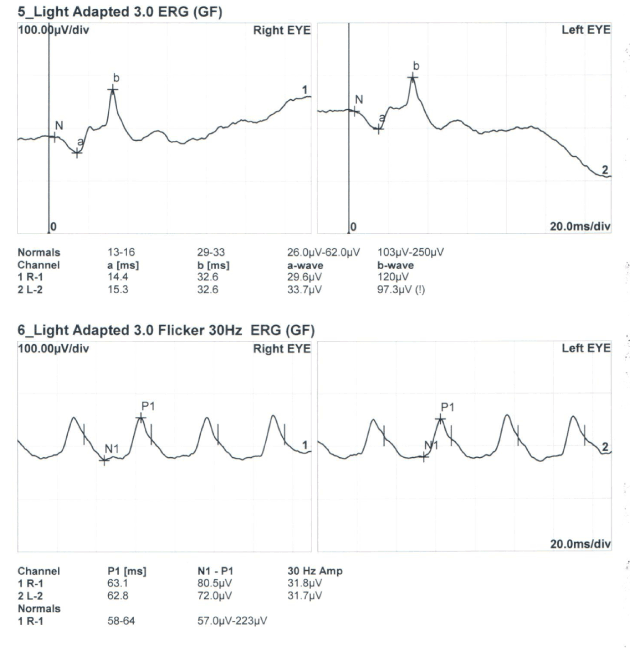
Figure 4. Light adapted and cone flicker ERG findings.
Familial ILM dystrophy, synonyms of which are Muller cell sheen dystrophy and retinal sheen dystrophy, was first described by Polk et al [1]. In their report, five of 14 individuals of a single family were diagnosed with a retinal dystrophy characterized by a glistening inner retinal surface throughout the posterior pole. Visual loss occurred in three affected patients in later life owing to superficial polycystic retinal edema and retinal folds. Electroretinography showed a selective diminution of the b wave. Pathologic examination revealed an abnormal internal limiting membrane with schisis cavities in the inner retina. Endothelial cell swelling, pericyte degeneration, and basement membrane thickening were present in retinal capillaries. They reported autosomal dominant mode of inheritance and concluded that the main pathology was in Müller cells. They reported that there was no effective treatment for the associated visual loss. Renner et al reported similar clinical findings. Nevertheless, they reported ILM peeling to lead BVCA increase and anatomic improvement [2].
Michaelides et al. published a comprehensive review of cone dysfunction syndromes in 2004 [4]. In their review, they classified dyschromatopsia as rod monochromatism, cone monochromatism, and blue cone monochromatism. Rod monochromatism signifies an absence of functioning cone photoreceptors with spared rods. In consequence, there is a markedly reduced VA and total loss of color vision in rod monochromats. Conversely, cone monochromats have normal VA [5,6]. Cone monochromatism is a very rare disorder (estimated incidence: 1/100 million). Inheritence pattern is unknown. Unlike rod monochromatism, it has not diagnosed in more than one member of a single family yet [5]. In cone monochromatism, colour vision loss may be in various severities for certain colours. It may also correlate with the size of the color plate and the level of luminance [5-7]. Unlike other dyschromatopsia syndromes, ERG remains normal. This difference designates the suspect of post-receptoral defect proximal to the retinal photoreceptors and bipolar cells [8,9]. Presence of red and green sensitive pigments at the fovea and the ability of cone monochromats to accommodate by the help of ocular chromatic aberration support this suspect [6,10]. Also, Gibson reported evidence of colour mediating mechanisms in the cone monochromats by demonstrating some mechanism sensitivity that are like those of normal individuals [11]. Cone monochromats have normal VA. Absence of nystagmus and photophobia is important in differential diagnosis. Unlike monochromatic deficiency, trichromacy is a relatively frequent color vision deficiency. Most individuals with congenital color defects are anomalous trichromats and use abnormal proportions of the three primary colors to match those in the light spectrum. These individuals can show protanomalous, tritanomalous, or as in this case deuteranomalous color vision deficiency. Incidences of protanomaly, deuteranomaly, and tritanomaly are 1.08%, 4.63%, and 0.2% respectively. Protanomaly and deuteranomaly have X-linked recessive inheritance, whereas tritanomaly has autosomal dominant inheritance. Incidence of dichromacy is 2.30% and inheritance is same as trichromatic deficiency [12].
The presenting case has a normal VA and defective color vision. Color vision deficiency may cause visual impairment depending on the type of the cone dysfunction. In this case, deuteranomalous trichromacy did not affect the VA as expected. Familial ILM dystrophy causes VA decrease in the late period. The presenting case has only glistening wrinkled inner retina surface. The separation of inner retinal layers and cystic spaces has not formed yet. Therefore, normal VA and ERG findings can be due to initial stage of ILM dystrophy. To our knowledge, there is no coincidence of these two rare disorders in the literature and a little probability has been occurred in this case.
- Polk TD, Gass JD, Green WR, Novak MA, Johnson MW (1997) Familial internal limiting membrane dystrophy. A new sheen retinal dystrophy. Arch Ophthalmol 115: 878-85. [Crossref]
- Renner AB, Radeck V, Kellner U, Jägle H, Helbig H (2014) Ten-year follow-up of two unrelated patients with Müller cell sheen dystrophy and first report of successful vitrectomy. Doc Ophthalmol 129: 191-202.
- Goodman G, Ripps H, Siegel IM (1963). Cone dysfunction syndrome. Arch Ophthalmol 70: 214-231
- Michaelides M, Hunt DM, Moore AT (2004). The cone dysfunction syndromes. Br J Ophthalmol 88: 291-297. [Crossref]
- Weale RA (1953) Cone monochromatism. J Physiol 121: 548-569. [Crossref]
- Weale RA (1959) Photosensitive reactions in foveae of normal and cone monochromatic observers. Opt Acta 6: 158-174.
- Crone RA (1956) Combined forms of congenital colour defects: a pedigree with atypical total colour blindness. Br J Ophthalmol 40: 462-472. [Crossref]
- Ikeda H, Ripps H (1966) The electroretinogram of a cone-monochromat. Arch Ophthalmol 75: 513-517.
2021 Copyright OAT. All rights reserv
- Krill AE, Schneiderman A (1966). Retinal function studies, including the electroretinogram, in an atypical monochromat. Clinical Electroretinography, Supplement to Vision Research. New York: Oxford Press: 351-361.
- Fincham E (1953) Defects of the colour-sense mechanism as indicated by the accommodation reflex. J Physiol 121: 570-580. [Crossref]
- Gibson IM (1962) Visual mechanisms in a cone-monochromat. J Physiol 161:10-11.
- Simunovic MP (2010) Colour vision deficiency. Eye 24: 747-755