Introduction
Fas [Apo 1/ CD 95/ tumor necrosis factor superfamily 6 (TNFRSF 6)/APT 1] is a transmembrane receptor expressed in particular in brain, heart, kidney, liver, pancreas, thymus and lymphoid tissues. It belongs to the death receptor family, a subgroup of the TNF/nerve growth factor (NGF) receptor superfamily [1,2], and acts as the target of cell death-inducing antibodies [3].
These cell surface cytokine receptors are able to initiate an apoptotic signaling cascade after binding a group of structurally related ligands or specific antibodies. In addition to its apoptotic function, it has other cellular responses including migration, invasion, inflammation, and proliferation. The members of this family are type I transmembrane proteins with a C-terminal intracellular tail, a membrane-spanning region, and an extracellular N-terminal domain rich in cystein. Through interaction with the N-terminal domain, the receptors bind their cognate ligands (called death ligands) [4,5]. Although soluble forms of the receptor also exist, whose functions are still largely unknown, the membrane-bound form is largely predominant and highly biologically active [6].
Fas is one of the members of the tumor necrosis factor receptor (TNFR) superfamily, currently comprising 29 receptors that are mirrored by only 19 ligands, representing the cognate TNF ligand superfamily. This already indicates that a single ligand might be capable to bind to more than one receptor and/or that there still exist orphan receptors [7].
Activation of CD95-associated intracellular signaling pathways is not a simple consequence of ligand binding but is the fine-tuned result of a complex interplay of various molecular mechanisms that eventually determine the strength and quality of the CD95 response [3].
In order to avoid unnecessary activation of the apoptotic pathway, Fas expression and localization are tightly regulated through a variety of mechanisms. The minimum amount of Fas is expressed on the plasma membrane in un-stimulated cells (whereas the majority of the receptor localizes in the cytosol, in particular, in the Golgi complex and the trans-Golgi network) [8]. Then, after a pro-apoptotic stimulus, Fas- containing vesicles translocate to the cell surface, increasing Fas expression on the plasma membrane and initiating the apoptotic signal. This mechanism provides an effective tool to regulate the plasma membrane density of the death receptor, and avoid its spontaneous activation [8,9]
Fas can also be modulated at a post-translational level, by glycosylation of the receptor [10], as well as at the transcriptional level, by direct regulation of Fas expression. A composite binding site for the transcription factor nuclear factor KB (NF-κB) is located in the Fas gene promoter [11], and a p53-responsive element has been identified within the first intron of the Fas gene, which co-operates with three sequences in the promoter to up-regulate Fas receptor expression [12].
In eczema, spongiosis is predominantly located in suprabasal epidermal layers, suggesting an anti-apoptotic mechanism protecting basal KCs. CD95 is slightly up-regulated on KCs throughout all epidermal layers in eczematous dermatitis as compared with healthy skin [13,14]. Thus, differential CD95 expression may basically account for the increased susceptibility of KCs to CD95-mediated apoptosis in eczema, but does not explain the apoptosis resistance of basal KCs. The differential expression of pro- and anti-apoptotic factors, which may influence the susceptibility to CD95-mediated apoptosis, might provide an explanation for the restriction of spongiosis to suprabasal epidermal layers [15].
Mechanism
In non-stimulated cells Fas pre-aggregates in the form of complexes due to its amino-terminal preligand-binding assembly domain [11,16].
Fas death receptor is physiologically activated through binding to its cognate ligand, Fasligand (FasL). Fas/FasL interaction induce oligomerization and aggregation of Fas receptor, leading eventually to apoptosis after protein-protein interactions with adaptor and effector proteins [17]. Binding of CD95 or agonistic antibodies to CD95 leads to formation of a receptor complex at the cellular membrane, which was named death-inducing signal complex (DISC) [18].
The DISC consists of oligomerized receptors, the death domain (DD) -containing adaptor molecule fas-associated death domain/ mediator of receptor-induced toxicity (FADD/MORT1), procaspase-8 ]FADD-like interleukin-1 beta-converting enzyme (FLICE), MACHa, Mch5[, procaspase-10 and the cellular Flice like inhibitory protein (c-FLIP) (Figure 1) . The DD of the receptor interacts with the DD of FADD, whereas the death effector domain (DED) of FADD interacts with the N-terminal tandem DEDs of procaspase-8, procaspase -10 and c-FLIP. As a result of DISC formation procaspase-8 is activated at the DISC resulting in the formation of the active caspase-8, which leads to apoptosis [19].
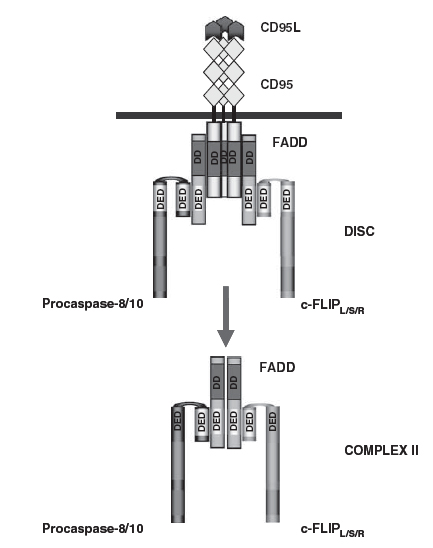
Figure 1. The CD95 DISC and complex II. The DISC consists of CD95, FADD, procaspase-8/procaspase-10 and c-FLIP. Complex II comprises FADD, procaspase-8/10 and c-FLIP. The interactions between the molecules at the DISC and complex II are based on homotypic contacts. The DD of CD95 interacts with the DD of FADD while the DED of FADD interacts with the N-terminal tandem DEDs of procaspase-8, procaspase-10 and c-FLIP [19].
While FADD binds directly to the DD of Fas by its own C-terminal DD, procaspase-8 is indirectly recruited to Fas via FADD, which interacts via its N-terminal DED with the corresponding structure in procaspase-8. This FADD-mediated recruitment into the DISC allows the transient formation of enzymatically active procaspase-8 dimers that convert by autoproteolytic processing to mature active caspases-8 heterotetramers, mature caspase-8 is released from the DISC and, dependent on the cell type, can trigger the execution phase of apoptosis by two pathways, the extrinsic & the intrinsic pathway [20].
Extrinsic pathway of apoptosis is induced by the signal molecules – known as ligands – which are released by other cells, and which bind to the trans-membrane death receptors of the target cell. For example, the immune system’s natural killer cells possess the FasL on their surface: the binding of the FasL to Fas receptors (Fas-R) (a death receptor) on various target cells will trigger the aggregation of multiple receptors on the surface of that target cell [21]. The aggregation of these receptors then leads to the recruitment of an adapter protein, known as FADD, on the cytoplasmic side of the receptors. FADD, in turn, recruits caspase-8 (an initiator protein), forming the DISC [22].
The intrinsic pathway is triggered by cellular stress – specifically, mitochondrial stress caused by various factors, such as deoxy ribonucleic acid (DNA) damage. The stress signal will cause the pro-apoptotic proteins found in the cytoplasm – BAX (pro-apoptotic, cytoplasmic protein) and BID – to bind to the outer membrane of the mitochondria and signal the release of the internal mitochondrial content. However, the signal of BAX and BID is not enough to trigger a full release of the mitochondrial content: BAK, a pro-apoptotic protein found in the mitochondria, is also needed to fully promote the mitochondrial release; it is important to note that the mitochondrial content also includes cytochrome C (Figure 2). Besides cytochrome C, the mitochondrial content released also contains the apoptosisinducing factor (AIF) which facilitates DNA fragmentation, preventing the activity of the inhibitors of apoptosis (IAP) [23,24].
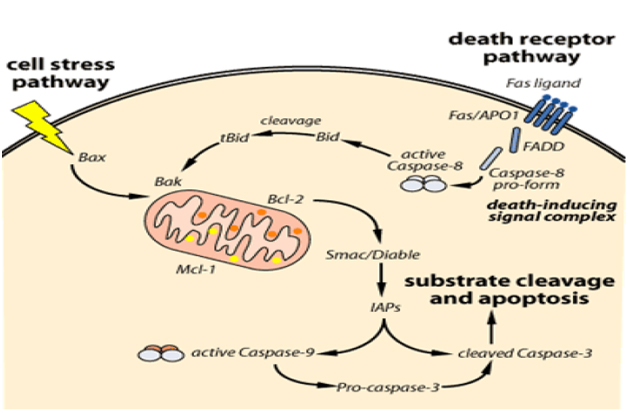
Figure 2. The intrinsic and extrinsic pathways leading to apoptosis [23].
The ligation of Fas by FasL causes the activation of target cell enzymes to degrade target cell nuclear DNA with concomitant fragmentation of the target cell nucleus, leading to programmed cell death. When FasL binds Fas, Fas trimerizes and activates its “death” domain that interacts with the “death” domains of several cytosolic proteins including FADD. Activation of FADD triggers the activation of a series of cysteine proteases known as caspases, resulting in apoptosis of the cell with the consequent morphological changes (reduction of cell volume, plasma membrane blebbing, condensation of chromatin, and fragmentation of DNA) [2,24-26].
Phosphatidylserine, normally found on the cytosolic surface of plasma membranes, is redistributed during the process of apoptosis to the EC surface. Phagocytic cells recognize this aberrant placement and remove the dying cells without the induction of inflammation. Cell removal is followed by a reset of the activated T lymphocytes to initiate another Fas/FasL interaction [1,2,24,25].
Fas Ligand (FasL)
FasL (CD 178/TNFRSF6/APTILG1) is a trimeric type II transmembrane protein that plays an important immune-regulatory role in limiting the host immune response [27].
The human FasL gene was mapped on chromosome lq23 by. The FasL gene consists of approximately 8.0 kb and is split into four exons [28].
FasL is predominandy expressed on activated T lymphocytes and natural killer cells but also at immune privileged sites. Similar to many other members of the TNF ligand superfamily, FasL can be cleaved by metalloproteinases in its EC domain to release the soluble trimeric ligand [27]. Both forms are capable to bind to Fas, but only membrane bound FasL is efficient in induction of cytotoxicity [29].
As soluble FasL competes with the membrane-bound counterpart, however, it can act even as an antagonist preventing apoptosis induction by the membrane integrated form of the ligand [27,30]. On the other hand, soluble FasL binds effectively to fibronectin of the EC matrix, which results in retention of the molecule and an enhanced capacity to induce apoptosis [31]. Besides its role in the blockage of apoptosis the soluble form of FasL has been shown to function as a strong chemoattractant, and to enhance neutrophil and phagocyte migration to inflammatory sites [32,33] (Figure 3).
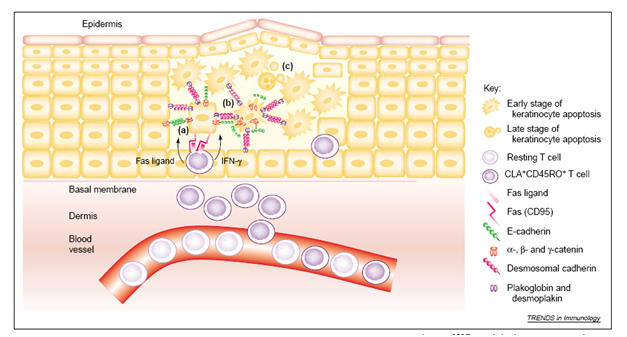
Figure 3. Schematic representation of Fas and FasL. Fas is a type I and FasL a type II transmebrane protein.The upper parts of both molecules represent the extracellular domains. CRD = cysteine rich domain; TM= transmembrane domain; DD = death domain; PRD = proline rich domain. Small arrows indicate aminoacid numbers, the larger arrow the putative metalloproteinase cleavage site.
Binding of membrane FasL to Fas triggers the re-organization of these complexes into signaling competent ligand-receptor aggregates. These aggregates are capable to interact with cytoplasmic signaling molecules. In sensitive cells this "active" Fas complex inducing apoptosis has been named DISC [34].
Role of Fas
Fas/FasL: Key role in autoimmunity
Normally, in order to eliminate auto-reactive T-cells (mature T lymphocytes that recognize self-antigens), interaction between auto reactive T-cell Fas and activated T lymphocytes FasL induces apoptosis of the auto reactive T-cells [35]. Inadequate removal of self-reactive T lymphocytes permits the production of pathogenic autoantibodies that characterize autoimmune diseases [35].
This negative regulation of T-cells contributes to the elimination of T lymphocyte-activated autoreactive B cells, in the absence of presentation of the auto-antigen by the auto reactive T-cell [24].
By virtue of elimination of these cells, the immune system remains safe and effective. Thus, normal Fas/FasL function results in normal lymphocyte homeostasis and controlled auto reactivity. An alteration in Fas/FasL structure results in impaired immune tolerance and in uncontrolled lymphoproliferation [36,37].
Fas-mediated apoptosis plays a critical role in the removal of mature autoreactive B and T lymphocytes as well as in the elimination of infected or malignant cells [24]. A dysfunctional apoptotic pathway may lead to the development of cancers. Due to the sensitivity of the intrinsic pathway, tumors arise more often through the intrinsic pathway dysfunction than the extrinsic pathway [38].
Fas/Fasl: Role in tumor surveillance
It has been assumed that Fas and FasL works as tumor suppressors, since mutations that down regulate normal function of Fas have been proposed as a mechanism by which tumor cells avoid apoptosis or destruction by the immune system [26].
However, a single point mutation of the Fas gene can change Fas/FasL interaction from tumor suppression to tumor promotion by induction of pro-survival genes through non-apoptotic pathways. This dual role of Fas/FasL is found in advanced cancer in humans, resulting in apoptosis resistance and activation of tumorigenic pathways [39].
Stimulation of CD95 has been also reported to trigger non-apoptotic pathways [40]. However, details of CD95- mediated non-apoptotic pathways remain largely unknown. Importantly, it has been shown that membrane-bound CD95L is essential for the cytotoxic activity, whereas soluble CD95L appears to promote autoimmunity and tumorigenesis via induction of non-apoptotic pathways, in particular NF-kB [41]. Future studies are needed to show more details on the mechanism of non-apoptotic action of CD95 [42].
Fas/FasL: Role in inflammation
Inflammatory biomarkers might help to identify specific inflammatory disturbances. Therefore, targeting specific biomarkers of inflammation might represent new therapeutic approaches [43]. Both Fas and FasL proteins exert a wide range of pro-inflammatory functions by inducing secretion of cytokines and chemokines [44].
Beyond activating apoptosis, the activation of the Fas “DD” can initiate multiple non-apoptotic signaling pathways, including inflammatory responses [2,26]. Furthermore, Fas/FasL increases cell removal from areas of chronic inflammation [26], through its role in the blockage of apoptosis the soluble form of FasL has been shown to function as a strong chemoattractant and to enhance neutrophil and phagocyte migration to inflammatory sites [32].
Activation-induced cell death (AICD) down-modulates the immune response [45] and, therefore, plays a key role in the prevention of inflammatory and autoimmune responses. AICD of T cells, B cells, and macrophages is mediated by Fas [47] (Figure 4).

Figure 4. T cells attack keratinocytes (KCs) in the elicitation phase of eczematous dermatitis. The infiltration of activated CD4+ and CD8+ T cells into the dermis and epidermis results in eczematous changes to the epidermis. The apoptosis of KCs is characterized by impairment of the cohesion between KCs (spongiosis). The key pathogenic steps are as follows. (a) Interferon γ (IFN-γ) secreted by activated T cells (CLA+CD45RO+) enhances the expression of Fas on KCs. Membrane-bound and soluble Fas ligand produced by activated T cells triggers Fas on the KCs. (b) During the early stages of the apoptosis of KCs, E-cadherin is cleaved by caspases that remove the β-catenin-binding domain from its cytoplasmic tail. By contrast, desmosomal cadherins (e.g. desmogleins and desmocollins) remain intact. The intracellular domain of E-cadherin is linked to actin microfilaments through its association with α-catenin, β-catenin and γ-catenin (plakoglobin). Desmosomal cadherins bind to the cytoplasmic proteins plakoglobin and desmoplakin, and are linked to keratin intermediate filaments13. (c) Finally, DNA is fragmented and apoptotic bodies form. Abbreviation: CLA, cutaneous lymphocyte-associated antigen.
Role in eczematous dermatitis
The clinical features of eczema are related to increased blood flow in the vessels (erythema), augmented vascular permeability (edema), invasion of T cells into the tissue (infiltration), epidermal spongiosis (vesiculation), and a release of mediators (pruritus). During these eczematous diseases, the resident structural elements in the skin (for example, KCs, fibroblasts, endothelial cells) tightly interact with cells that are actively recruited from the blood in response to inflammatory stimuli. A complex interaction of numerous chemokines controls the recruitment of T cells from the blood vessels and their migration into the dermis and epidermis [48].
The early acute phase of AE is characterized by a Th2 immune response with a distinct cytokine profile . As the disease progresses, there is a shift to a Th1 response, characterized by CD4+ Th lymphocytes and the release of high levels of pro-inflammatory cytokines such as interferon-gamma (IFN- ɤ), which also helps modulate the transition from acute to chronic inflammation [49]. The Th1 cytokines released by T cells in skin, including IFN- ɤ, can up-regulate Fas expression and increase KC susceptibility to apoptosis [50]. Additionally, IFN- ɤ works synergistically with TNF-related apoptosis-inducing ligand (TRAIL) receptor 2 antibody, soluble TRAIL and TNF- α [51]. In AE, it has been found that T cells induce the expression of death receptor Fas on the surface of KCs [13].
FasL, either secreted from activated T cells or present on their surface, interacts with up-regulated Fas on KCs resulting in apoptosis. KC apoptosis induced by T cells disrupts the integrity of the skin leading to altered barrier function that favours invasion of allergens, with subsequent production of inflammatory cytokines and amplification of epithelial damage [52].
The eczematous inflammation of the epidermo-dermal unit is caused by the intricate interaction of T cells and KCs with T cell-derived inflammatory cytokines, IFNs, and other immunoregulatory mediator produced by KCs. Thus the local response of KCs together with the reaction of endothelial cells, T cells, mast cells, and dendritic cells finally leads to the characteristic clinical and histological symptoms of eczema. It has been suggested that the death ligand Fas expressed on activated T cells plays a crucial role in KC death during the elicitation phase of eczematous dermatitis [13]. However, only single KC undergo apoptosis in acute eczema; T-cell-mediated apoptosis of single KCs is a key feature of epidermal pathology in acute eczematous dermatitis [15]. This supports a concept in which the resistance to death ligand mediated apoptosis may rather define an alternative response of KCs to death receptor ligation.
Damage of KCs decreases the effectiveness of the epidermis as a barrier against invasion by infectious agents. Furthermore, damage to the epidermis might allow greater access of allergens and superantigens to Langerhans cells, dermal dendritic cells and T cells, serving to amplify the inflammatory process. Thus, the apoptosis of KCs and damage to the epidermis might play a pivotal role in the development of chronic eczema [48].
In the skin of patients with eczema, the basal layer of KCs and the basement membrane are morphologically intact. It seems probable that during the course of eczema, KC stem cells located directly at the basal membrane are protected from T-cell induced apoptosis. It has been demonstrated that postmitotic, suprabasal KCs are damaged relatively easily, whereas basal stem cells have strong anti-apoptotic defense mechanisms [53].
CD95 is not only needed for a silent end of the life of KCs during eczema but may rather contribute to a “going out with an (inflammatory) bang” intracellular inhibition of effector caspases or mitochondrial signaling pathways of apoptosis downstream of caspase-8 activation by Bcl-2 family members or inhibitor-of-apoptosis proteins (IAPs) [54] might interfere with apoptotic, but not non-apoptotic, signals by death receptors. This scenario may ultimately result in an uncontrolled activation of CD95-mediated inflammation in the skin, but this hypothesis awaits further experimental studies.It will be interesting to determine under which conditions apoptosis may predominate over CD95-mediated inflammation in KCs. This difference subtle at first glance of a death receptor-mediated signal might prove to be highly relevant to the quantitative response in the skin as the target organ of eczematous inflammation.
A common histopathological feature of 2021 Copyright OAT. All rights reserv epidermal vesicles that are disruptive to the normal barrier function of the skin. Although vesicle formation in eczemas has been largely attributed to rupturing of KCs attachments as a result of intercellular edema (spongiosis) recent findings suggest that KC death plays a major role in vesicle formation [13]. This KCs death appears to be apoptotic and to be mediated by FasL, delivered to the epidermis by infiltrating T lymphocytes and acting on Fas whose expression on the surface of KCs is induced by T lymphocyte-derived IFN-ɤ [13].
These findings clearly demonstrated the important role of FasL in the epidermal destruction in inflammatory skin diseases. However, whether FasL is directly involved in the inflammatory process is not known. We demonstrate here that FasL elicits a pro-inflammatory reaction in human KCs by triggering the expression of stress-responsive transcription factors, inflammatory cytokines, chemokines, and the adhesion molecule ICAM-1. KC; as a target of Fas-induced apoptosis, provides evidence that this form of cell death contributes to the pathogenesis of eczematous dermatitis. KCs normally express low levels of Fas, but IFN-ɤ up-regulates Fas on these cells [55]. Secretion of IFN- ɤ by T lymphocytes, which promotes Fas up-regulation in KCs, is a crucial early step in this pathway. Therefore, in this case KC apoptosis occurs only in association with an inflammatory reaction; but it is important to mention that the inflammatory infiltrate is not the consequence but the cause of apoptosis.
From the pathophysiologic point of view, it is interesting that the same mechanism is demonstrated in AD and ACD, since these dermatoses are usually regarded as mutually exclusive, AD being a classic example of a Th2-mediated and ACD of a Th1-mediated process [56].
Some authors speculated that IFN-ɤ expression via up-regulation of the intercullar adhesion molecule1(ICAM-1) contributes to the subsequent accumulation of inflammatory cells. Consistent with this suggestion, the present findings [13]. imply that the high expression of IFN- ɤ also propagates the inflammatory process via disruption of the epidermal barrier. Interestingly, in histologic sections of AD and ACD lesions alike, the majority of apoptotic KCs were found not in spongiotic regions, but in areas that retained normal cohesion of epidermal cells. Hence, one can speculate that apoptosis precedes spongiosis and, further, that apoptotic death of KCs promotes spongiosis by enabling the influx of EC fluid into the epidermis.
Activated transcription factors, such as nuclear factor κB (NF-κB), activator protein 1 (AP-1), nuclear factor of activated T cells(NF-AT) and signal transducer and activator of transcription (STAT) factors, induce inflammation and favor the recruitment of CLA+ T cells to the skin through chemokines and cell-adhesion molecules (e.g. vascular-cell adhesion molecule 1 and E-selectin). During the final process of migration, some T cells penetrate the basal membrane and reach the intercellular space of epidermal KCs.Superfluous and damaged T cells are removed by apoptosis, promoting the resolution, rather than progression, of inflammation. An increased level of apoptosis controls the number of activated, skin-homing T cells in peripheral blood, but cytokines (e.g. IL-2, IL-4 and IL-15) and components of the EC matrix (e.g. fibronectin, laminin, tenascin and collagen IV) in eczematous skin prevent T-cell apoptosis. Therefore, the prolonged survival of T cells, due to components of the skin micro-environment, causes more-pronounced tissue damage and might contribute to chronicity in eczema [48].
References
- Elgert KD (2009) Immunologic tolerance and autoimmunity. In: Immunology: understanding the immune system. Wiley, New York. 431-460.
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE (2009) Cell death. N Engl J Med 361: 1570-1583. [Crossref]
- Wajant H (2014) Principles and mechanisms of CD95 activation. Biol Chem 395: 1401-1416. [Crossref]
- Ashkenazi A, Dixit VM (1999) Apoptosis control by death and decoy receptors. Curr Opin Cell Biol 11: 255-260. [Crossref]
- Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, et al. (1991) The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66: 233-243. [Crossref]
- Cascino I, Fiucci G, Papoff G, Ruberti G (1995) Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J Immunol 154: 2706-2713. [Crossref]
- Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3: 745-756. [Crossref]
- Sodeman T, Bronk SF, Roberts PJ, Miyoshi H, Gores GJ (2000) Bile salts mediate hepatocyte apoptosis by increasing cell surface trafficking of Fas. Am J Physiol Gastrointest Liver Physiol 278: G992-999. [Crossref]
- Feng G, Kaplowitz N (2000) Colchicine protects mice from the lethal effect of an agonistic anti-Fas antibody. J Clin Invest 105: 329-339. [Crossref]
- Peter ME, Hellbardt S, Schwartz-Albiez R, Westendorp MO, Walczak H, et al. (1995) Cell surface sialylation plays a role in modulating sensitivity towards APO-1-mediated apoptotic cell death. Cell Death Differ 2: 163-171. [Crossref]
- Chan H, Bartos DP, Owen-Schaub LB (1999) Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50-p65 recruitment. Mol Cell Biol 19: 2098-2108. [Crossref]
- Müller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, et al. (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 188: 2033-2045. [Crossref]
- Trautmann A, Akdis M, Kleemann D, Altznauer F, Simon HU, et al. (2000) T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest 106: 25-35. [Crossref]
- Simon D, Lindberg RL, Kozlowski E, Braathen LR, Simon HU (2006): Epidermal caspase-3 cleavage associated with interferon-gamma-expressing lymphocytes in acute atopic dermatitis lesions. Exp Dermatol 15: 441-446. [Crossref]
- Armbruster N, Trautmann A, Bröcker EB, Leverkus M, Kerstan A (2009) Suprabasal spongiosis in acute eczematous dermatitis: cFLIP maintains resistance of basal keratinocytes to T-cell-mediated apoptosis. J Invest Dermatolol 129: 1696-1702. [Crossref]
- Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, et al. (2000) Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 288: 2354-2357. [Crossref]
- Mollinedo F, Gajate C (2006) FasL-Independent Activation of Fas. Fas Signaling, University of Wuerzburg Wuerzburg, Germany, FEBS J 276: 6912-6927. [Crossref]
- Krammer PH, Arnold R, Lavrik IN (2007) Life and death in peripheral T cells. Nat Rev Immunol 7: 532-542. [Crossref]
- Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, et al. (2002) Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signaling complexes in a FADD-dependent manner but cannot functionally substitute caspase-8. EMBO J 21: 4520-4530. [Crossref]
- Donepudi M, Mac Sweeney A, Briand C, Grütter MG (2003) Insights into the regulatory mechanism for caspase-8 activation. Mol Cell 11: 543-549. [Crossref]
- Saratoy S, Badrawy T and Koth S (2002) Expression of CD95 (FAS/APO-1) in Non-Hodgkin Lymphoma. E J Surg 21: 963- 969.
- Udristioiu A, Iliescu R, Udristioiu L, Cojocaru M (2011) A new approach of abnormal apoptosis as a cause of autoimmunity and malignancy. Biotechnology and Molecular Biology Review 6: 166-171.
- Warrell DA, Cox TM, John D (2003) Apoptosis in Health and Diseases. Andrew H, Wyllie J, Mark J. Oxford textbook of Medicine. Oxford Medical 4: 121-128.
- Elgert KD (2009) Tumor immunology. In: Immunology: understanding the immune system. Wiley, New York. 489-528.
- Elgert KD (2009) Cellular interactions: development of effector functions and their regulation. In: Immunology: understanding theimmune system. Wiley, New York. 321-368.
- Yap AS, Brieher WM, Gumbiner BM (1997) Structure and functions of classical cadherins. Ann Rev Cell Dev Biol 13: 119-146.
- Guicciardi ME, Gores GJ (2009) Life and death by death receptors. FASEB J 23: 1625-1637. [Crossref]
- Tanaka M, Suda T, Takahashi T, Nagata S (1995) Expression of the functional soluble form of human fas ligand in activated lymphocytes. EMBO J 14: 1129-1135. [Crossref]
- Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, et al. (1994) Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 76: 969-976. [Crossref]
- Shudo K, Kinoshita K, Imamura R, Fan H, Hasumoto K, et al. (2001) The membrane-bound but not the soluble form of human Fas ligand is responsible for its inflammatory activity. Eur J Immunol 31: 2504-2511. [Crossref]
- Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, et al. (1998) Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med 187: 1205-1213. [Crossref]
- Aoki K, Kurooka M, Chen JJ, Petryniak J, Nabel EG, et al. (2001) Extracellular matrix interacts with soluble CD95L: retention and enhancement of cytotoxicity. Nat Immunol 2: 333-337. [Crossref]
- Ottonello L, Tortolina G, Amelotti M, Dallegri F (1999) Soluble Fas ligand is chemotactic for human neutrophilic polymorphonuclear leukocytes. J Immunol 162: 3601-3606. [Crossref]
- Sekine C, Yagita H, Kobata T, Hasunuma T, Nishioka K, et al. (1996) Fas-mediated stimulation induces IL-8 secretion by rheumatoid arthritis synoviocytes independently of CPP32-mediated apoptosis. Biochem Biophys Res Commun 228: 14-20. [Crossref]
- Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, et al. (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 14: 5579-5588. [Crossref]
- Strasser A, Pellegrini M (2004) T-lymphocyte death during shutdown of an immune response. Trends Immunol 25: 610-615. [Crossref]
- Hughes R, Collins P, Rogers S (2008) Further experience of using azathioprine in the treatment of severe atopic dermatitis. Clin Exp Dermatol 33: 710-711. [Crossref]
- Turbyville JC, Rao VK (2010) The autoimmune lymphoproliferative syndrome: A rare disorder providing clues about normal tolerance. Autoimmun Rev 9: 488-493. [Crossref]
- Martin R (1998) Cell suicide for beginners. Nature 396: 119-122. [Crossref]
- Peter ME, Legembre P, Barnhart BC (2005) Does CD95 have tumor promoting activities? Biochim Biophys Acta 1755: 25-36. [Crossref]
- Neumann L, Pforr C, Beaudouin J, Pappa A, Fricker N, et al. (2010) Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol Syst Biol 6: 352. [Crossref]
- O' Reilly LA, Tai L, Lee L, Kruse EA, Grabow S, et al. (2009) Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature 461: 659-663. [Crossref]
- Lavrik IN, Krammer PH (2012) Regulation of CD95/Fas signaling at the DISC. Cell Death Differ 19: 36-41. [Crossref]
- Schütz C, Oelke M, Schneck JP, Mackensen A, Fleck M (2010) Killer artificial antigen-presenting cells: the synthetic embodiment of a 'guided missile'. Immunotherapy 2: 539-550. [Crossref]
- Kay RA (2003) The principles of adaptative immunity. Rheumatology. Edinburgh: Mosby. 113-126.
- Kiener PA, Davis PM, Starling GC, Mehlin C, Klebanoff SJ, et al. (1997) Differential induction of apoptosis by Fas-Fas ligand interactions in human monocytes and macrophages. J Exp Med 185: 1511-1516. [Crossref]
- Lenardo MJ (1996) Fas and the art of lymphocyte maintenance. J Exp Med 183: 721-724. [Crossref]
- Goebeler M, Trautmann A, Voss A, Bröcker EV, Toksoy A, et al. (2001) Differential and sequential expression of multiple chemokines during elicitation of allergic contact hypersensitivity. Am J Pathol 158: 431-440. [Crossref]
- Thepen T, Langeveld-Wildschut EG, Bihari IC, van Wichen DF, van Reijsen FC, et al. (1996) Biphasic response against aeroallergen in atopic dermatitis showing a switch from an initial TH2 response to a TH1 response in situ: an immunocytochemical study. J Allergy Clin Immunol 97: 828-837. [Crossref]
- Konur A, Schulz U, Eissner G, et al. (2005) Interferon (IFN)- gamma is a main mediator of keratinocyte (HaCaT) apoptosis and contributes to autocrine IFN-gamma and tumour necrosis factoralpha production. Br J Dermatol 152: 1134-1142. [Crossref]
- de Vries IJ, Langeveld-Wildschut EG, van Reijsen FC, Dubois GR, van den Hoek JA, et al. (1998) Adhesion molecule expression on skin endothelia in atopic dermatitis: effects of TNF-alpha and IL-4. J Allergy Clin Immunol 102: 461-468. [Crossref]
- Sayama K, Yonehara S, Watanabe Y, Miki Y (1994) Expression of Fas antigen on keratinocytes in vivo and induction of apoptosis in cultured keratinocytes. J Invest Dermatol 103: 330-334. [Crossref]
- Norris DA, Middleton MH, Whang K, Schleicher M, McGovern T, et al. (1997) Human keratinocytes maintain reversible anti-apoptotic defenses in vivo and in vitro. Apoptosis 2: 136-148. [Crossref]
- Vaux DL, Silke J (2005) IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol 6: 287-297. [Crossref]
- Matsue H, Kobayashi H, Hosokawa T, Akitaya T, Ohkawara A (1995) Keratinocytes constitutively express the Fas antigen that mediates apoptosis in IFN gamma-treated cultured keratinocytes. Arch Dermatol Res 287: 315-320. [Crossref]
- Romagnani S (1997) The Th1/Th2 paradigm. Immunol Today 18: 263-266. [Crossref]