Abstract
Heart failure (HF) is a major public health problem that is defined as a clinical syndrome that results when the heart is unable to provide sufficient blood flow to meet metabolic requirements or accommodate systemic venous return. Some systems participate as compensatory mechanisms, these include neurohumoral, Cellular immune activation and renin angiotensin system changes that lead to maladaptive condition. Several treatment strategies have been developed based upon the understanding of these compensatory mechanisms. In this paper, we review the general compensatory mechanisms, they lead to a maladaptive condition and the role that plays each component of renin angiotensin aldosterone system play in the development of heart failure.
Key words
heart failure, ACE, angiotensin II, ACE2, angiotensin (1-9), angiotensin (1-7), apelin
Renin angiotensin system role on heart failure
During the past two decades we have witnessed the advances of knowledge over renin angiotensin system. Although originally perceived as a classical humoral system in which biologically active molecules are secreted from a dedicated gland to reach their target tissue via the bloodstream, a number of observations have indicated that local renin angiotensin systems are complete with respect to all the components of the enzymatic pathway, may reside within individual organs or tissues. This is due in part to the advent of molecular biological methods that for the first time, allowed unequivocal confirmation of local synthesis of the elements have demonstrated the importance of a RAS in the brain, heart, peripheral blood vessels, adrenal glands, and kidney.
Angiotensinogen
It has been shown that a marked increase of ventricular angiotensinogen mRNA levels in pressure overload-induced cardiac hypertrophy as well as in the heart after myocardial infarction have been interpreted as indicating that increased wall stress may represent a trigger for angiotensinogen gene expression. In rats with chronic pressure overload and in rats with left ventricular failure early after coronary artery ligation of following tachypacing, cardiac angiotensinogen gene expression is enhanced, suggesting that the local generation of angiotensinogen within the myocardium may become more important during cardiac hypertrophy and failure [1-3].
Renin
Although cardiac renin mRNA expression is extremely low, renin activity is readily detectable in the heart [4] but increased expression of renin mRNA has been detected in the border zone of the infarcted left ventricle in rats, indicating that cardiac renin expression may be induced under pathological conditions [5]. Also renin has been demonstrated in isolated ventricular myocytes using a specific renin antibody [6] suggesting that synthesis occurs in cardiomyocytes. Renin has been thought to have no direct biological action. The renin receptor is a 350-amino-acid protein with a single trans membrane domain that specifically binds both renin and prorenin. Binding induced the activation of the extracellular signal-related MAPKs (ERK1 and ERK2) associated with serine and tyrosine phosphorylation and a 4-fold increase in the catalytic conversion of AGT to AG I. The receptor was localized in the glomerular mesangium and the sub endothelial layer of both coronary and renal arteries, associated with vascular smooth muscle cells and colocalized with renin [7]. A direct functional role of the renin/prorenin receptor might contribute to the generation of tissue ANGs in the heart, kidney, and/or peripheral blood vessels.
Ang II
Ang II is produced by the convertion locally rather than blood-derived Ang I [8]. In addition of the effects of Ang II on vasoconstriction and hypertension, cardiac hypertrophy and remodeling, recent evidence suggests that Ang II can also affect cardiac energy metabolism in heart failure [9,10]; decreased cardiac efficiency can contribute to the severity of the energy deficit. It has shown that chronic elevations in Ang II alter the relationship between fatty acid and carbohydrate oxidation in the heart, resulting in a shift from carbohydrate oxidation towards fatty acid β-oxidation [11]. While glycolysis may be increased in heart failure, the subsequent oxidation of the pyruvate (glucose oxidation) can be decreased, a finding similar to what occurs following Ang II treatment. In support of this, a recent study in pigs with dilated cardiomyopathy due to rapid pacing showed that myocardial glucose oxidation rates were impaired [12]. Ang II mediated damage of mitochondrial respiratory enzymes might cause further production of ROS, resulting in the further damage to the mitochondria, then may eventually lead to the reduction of overall mitochondrial oxidative phosphorylation. In an Ang II induced hypertensive model, diastolic dysfunction precedes systolic dysfunction and is accompanied by mitochondrial damage [13]. Furthermore, perturbation in cardiac energy metabolism, accompanied by cardiac hypertrophy, precedes the development of diastolic dysfunction. In contrast, in Tacrolimus-induced hypertensive rats (TAC-induced hypertensive rats), diastolic dysfunction is not associated with mitochondrial dysfunction, and mitochondrial damage does not occur until the development of systolic dysfunction [14].
ACE
Myocardial expression of ACE is increased in rats with pressure overload ventricular hypertrophy and in the viable myocardium following experimental myocardial infarction [15-19]. Moreover ACE has been shown to be upregulared in the myocardium of patients with end stage heart failure [20]. Increased ACE activity has been detected in the left ventricular aneurysms of patients after myocardial infarction [21]. A report from the same group indicated that ACE was expressed in fibroblasts, macrophages and endothelial cells present in the scar tissue and in the cardiac myocytes in the region adjacent to the infarct scar [22]. On the other hand some studies have challenged the notion that ACE represents the mayor Ang II forming enzyme in the human heart. Urata and coworkers [23,24] demonstrated that ACE inhibitors could block only the 10-20 % of total Ang I to Ang II conversion in left ventricular membranes prepared from human hearts.
AT1 Receptor
Two AT1 receptor subtypes, designated AT1A and AT1B, have been identified in rodents. The AT1A and AT1B receptor subtypes are encoded by distinct genes, but share a 96% homology at the amino acid level [25]. On the mRNA level, the AT1A receptor subtype is much more abundant than the AT1B receptor subtype in most tissues, including the heart [26]. Meggs and coworkers [27] demonstrated that cardiac myocytes isolated from rats with post-infarction left ventricular failure exhibit an increase in AT1 receptor density, which may be associated with an enhanced susceptibility to Ang II in vitro. In the same study it was shown that hypertrophied myocytes as those from control heart, do not express AT2 receptor binding sites. Haywood [28] demonstrated that AT1 receptor mRNA expression is reduced, whereas AT2 receptor mRNA expression is unchanged in failing human hearts. A similar conclusion was reached by Asano [29] who analysed the binding of radiolabeled Ang II to cardiac membranes prepared from terminally failing and control hearts: in the failing heart, AT1 receptor density was downregulated, whereas AT2 receptor density was unaltered. The NFkB pathway has been shown to mediate an increase in sympathetic nerve activity since its blockade reduces sympathetic outflow, AT1 expression and oxidative stress in rats with Cronic Heart Failure (CHF) and hypertension [30,31]. Also Liu et al. showed that Exercise training (ExT) reduced AT1 expression in the Rostral Ventrolateral medulla and reduced plasma Ang II. Similar results have been reported in rats with MI-induced CHF [32]. Exercise training in experimental CHF has been shown to reduce central AT1 and oxidative stress while at the same time increasing the sympatho-inhibitory effects of nitric oxide and improving baroreflex function [33]
AT2 Receptor
The cell signaling pathways involved in the activation of the AT2 receptor are not fully clarified, but appear to involve G protein-dependent and -independent pathways [33,34]. Recent evidence indicates that the receptor is G protein-coupled via Giα2 and Giα3 [35]. Current evidence suggests that AT2 receptor stimulation activates phosphotyrosine phosphatases, especially serine/threonine phosphatase 2A, protein kinase and phosphatase, and SHP-1 tyrosine phosphatase, resulting in the inactivation of MAPKs, specifically p42 and p44 MAPKs or ERKs [36]. A physiological role for the AT2 receptor in the cardiovascular system was first suggested by the observations that mice lacking the AT2 receptor have a slight but significant increase in baseline blood pressure [37,38]. The AT2 receptor was subsequently shown to mediate vasodilation by stimulating the production of BK, NO, and cGMP [39,40]. In the heart, the AT2 receptor inhibits growth and remodeling, induces vasodilation, and is up-regulated in pathological states [41,42]. Conflicting data on its antigrowth effects emerged from studies of mice lacking the AT2 receptor [43,44]. However, recent studies have helped to clarify the role of the AT2 receptor in cardiac remodeling post myocardial infarction and in cardiac hypertrophy and fibrosis due to Ang II infusion in mice overexpressing the AT2 receptor selectively in the myocardium. After myocardial infarction, AT2 receptor overexpression resulted in preservation of left ventricular global and regional function, indicating a beneficial role for the AT2 receptor in volume-overload states, including post-myocardial infarction remodeling. Overexpression of the AT2 receptor in cardiomyocytes was demonstrated to attenuate ANG II-induced cardiac interstitial fibrosis through BK/NO/cGMP pathway without effect on cardiomyocyte hypertrophy [45]. A very interesting information is that the AT1 receptor and the BK B2 receptor can communicate directly with each other. These two receptors can physically associate to form stable heterodimers in the cell membrane, resulting in the increased activation of G proteins Gαq and Gαi, the major signaling proteins mediating AT1 receptor responses [45]. The AT1 and AT2 receptors also have been shown to heterodimerize [46]. Thus, the AT2 receptor binds directly to the AT1 receptor, thereby antagonizing the signaling pathways and functions of the AT1 receptor. The direct inhibition of the AT1 receptor by the AT2 receptor binding does not depend on AT2 receptor-stimulated G protein activation. Furthermore, increased AT1/AT2 receptor heterodimerization in myometrial cells of pregnant women correlated with decreased ANG II responsiveness. The results of these studies were consistent with the concept that the AT2 receptor stabilizes AT1 receptor structurally so that it can no longer undergo the requisite conformational changes to activate G proteins. Thus, it appears that the AT2 receptor can be a direct AT1 receptor-specific antagonist by heterodimerization [47].The angiotensin AT2 receptor is an AT1 receptor antagonist.
ACE2
In the year 2000, ACE2 was discovered and was characterized as an enzyme similar to ACE [48]. Insight to the functional significance of ACE2 in the cardiac and intrarenal RAS has recently been provided using genetic approaches. The gene for ACE2 has been mapped to the X chromosome in humans, to the region that has been previously shown to be a quantitative trait locus for several rat models of hypertension [49]. Indeed, ACE mRNA and protein levels were down-regulated in the kidneys of these rat models, indicating that ACE 2 may be a candidate gene for the quantitative trait locus on the X chromosome. The increase in cardiac and plasma ACE2 activity noted after experimental MI is consistent with studies in patients with heart failure, where both cardiac ACE2 gene [50] and protein are increased. ADAM 17 (metallopeptidase domain 17), also known as tumor necrosis factor-α-converting enzyme (TACE) is a major protease that cleaves ACE2 from the cellular membrane to allow its shedding as a fully active soluble glycoprotein [68]. Donogue et al. reported that ACE gene ablation did not alter blood pressure, but severely impaired cardiac contractility and caused mild ventricular dilation and increased ANG II levels, suggesting that ACE2 may nullify the physiological actions of ACE. Ablation of both the ACE and ACE2 genes completely prevented the cardiac abnormalities and the increase in ANG II production [49]. These observations suggest a direct effect of ANG II on cardiac function and indicate that ACE2 probably counterbalances the enzymatic actions of ACE because it provides a potential major pathway for ANG (1-7)production. ACE2 is highly efficient in the conversion of ANG II to ANG [1–7], over 400-fold greater than for the conversion of ANG I to ANG [1–9]. The recent biosynthesis of a potent, selective ACE2 inhibitor will afford a means to determine the role of ACE2 in cardiovascular and renal function and disease [52]. On the other hand Xiao et al. [53] examined the sympathetic nervous effects in transgenic and wild type mice subjected to a chronic myocardial infarction and the subsequent development of CHF. While there were no major differences in cardiac function in both groups of mice, transgenic mice exhibited an improvement in arterial baroreflex function. Mice that overexpressed central ACE2 were able to suppress renal sympathetic nerve activity (RSNA) to zero during increases in blood pressure in contrast to wild type mice with CHF who could not lower RSNA in response to an increase in blood pressure. Examination of the spontaneous baroreflex control of heart rate also indicated an enhanced sympatho inhibitory process in these mice. In a recent study by Zheng et al. [54] it was shown that viral overexpression of ACE2 reduced RSNA in a rat CHF model. This effect was apparently mediated by an increase in nitric oxide. Another component that opposite the axis of AngI-ACE-AngII-AT1 receptor is the sACE2 (ACE2 soluble) that may provide a vasoprotective/antiproliferative mechanism resulting in the counter-regulation of the RAS [55]. The sACE2 was also implicated as a biomarker of definitive heart failure in animal models where absence of sACE2 caused severely impaired cardiac function. There is a wealth of information in human heart failure where myocardial sACE2 gene expression increased in patients with left ventricular dysfunction including thinning of the left ventricle and severe reduction in cardiac contractility. Burrell et al. observed an increase in sACE2 expression in failing human hearts independently of AT1R blockade [51]. There have been no reports on the combination of RAS blockade with either ACE2 overexpression or an ACE2 activator. Such studies are needed to test the hypothesis that augmentation of ACE2 over and above that due to cardiac injury itself would have further beneficial effects. Although there is great interest in this novel approach to cardiovascular diseases, further long-term studies are needed in experimental disease models as ACE2 overexpression may not always be beneficial. In ACE2 transgenic mice, there was a high incidence of sudden death that correlated with transgene expression levels and sustained overexpression of ACE2 in the heart of stroke-prone SHRs led to severe fibrosis and impaired cardiac function. Also, it has been reported that the overexpression of ACE2 in paraventricular nucleus attenuated Ang II-induced increase in the expression of TNF-a, IL-1b and IL-6. Up-regulation of IL-10, ACE2 and Mas expression was also reported in spontaneous hypertensive rats subjected to exercise of moderate intensity [55].
APELIN
Apelin is a second catalytic substrate for ACE2 and functions as an inotropic and cardioprotective peptide. While an antagonistic relationship between the RAS and apelin has been proposed, such functional interplay remains elusive. It was found that ACE2 was downregulated in apelin-deficient mice. Apelin is an endogenous peptide with a potent positive inotropic activity [56,57]. The physiological effects of apelin are exerted through binding to its receptor APJ, a G protein–coupled receptor that shares significant homology with the angiotensin II type 1 receptor (AT1 receptor) [58,59]. Under pathological settings, apelin signaling regulates cardiovascular functions including blood pressure, cardiac contractility, and fluid balance [60,61]. Apelin exerts load-independent positive inotropy and increases coronary blood flow by vascular dilation, thereby providing beneficial effects in failing hearts. In gene-targeting studies of apelin and APJ, Ashley EA, et al. and others have demonstrated that the endogenous apelin-APJ axis regulates heart contractility associated with aging, exercise, and pressure overload; in the absence of apelin or APJ expression, mutant mice show reduced contractile cardiac functions [62,63]. Ligand-independent but stretch-dependent APJ signaling has recently been shown to promote pathological cardiac hypertrophy [65-66]. However, the precise role of endogenous apelin signaling in heart function remained elusive.
Ang (1-7)
The ANG (1-7)heptapeptide fragment of ANG II was first discovered to have biological activity in 1988 (51). Since that time, several studies have documented that ANG (1-7) is a major biologically active peptide product of the RAS. ANG (1-7) can be formed directly from ANG I by the action of several peptidases, including neutral-endopeptidase (NEP) 24.11 or prolyl-endopeptidase (PEP) or from ANG II via PEP or prolyl-carboxypeptidase. Current data suggest that NEP 24.11 plays a major role in both circulating and tissue ANG (1–7) formation [67] . The first Ang (1-7) analogue, AVE0991, can promote vasorelaxation, lower blood pressure, and attenuate remodeling in a post-infarction animal model. Similarly, Ang (1-7) peptide was shown to rescue systolic dysfunction in ACE2-null mice in a pressure-overloaded induced HF model [68]. It was shown that by Yamamoto et al. that the levels of Ang (1-7) increase so 25 times after inhibition with an ACE inhibitor or an ARB. ACE2 is particularly abundant in the coronary circulation, playing an important role in the generation of Ang (1-7) [67]. The actions of Ang (1-7) are mainly antiproliferative, inhibits protein synthesis, and amplify the vasodilator effect of bradykinin and nitric oxide signaling stimulates cGMP/ protein kinase G also acts as an antiarrhythmic agent and protects infarction of the consequences of cardiac reperfusion injury [69]. De Mello in 2004 [70], showed that angiotensin (1-7) activates the sodium pump, hyperpolarizes heart cells and restores the arrhythmias because of ischemia by reperfusion. Also has been reported that angiotensin (1-7) can inhibit oxidative stress, stimulating the tissue plasminogen activator 1 platelet aggregation and can act as an agent anti-inflammatory. Fulvia et al. evaluated the chronic cardiac effects of a formulation developed by including angiotensin Ang (1–7) in hydroxypropyl β-cyclodextrin (HPβCD), in infarcted rats. Myocardial infarction (MI) was induced by left coronary artery occlusion and found once-a-day oral HPβCD/Ang (1–7) administration improved the cardiac function and reduced the deleterious effects induced by MI on TGF-β and collagen type I expression, as well as on the velocity and displacement of myocardial fibers. These findings confirm cardioprotective effects of Ang (1–7) and indicate HPβCD/Ang (1–7) as a feasible formulation for long-term oral administration of this heptapeptide. HPβCD/Ang (1–7) was administered for 60 days (76 μg/Kg/once a day/gavage) starting immediately before infarction [71], in keeping with previous studies showing beneficial effects of chronic Ang (1–7) administration in different models of cardiovascular diseases [72]. Chronic Ang (1–7) administration improved LV function of Wistar rats [73] and of diabetic spontaneously hypertensive rats (SHRs) after global ischemia, attenuated the heart failure induced by MI, prevented the development of severe hypertension and end-organ damage in SHR treated with L-NAME, and reduced the cardiac remodeling in DOCA-salt and in Ang II-infused rats. In addition, an antifibrotic effect was observed in transgenic animals, which chronically present an increased plasma Ang (1–7) levels [74]. In 2007, Chappel describes the signaling pathway of Ang (1-7) according to the effects it produces. Ang (1-7) participates in oxidative stress by inhibiting NADPH oxidase (NOX) which is the main enzyme generating reactive oxygen species such as superoxide anion [74-97] . Also inhibits the mitogen activated protein kinases (MAPK) (ERK 1 and 2), which promotes cell proliferation. More recently, several studies have shown that the counter-regulatory ACE2/Ang (1-7)/Mas axis may also influence inflammatory responses. Indeed, there is now much evidence demonstrating that Ang (1-7) modulates negatively leukocyte migration, cytokine expression and release, and fibrogenic pathways [97-106]. For example, studies by Sukumaran and co-workers have shown that the ACE2/Ang (1-7)/Mas axis is activated and is relevant for the anti-inflammatory effects of the ARBs Telmisartan and Olmesartan in a rat model of autoimmune myocarditis. ARBs increased ACE2, Ang (1-7) and Mas expression in line with reduction of pro-inflammatory cytokines as TNF-α, IFN-ϒ, IL-1b, IL-6 and increase of the anti-inflammatory cytokine, IL-10 [76]. These anti-inflammatory effects were associated with less myocardial fibrosis and down-regulation of PI3K, phospho-Akt, phospho-p38 MAPK, phospho-JNK, phospho-ERK and phospho-MAPK-2, but formal demonstration of the role of the ACE2/Ang (1-7)/Mas axis was not provided. The anti-proliferative and anti-fibrotic effect of Mas activation in the heart may also involve modulation of several extracellular matrix proteins. Neonatal and adult Mas-deficient mice showed significantly higher levels of collagen types I and III and fibronectin and reduced levels of collagen IV in both right ventricle and AV valves. In mice lacking the angiotensin receptors, Ang (1–7) reduced mean arterial pressure, suggesting that the AT2R may not be responsible for the vasodepressor effect of Ang (1–7) [77]. As well as cardioprotective effects with regards to fibrosis and hypertrophy. Ang (1–7) has recently been shown to promote angiogenesis in infarcted rat hearts in a Mas-dependent manner via the up-regulation of VEGF-D (vascular endothelial growth factor D) and MMP-9 (matrixmetalloproteinase-9) (78). Ang (1–7) has also been demonstrated to reduce the production of cardiac ROS (reactive oxygen species) in response to cardiac injury, as well as increased NO production, thus further providing a cardioprotective effect [79]. Beneficial functional effects of Ang (1–7) were also observed in ACE2-knockout mice in pressure-overload-induced heart failure. Ang (1–7)-infused animals showed normalization of fractional shortening, LVEDPs (LV end-diastolic pressures) and + dP/dtmax. This was accompanied by normalization of heart weight and hypertrophy marker expression levels, as well as attenuated NADPH oxidase activation. Moreover, the benefits observed were comparable with those observed in animals treated with the AT1R blocker irbesartan [80]. Many studies on the electrical properties of Ang (1–7) on the heart have been performed using ex vivo whole-heart Langendorff preparations, where anti-arrhythmogenic effects inischaemia/reperfusion injury has been clearly demonstrated [70,81,82]. Ang (1–7) delivered via an osmotic mini-pump in a dog pacing model of AF (atrial fibrillation) reduced interstitial fibrosis and, as a result, showed reduced susceptibility to and duration of induced AF. Again, this effect was believed to be modulated by a reduction in ERK1/2 signaling. In this model, Ang-(1–7) also attenuated the decrease in action potential duration, characteristically observed in atrial myocytes during AF, as well as preventing the decrease in expression of ICaL (L-type calcium channel) and ITO (outward potassium channel) observed in the model; however, the mechanism by which this occurs remains to be clarified [83-86]. This demonstrates that Ang (1–7) not only has potential benefits for cardiac function through anti structural remodeling, but also that it has the potential to alter ion channel and calcium-handling protein expression, resulting in modulation of cardiac function (Figure 1).
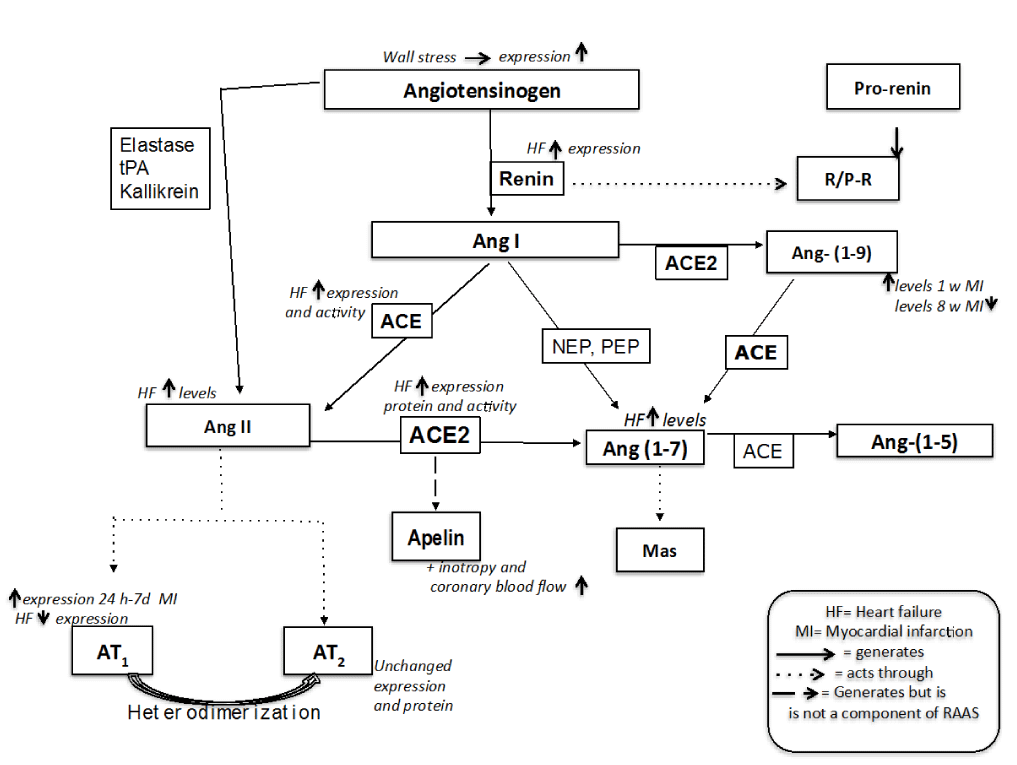
Figure 1. Renin Angiotens in Sstem and its regulation through the evolution of Miocardial in farction (MI) until Heart Failure (HF)
Ang (1-9)
Ang (1-9) is the product of the Ang I metabolism by ACE2. The first evidence to suggest that Ang (1–9) elicits independent biological effects in the heart was reported by Ocaranza et al. [87], using a rat coronary artery ligation model of MI, where cardiac hypertrophy and dysfunction are present at 8 weeks, and in which they assessed peptide and enzyme levels. It was found that, 1 week post-MI, circulating levels of Ang (1–9), along with levels of AngII, ACE, and ACE2, were increased compared with control animals; however, at 8 weeks, only Ang II and ACE remained high, with circulating levels of ACE2 and Ang (1–9) diminishing to levels lower than the control group. Treatment of the animals with the ACE inhibitor enalapril prevented the changes observed at 8 weeks, suggesting the generation of Ang (1–9) via ACE2 is able to counter-regulate Ang II-mediated actions. Chronic infusion of Ang (1–9) has been demonstrated by echocardiography to prevent LV wall thickening, LVESV (LV end-systolic volume) and LVEDV (LV end-diastolic volume) in comparison with the MI animals [88]. However, there was no change observed in cardiac function between MI and Ang (1–9)-infused MI animals as measured via LVEF (LV ejection fraction) and LVFS (LV fractional shortening). Ang (1–9) has also been reported to indirectly contribute to improved vascular function by stimulating bradykinin release in endothelial cells and enhancing the effects of bradykinin by augmenting NO and arachidonic acid release. Although Ang II is largely accepted to be pro-thrombotic and Ang (1–7) anti-thrombotic, evidence for Ang (1–9) is inconclusive. Florez-Munoz et al. [89] provided the first evidence to suggest a direct beneficial effect of Ang (1–9) in vascular function. Ang (1–9) infusion in the SHRSP improved aortic vasorelaxation and NO bioavailability via the AT2R (117). Although the mechanisms involved are currently unknown, it is possible that Ang (1–9) may increase NO bioavailability by stimulating bradykinin release, as described previously in cardiac endothelial cells, or by enhancing the activity of eNOS, as has been shown for Ang (1–7). Additionally, Ang (1–9) infusion and AT2 receptor stimulation resulted in an increase in aortic expression of NOX4 (NADPH oxidase 4), which has been demonstrated previously to promote vasodilation via the production of H2O2. However, this protective effect of NOX4 is vascular-bed-specific and therefore further investigation is required to fully assess the involvement of increased NOX4 levels in the aorta in response to Ang (1–9) infusion [90].
Grants
This study was supported by grants from DGAPA PAPIIT IN224310-3, IN 212213-3 UNAM CATEDRA CONS-26 FES Cuautitilán, Universidad Nacional Autónoma de México.
Disclosures
Authors declare to have no conflicts of interest
References
- Baker KM, Chernin MI, Wixson SK, Aceto JF (1990) Renin-angiotensin system involvement in pressure-overload cardiac hypertrophy in rats. Am J Physiol 259: H324-332. [Crossref]
- Lindpaintner K, Lu W, Neidermajer N, Schieffer B, Just H, et al. (1993) Selective activation of cardiac angiotensinogen gene expression in post-infarction ventricular remodeling in the rat. J Mol Cell Cardiol 25: 133-143. [Crossref]
- Finckh M, Hellman W (1991) Enhanced cardiac angiotensinogen gene expression and angiotensin converting enzyme activity in tachypacing- induced heart failure in rats. Basic Res Cardiol 86: 303-316
- Danser AH, van Kats JP, Admiraal PJ, Derkx FH, Lamers JM, et al. (1994) Cardiac renin and angiotensins. Uptake from plasma versus in situ synthesis. Hypertension 24: 37-48. [Crossref]
- Passier RC, Smits JF, Verluyten MJ, Daemen MJ (1996) Expression and localization of renin and angiotensinogen in rat heart after myocardial infarction. Am J Physiol 271: H1040-1048. [Crossref]
- Re R (1987) The myocardial intracellular renin-angiotensin system. Am J Cardiol 59: 56A-58A. [Crossref]
- van Kesteren CAM, Danser AHJ, Derkx FHM, Dekkers DHW, Lamers JMJ, et al. (1997) MADH. Mannose-6-phosplate receptor mediated internalization and activation of prorenin by cardiac cells. Hypertension 30: 1389–1396
- Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85: 1093-1129. [Crossref]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC (2010) Myocardial fatty acid metabolism in health and disease. Physiol Rev 90: 207-258. [Crossref]
- Mori J, Basu R, McLean BA, Das SK, Zhang L, Patel VB, et al.( 2012) Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ Heart Fail 5: 493–503.
- Lewis GD, Asnani A, Gerszten RE (2008) Application of metabolomics to cardiovascular biomarker and pathway discovery. J Am Coll Cardiol 52: 117-123. [Crossref]
- Turer AT1 (2013) Using metabolomics to assess myocardial metabolism and energetics in heart failure. J Mol Cell Cardiol 55: 12-18. [Crossref]
- SchroederMA, Lau AZ, Chen AP,Gu Y,Nagendran J, Barry J, et al. (2013) Hyperpolarized (magnetic resonance reveals early- and late-onset changes to in vivo pyruvate metabolism in the failing heart. Eur J Heart Fail 15: 130–40.
- Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, et al. (2011) Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 108: 837–846.
- Schunkert H, Dzau VJ, Tang SS, et al.(1990) Increased rat cardiac angiotensin converting enzyme activity and mRNA expression in pressure overload left ventricular hypertrophy. Effects on coronary resistance, contractility, and relaxation. J Clin Invest 86: 1913-1920
- Challah M, Nicoletti A, Arnal JF, et al. (1995) Cardiac angiotensin converting enzyme overproduction indicates interstitial activation in renovascular hypertension. Cardiovasc Res 30: 231–239
- Falkenhahn M, Franke F, Bohle RM, et al. (1995) Cellular distribution of angiotensin-converting enzyme after myocardial infarction. Hypertension 25: 219–226.
- Hirsch AT, Talsness CE, Schunkert H, Paul M, Dzau VJ (1991) Tissue-specific activation of cardiac angiotensin converting enzyme in experimental heart failure. Circ Res 69: 475–482
- Wollert KC, Studer R, von Bülow B, Drexler H (1994) Survival after myocardial infarction in the rat. Role of tissue angiotensin-converting enzyme inhibition. Circulation 90: 2457–2467.
- Studer R, Reinecke H, Müller B, Holtz J, Just H, et al. (1994) Increased angiotensin-I converting enzyme gene expression in the failing human heart. Quantification by competitive RNA polymerase chain reaction. J Clin Invest 94: 301-310. [Crossref]
- Hokimoto S, Yasue H, Fujimoto K, Sakata R, Miyamoto E (1995) Increased angiotensin converting enzyme activity in left ventricular aneurysm of patients after myocardial infarction. Cardiovasc Res 29: 664–669.
- Hokimoto S, Yasue H, Fujimoto K, et al. (1996) Expression of angiotensin-converting enzyme in remaining viable myocytes of human ventricles after myocardial infarction. Circulation 94:1513–1518
- Urata H, Healy B, Stewart RW, Bumpus FM, Husain A (1990) Angiotensin II-forming pathways in normal and failing human hearts. Circ Res 66: 883–890.
- Urata H, Kinoshita A, Perez DM, Misono KS, Bumpus FM, et al. (1991) Cloning of the gene and cDNA for human heart chymase. J Biol Chem 266: 17173-17179. [Crossref]
- Burson JM, Aguilera G, Gross KW, Sigmund CD (1994) Differential expression of angiotensin receptor 1A and 1B in mouse. Am J Physiol 267: E260-267. [Crossref]
- Nio Y, Matsubara H, Murasawa S, Kanasaki M, Inada M (1995) Regulation of gene transcription of angiotensin II receptor subtypes in myocardial infarction. J Clin Invest 95: 46-54. [Crossref]
- Meggs LG, Coupet J, Huang H, Cheng W, Li P, et al. (1993) Regulation of angiotensin II receptors on ventricular myocytes after myocardial infarction in rats. Circ Res 72: 1149-1162. [Crossref]
- Haywood GA, Gullestad L, Katsuya T, Hutchinson HG, Pratt RE, et al. (1997) AT1 and AT2 angiotensin receptor gene expression in human heart failure. Circulation 95: 1201-1206. [Crossref]
- Berry C, Touyz R, Dominiczak AF, Webb RC, Johns DG (2001) Angiotensin receptors: signaling, vascular pathophysiology, and interactions with ceramide. Am J Physiol Heart Circ Physiol 281: H2337-2365. [Crossref]
- Guggilam A, Cardinale JP, Mariappan N, Sriramula S, Haque M, et al. (2011) Central TNF inhibition results in attenuated neurohumoral excitation in heart failure: a role for superoxide and nitric oxide. Basic Res Cardiol 106: 273-286. [Crossref]
- Liu JL, Kulakofsky J, Zucker IH (2002) Exercise training enhances baroreflex control of heart rate by a vagal mechanism in rabbits with heart failure. J Appl Physiol 92: 2403-2408. [Crossref]
- Zheng H, Sharma NM, Liu X, Patel KP (2012) Exercise training normalizes enhanced sympathetic activation from the paraventricular nucleus in chronic heart failure: role of angiotensin II. Am J Physiol Regul Integr Comp Physiol 303: R387–394
- Hansen JL, Servant G, Baranski TJ, Fujita T, Iiri T, et al. (2000) Functional reconstitution of the angiotensin II type 2 receptor and G(i) activation. Circ Res 87: 753-759. [Crossref]
2021 Copyright OAT. All rights reserv
- Horiuchi M, Akishita M, Dzau VJ (1999) Recent progress in angiotensin II type 2 receptor research in the cardiovascular system. Hypertension 33: 613-621. [Crossref]
- Hein L, Barsh GS, Pratt RE, Dzau VJ, Kobilka BK (1995) Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor in mice. Nature 377: 744-747. [Crossref]
- Ichiki T, Labosky PA, Shiota C, Okuyama S, Imagawa Y, et al. (1995) Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type-2 receptor. Nature 377: 748-750. [Crossref]
- Siragy HM, Carey RM The subtype-2 (AT2) angiotensin II receptor regulates renal guanosine cyclic 3', 5' monophosphate and AT1 receptor-mediated prostaglandin E2 production in conscious rats. J Clin Invest 97: 1978–1982
- Siragy HM, Carey RM (1997) The subtype 2 (AT2) angiotensin receptor mediates renal production of nitric oxide in conscious rats. J Clin Invest 100: 264-269. [Crossref]
- Schneider MD, Lorell BH (2001) AT(2), judgment day: which angiotensin receptor is the culprit in cardiac hypertrophy? Circulation 104: 247-248. [Crossref]
- Ohkubo N, Matsubara H, Nozawa Y, Mori Y, Murasawa S, et al. (1997) Angiotensin type 2 receptors are reexpressed by cardiac fibroblasts from failing myopathic hamster hearts and inhibit cell growth and fibrillar collagen metabolism. Circulation 96: 3954-3962. [Crossref]
- Ichihara S, Senbonmatsu T, Price Jr E, Gaffney FA, Inagami T (2001) Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation 104: 346–351
- Senbonmatsu T, Ichihara S, Price Jr E, Gaffney FA, Inagami T (2000) Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J Clin Invest 106: R25–R29
- Yang Z, Bove CM, French BA, Epstein FH, Berr S, et al. (2002) Angiotensin II type 2 receptor over-expression preserves left ventricular function after myocardial infarction. Circulation 106: 106–111
- Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, et al. (2003) Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension 41: 99-107. [Crossref]
- AbdAlla S, Lother H, Quitterer U (2000) AT1 receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 407: 94–98
- AbdAlla S, Lother H, el Massiery A, Quitterer U (2001) Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat Med 7: 1003-1009. [Crossref]
- AbdAlla S, Lother H, Quitterer U (2000) AT1 receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 407: 94-98
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ (2000) A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275: 33238–33243
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, et al. (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE 2) converts angiotensin I to angiotensin (1–9). Circ Res 87: 1–9
- Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, et al. (2002) Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417: 822–828. [Crossref]
- Schiavone MT, Santos RA, Brosnihan KB, Khosla MC, Ferrario CM (1988) Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1-7) heptapeptide. Proc Natl Acad Sci U S A 85: 4095-4098.
- Xiao L, Gao L, Lazartigues E, Zucker IH (2011) Brain-Selective Overexpression of Angiotensin- Converting Enzyme 2 Attenuates Sympathetic Nerve Activity and Enhances Baroreflex Function in Chronic Heart Failure. Hypertension 58: 8.
- Zheng H, Liu X, Patel KP (2011) Angiotensin-converting enzyme 2 overexpression improves central nitric oxide-mediated sympathetic outflow in chronic heart failure. Am J Physiol Heart Circ Physiol 301: H2402–2412.
- Epelman S, Shrestha K, Troughton RW, Francis GS, Sen S, et al. (2009) Soluble angiotensin-converting enzyme 2 in human heart failure: relation with myocardial function and clinical outcomes. J Card Fail 15: 565–571
- Ferrario CM1 (2011) ACE2: more of Ang-(1-7) or less Ang II? Curr Opin Nephrol Hypertens 20: 1-6. [Crossref]
- Szokodi I, Tavi P, Földes G, Voutilainen-Myllylä S, Ilves M, et al. (2002) Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res 91: 434-440. [Crossref]
- Berry MF, Pirolli TJ, Jayasankar V, Burdick J, Morine KJ, et al. (2004) Apelin has in vivo inotropic effects on normal and failing hearts. Circulation 110: II187-193. [Crossref]
- O'Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, et al. (1993) A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 136: 355-360. [Crossref]
- Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, et al. (1998) Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun 251: 471-476. [Crossref]
- Santos RA, Campagnole-Santos MJ, Andrade SP (2000) Angiotensin-(1-7): an update. Regul Pept 91: 45-62. [Crossref]
- Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, et al. (2000) Characterization of apelin, the ligand for the APJ receptor. J Neurochem 74: 34-41. [Crossref]
- Reaux A, De Mota N, Skultetyova I, Lenkei Z, El Messari S, et al. (2001) Physiological role of a novel neuropeptide, apelin, and its receptor in the rat brain. J Neurochem 77: 1085-1096. [Crossref]
- Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, et al. (2005) The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res 65: 73-82. [Crossref]
- Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, et al. (2010) Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure. Circulation 121: 1818-1827. [Crossref]
- Kuba K, Zhang L, Imai Y, Arab S, Chen M, et al. (2007) Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ Res 101: e32-42. [Crossref]
- Charo DN, Ho M, Fajardo G, Kawana M, Kundu RK, et al. (2009) Endogenous regulation of cardiovascular function by apelin-APJ. Am J Physiol Heart Circ Physiol 297: H1904-1913. [Crossref]
- Yamamoto K, Chappell K, Broshinan KB, Ferrario CM (1992) In vivo metabolism of angiotensin I by neutral endopeptidase (EC3.4.24.11) in spontaneously hypertensive rats. Hypertension 19: 1202–1211
- Patel VB, Bodiga S, Fan D, et al. (2012) Cardioprotective effects mediated by angiotensin II type 1 receptor blockade and enhancing angiotensin 1-7 in experimental heart failure in angiotensinconverting enzyme 2-null mice. Hypertension 59: 1195–1203.
- Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF,Heringer-Walther S, et al. (2005) G-protein-coupledreceptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 111: 1806–1813.
- De Mello WC (2004) Angiotensin (1–7) re-establishes impulse conduction in cardiac muscle during ischaemiareperfusion. The role of the sodium pump. J Renin Angiotensin Aldosterone Syst 5: 203–208
- Fulvia D Marques, Marcos B Melo, Leandro E Souza, Maria Claudia C, Irigoyen 1 Ruben, D Sinisterra, et al. (2012) Beneficial effects of long term Aministration of an Oral Formulation of Angiotensin (1-7) in Infarcted Rats Int Journal of Hypert.
- Santiago NM, Guimarães PS, Sirvente RA, Oliveira LA, Irigoyen MC, et al. (2010) Lifetime overproduction of circulating Angiotensin-(1-7) attenuates deoxycorticosterone acetate-salt hypertension-induced cardiac dysfunction and remodeling. Hypertension 55: 889-896. [Crossref]
- Santos RA, Castro CH, Gava E, Pinheiro SV, Almeida AP, et al. (2006) Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor MAS knockout mice. Hypertension 47: 996-1002. [Crossref]
- Shenoy V, Grobe JL, Qi Y, Ferreira AJ, Fraga-Silva RA, Collamat Get al. (2009) 17beta-Estradiol modulates local cardiac renin-angiotensin system to prevent cardiac remodeling in the DOCA-salt model of hypertension in rats. Peptides 30: 2309–2315.
- Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, et al. (2007) Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1-7). Am J Physiol Heart Circ Physiol 292: H736-742. [Crossref]
- Sukumaran V, Veeraveedu PT, Gurusamy N, Yamaguchi K, Lakshmanan AP, et al. (2011) Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin-converting enzyme-2/angiotensin 1-7/mas receptor axis. Int J Biol Sci 7: 1077-1092. [Crossref]
- Sukumaran V, Veeraveedu PT, Gurusamy N, Lakshmanan AP, Yamaguchi K, et al. (2012) Telmisartan acts through the modulation of ACE-2/ANG 1-7/mas receptor in rats with dilated cardiomyopathy induced by experimental autoimmune myocarditis. Life Sci 90: 289-300. [Crossref]
- Gava E, de Castro CH, Ferreira AJ, Colleta H, Melo MB, et al. (2012) Angiotensin-(1-7) receptor Mas is an essential modulator of extracellular matrix protein expression in the heart. Regul Pept 175: 30-42. [Crossref]
- Gembardt F, van Veghel R, Coffman TM, Schultheiss HP, Danser AH, et al. (2012) Hemodynamic effects of vasorelaxant compounds in mice lacking one, two or all three angiotensin II receptors. Hypertens Res 35: 547-551. [Crossref]
- Zhong J, Basu R, Guo D, Chow FL, Byrns S, et al. (2010 Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 122: 717–728
- Zhao W, Zhao T, Chen Y, Sun Y1 (2015) Angiotensin 1-7 promotes cardiac angiogenesis following infarction. Curr Vasc Pharmacol 13: 37-42. [Crossref]
- Patel VB, Bodiga S, Fan D, Das SK, Wang Z, et al. (2012) Cardioprotective effects mediated by angiotensin II type 1 receptor blockade and enhancing angiotensin 1-7 in experimental heart failure in angiotensin-converting enzyme 2-null mice. Hypertension 59: 1195-1203. [Crossref]
- Ferreira AJ, Santos RA, Almeida AP (2001) Angiotensin-(1-7): cardioprotective effect in myocardial ischemia/reperfusion. Hypertension 38: 665-668. [Crossref]
- Liao X, Wang L, Yang C, He J, Wang X, et al. (2011) Cyclooxygenase mediates cardioprotection of angiotensin-(1–7) against ischemia/reperfusion-induced injury through the inhibition of oxidative stress. Mol Med Rep 4: 1145–1150
- Liu E, Yang S, Xu Z, Li J, Yang W, et al. (2010) Angiotensin-(1-7) prevents atrial fibrosis and atrial fibrillation in long-term atrial tachycardia dogs. Regul Pept 162: 73-78. [Crossref]
- Ocaranza MP, Godoy I, Jalil JE, Varas M, Collantes P, et al. (2006 Enalapril attenuates downregulation of Angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension 48: 572–578
- Ocaranza MP, Lavandero S, Jalil JE, Moya J, Pinto M, et al. (2010) Angiotensin-(1-9) regulates cardiac hypertrophy in vivo and in vitro. J Hypertens 28: 1054-1064. [Crossref]
- Jackman HL, Massad MG, Sekosan M, Tan F, Brovkovych V, et al. (2002) Angiotensin 1-9 and 1-7 release in human heart: role of cathepsin A. Hypertension 39: 976-981. [Crossref]
- Flores-Munoz M, Work LM, Douglas K, Denby L, Dominiczak AF, et al. (2012) Angiotensin-(1–9) attenuates cardiac fibrosis in the stroke-prone spontaneously hypertensive rat via the angiotensin type 2 receptor. Hypertension 59: 300–307
- Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, et al. (2011) Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol 31: 1368-1376. [Crossref]