Intensification in the frequency of diabetes and the associated vascular complications has been a root cause of blindness and visual impairment worldwide. One such vascular complication which has been the prominent cause of blindness; retinal vasculature, neuronal and glial abnormalities is diabetic retinopathy (DR), a chronic complicated outcome of Type 1 and Type 2 diabetes. It has also become clear that “genetic” variations in population alone can’t explain the development and progression of diabetes and its complications including DR. DR experiences engagement of foremost mediators of diabetes such as hyperglycemia, oxidant stress, and inflammatory factors that lead to the dysregulation of “epigenetic” mechanisms involving histone acetylation and histone and DNA methylation, chromatin remodeling and expression of a complex set of stress-regulated and disease-associated genes. In addition, both elevated glucose concentration and insulin resistance leave a robust effect on epigenetic reprogramming of the endothelial cells too, since endothelium associated with the eye aids in maintaining the vascular homeostasis. Furthermore, several studies conducted on the disease suggest that the modifications of the epigenome might be the fundamental mechanism(s) for the proposed ‘metabolic memory’ resulting into prolonged gene expression for inflammation and cellular dysfunction even after attaining the glycemic control in diabetics. Henceforth, the present review focuses on the aspects of genetic and epigenetic alterations in genes such as vascular endothelial growth factor and aldose reductase considered being associated with DR. In addition, we discuss briefly the role of the thioredoxin-interacting protein TXNIP, which is strongly induced by high glucose and diabetes, in cellular oxidative stress and mitochondrial dysfunction potentially leading to chromatin remodeling and ocular complications of diabetes. The identification of disease-associated genes and their epigenetic regulations will lead to potential new drugs and gene therapies as well as personalized medicine to prevent or slow down the progression of DR.
genetics, epigenetics, diabetic retinopathy, vascular complications, mitochondrial dysfunction
Diabetes, obesity and pre-diabetes are increasing enormously around the globe with multiplying of middle class families in developed as well as in developing countries together fueled by modern sedentary work-related physical inactivity and easy access to processed high calorie foods. Nonetheless, the disease initiation and progression of diabetes and its complications can’t be fully explained by known genetic mutation and polymorphisms alone in these diverse populations thereby advocating an environmental factor that influences the disease-associated gene functions. Thus, epigenetics is considered as a phenomenon that is beyond genetics, having its association with development involving interaction of numerous genes with each other and with the environment without changes in DNA sequences. Nonetheless, in the course of 50 years till date, the significance of the term “epigenetics” has itself suffered an evolution that equals our amplified knowledge of the molecular mechanisms essentially regulating the gene expression in eukaryotes [1]. Waddington in the year 1942 coined the term “epigenotype” to initiate the study of fundamental mechanisms like DNA methylation, RNA regulation and histone alterations [2]. These mechanisms alter the regular metabolic processes by heritable gene silencing and also do not cause any changes to nucleotide sequences [3]. In a study by Holliday, cytosine methylation was observed in DNA and consistent suppression of gene expression in higher order organisms [4] due to this epigenetic DNA modification implying its significant influence on tissue-specificity and the process of gene silencing and expression. Likewise, numerous nutritional and environmental studies exemplify the influence of epigenetic modifications in an organism. An African-American study demonstrated epigenetic factors similar to psychological stress and social context that are correlated with swelling and infection in heart diseases and strokes [5]. Previous studies validated the contribution of epigenomics in the therapeutics of breast cancer [6] in which it was observed that several dietary chemo-preventive agents (retinoids/Vitamin A, green tea, Vitamin D, etc.) acted on the miRNA signaling pathways, so as to obstruct the uncontrolled metabolic mechanisms of breast cancer. Certainly, literature available has also concluded that dietary supplements and environmental conditions have contributed to the diverse mechanistic patterns involved in epigenomics during the initial and advanced stage of obesity [7,8] as well.
In the same way, in early 90s Hales and Barker described a vital role of epigenetic modifications in the development of diseases, elucidating Type 2 diabetes (T2D) as the major breakthrough [9] since T2D has been a foundation of disability and morbidity associated to the vascular complications prevailing the onset and development of neuropathy, retinopathy, ischemic heart disease, nephropathy, and peripheral vasculopathy. This was the major hypothesis, deep-rooted by various epidemiological case studies further investigating the consequences of insulin resistance in postnatal, childhood and adulthood tenure [10-14]. Thus, the studies conducted so far towards an association between genetic and environmental factors in the development of diseases indicate epigenetic alterations [15, 16] and have an important role in sustained metabolic changes in diabetes leading to its complications.
Supplementary to the above studies, the stability of DNA is a question of great concern while epigenetic modifications are dynamic and reversible in nature and henceforth are the most potent and promising targets for the pharmacological mediations. According to the statistics of International Federation of Diabetes (IFD) [17,18] more than 382 million people were affected with diabetes in the year 2013, and the remaining were left undiagnosed. It has also being estimated that the number of cases is expected to rise to 592 million till 2035, this will thus suppress the health, life span and productivity of an individual. There will be an enormous burden on health costs across the globe and not only this; it will affect quality of life, socio-economic status, lifestyle and pathological manifestations of an entity. The progressive role of diabetes on macro- and microvascular complications has become a great concern for the scientific community. The hallmarks for diabetes are the defective insulin secretion/resistance, resulting into hyperglycemia. Recently, American Diabetes Association (ADA) had published its guidelines for diabetic care, which is contemplative over the use and exploration on the needs to individualize treatment objectives and plans [19]. Therefore, diagnosis, treatment, prevention on the genetic, phenotypic and clinical manifestations of a particular individual forms the foundation of personalized or precision medicines, with management strategies to combat the disease more effectively.
Over-and-above, ADA explained three subtypes of diabetes [19] viz., Type 1 diabetes (T1D), Type 2 diabetes, and gestational diabetes. Of these, T2D is the prime and prevalent diabetes covering 90% of all cases, thus causing indisposition and mortality in the developed and developing nations [17]. Also, the complications of this emerging disease is dreadful, consequences of which can be identified and diagnosed on endothelial tissues and cells of retina, peripheral neurons, cardiac and renal organs of an individual. The molecular mechanisms and glucose abundant pathways are complex but may include an elevated hexosamine pathway flux, polyol pathway, diacylglycerol PKC pathway, AGE-RAGE pathway and mitochondrial dysfunction, oxidative stress, and bioenergetics failure (Figure 1). In addition, the thioredoxin-interacting protein (TXNIP), which binds to thioredoxin (Trx) and inhibits it’s thiol reducing and oxidant scavenging capacity, has recently been shown to involve in cellular oxidative stress, NLRP3 inflammasome activation, inflammation, and apoptosis of pancreatic b cells and other cell types in diabetes suggesting a critical role for TXNIP in diabetes and its complications [20,21].
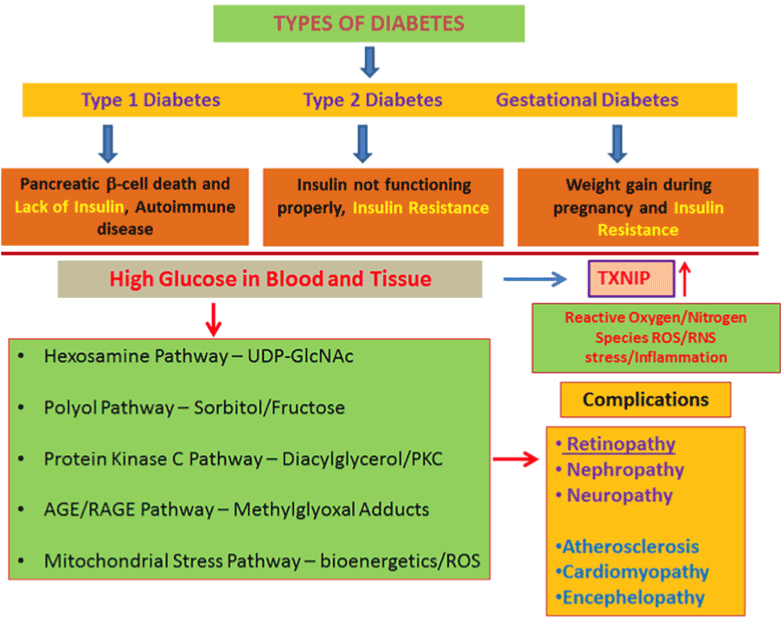
Figure 1. Hyperglycemia and potential metabolic pathways involved in the pathogenesis of diabetic complications including retinopathy. TXNIP is highly induced by diabetes and high glucose in most cell types examined thus far, and enhanced glucose metabolites such as glucose-6-phosphate, fructose-6-phosphate and glucosamine-6-phosphate as well as RAGE activation are known to activate TXNIP gene expression. TXNIP has been defined as a pro-oxidative stress, pro-inflammatory and pro-apoptotic protein involved in diabetes and its complications [20,21].
Moreover, for epigenetic mechanisms there has been various marks; one such major, countable and primary mark is DNA methylation, which involves the addition of a methyl group to the DNA fragment at nucleotide cytosine [16, 22,23]. DNA methyltransferases, DNMT1 and DNMT3A/B, use S-adenosylmethionine (SAM) as methyl donors to cytosine. Cytosine methylation, in general, represents repressive DNA via chromatin closing [24-27].
Second still prominent is histone modifications such as arginine methylation; lysine methylation and acetylation [28, 29] and alterations in ncRNAs (miRNAs, piwi RNAs, and long non-coding RNAs). These are the principle components involved in the epigenetic gene regulation of diabetes [30] and its complications. Histone acetyltransferases (HAT) adds an acetyl group to histone lysine using acetyl-coA as a substrate while histone deacetylases (HDACs) remove the acetyl group. Histone acetylation is a marker for chromatin opening and gene transcription.
Conversely, histone lysine or arginine can be alternatively methylated using histone methyltransferases and SAM as substrates. Histone methylation and DNA methylation condenses chromatin, making them inaccessible to transcription factors and co-factors, thereby inhibiting gene transcription or silencing [24]. Consitt et al. [31] and Liu et al. [32] studied interactions amidst epigenetic and environmental factors (lifestyle and principally dietary practices), in the progression of T2D and its complications.
However, diabetes-specific macro- and microvascular diseases in the glomerulus, retina, and vasa nervorum have comparable pathophysiological features. In the initial course of diabetes, intracellular hyperglycemic condition causes anomalies in blood flow thus increasing vascular permeability. This reveals reduced action of vasodilators such as nitric oxide, and amplified activity of vasoconstrictors like angiotensin II and endothelin-1, and amplification of the permeability factors such as vascular endothelial growth factor (VEGF). Similarly, the polymorphic activities in the promoter region of the VEGF gene along with aldose reductase (ALR) 2 gene run parallel with the pathogenesis of diabetic nephropathy [33] and might have its effect on retinopathy as well. In addition, both hypoxia and hyperglycemia enhances VEGF and its receptor expression, because of which elevated VEGF have been demonstrated in diabetic retinas leading to the chronic retinopathy complications [34-38]. Besides VEGF, some of the databases available also provides information that aldose reductase (aka aldehyde reductase) is expressed in most of the mammalian tissues and is found at high concentrations in sciatic nerve, retina, seminal vesicles, lens, and renal medulla [39], that could initiate the complications when hyperglycemia persists. So, there has been a considerable interest in the development and expansion of pharmacological inhibitors aiming these genes as a method of averting the complications linked with chronic hyperglycemia affecting visual dysfunction before the progress of retinopathy.
Henceforth, the intent of the review is to provide an overview of genetics and epigenetics involved in the metabolic pathway of diabetes and its complications prominently converging on diabetic retinopathy, leading to the notion of personalized medicines, concentrating over patient centric approach in conclusion. The article also comprises of the discussion of some candidate genes and their pathway connectivity. Previous findings suggest that the two candidate genes VEGF and ALR belong to the families that are closely associated (mutations/alterations/modifications) with diabetes and its complications (retinopathy). In addition, recent finding that TXNIP is strongly induced in pancreatic b cells and other tissues including the retina has proposed to be a potential target for diabetes and its complications. TXNIP has been defined as a pro-oxidative stress, pro-inflammatory and pro-apoptotic protein in diabetes and under hyperglycemic conditions. In short this article revolves around the polymorphic depiction of candidate genes, interaction with environment causing epigenetic changes, and their potent association with the susceptibility of retinopathy [40] and associated complications.
Capability of pancreatic β cells to secrete and produce insulin in response to glucose fluctuations is one of the main features to regulate glycemia in normal entities. Table 1 depicts some epigenetic molecules that aid the treatment and therapeutics of DR either being anti-inflammatory or delaying the onset of nephropathy and retinopathy or initiating or hindering β cell differentiation. Throughout the commencement and growth of DR, the need for insulin increases because of the increased insulin resistance in the body as seen in T2D or lack of insulin in T1D. Henceforward, insulin production, cell viability, and secretion potential are mechanisms that distress the pancreatic β cell and their functions. Studies also anticipated that the altered DNA methylation patterns (genome-wide) of human cells are obtained from the pancreatic islets of the deceased donors [41,42]. Current findings suggest that from a total of 1649 CpG sites corresponding to 853 genes, there have been alterations in the level of DNA methylation patterns in pancreatic islets from diabetic T2D patients versus non-diabetic individuals. Likewise, there were 102 genes presenting distinct DNA methylation that directed towards the conclusion of modified mRNA expression between the non-diabetic and diabetic patients (T2D), signifying epigenetic regulation of transcriptional activity [41,42].
Table 1. Epigenetic molecules of latent interest for diabetes treatment.
Epigenetic molecules |
Activity |
Effect |
Reference |
Trichostatin A |
HDACi |
Anti-inflammatory
Insulin sensitivity restoration
Nephropathy onset delay
Retinopathy onset delay
β-cell differentiation
Glucose uptake |
[162]
[163]
[164]
[165]
[166]
[167] |
Vorinostat (SAHA) |
HDACi |
Anti-inflammatory
Nephropathy progression |
[168]
[160] |
Givinostat (ITF2357) |
HDACi |
Anti-inflammatory
Increased insulin secretion |
[170]
[171] |
THS-78–5 |
HDACi |
Cyto-protective effect |
[172] |
Scriptaid |
HDACi |
Insulin sensitivity restoration |
[173] |
MS275 |
HDACi |
Insulin sensitivity restoration |
[174] |
Sodium butyrate |
HDACi |
β-cell differentiation |
[175] |
MC1568 |
HDACi |
β-cell differentiation |
[176] |
ANAC |
HATi |
Glucose uptake |
[177] |
Garcinol |
HATi |
Anti-inflammatory |
[178] |
Curcumin |
HATi |
Decreased ECM proteins diabetic vascular complication |
[179] |
SPV106 |
HATa |
Rescue of diabetic phenotype |
[180] |
5-Azacytidine |
DNMT inhibitor
Ngn3 inducer |
β-cell differentiation |
[181] |
Indolactam V |
Pdx1 inducer |
β-cell differentiation |
[182] |
Retinoic acid |
Ngn3 inducer |
β-cell differentiation |
[183] |
BRD7552 |
Pdx1 inducer |
β-cell differentiation |
[184] |
WS6 |
IκB kinase Activator |
β-cell proliferation |
[185] |
DR is becoming the foremost reason of blindness among the working individuals in the developed countries and among the elderly individuals in the developing countries. With the worldwide dominance of diabetes being anticipated to intensify to 438 million subjects by the year 2030, DR will undoubtedly pose as one of the major public health concerns [43,44]. The warning signs for the occurrence of DR are increased blood sugar levels, hazy vision, sudden loss of vision, etc. [45]. DR may lead to macular edema when blood and fluid leak into the retina caused by swelling of the central retina [46]. Clinically, the occurrence of DR is manifested by the advent of retinal microvascular lesions.
An initial change include hard exudates, intra-retinal microvascular abnormalities, hemorrhages, cotton wool spots, microaneurysms, and beading in the veins thus illustrating non-proliferative diabetic retinopathy (NPDR). The most severe form of DR is its proliferative form, as proliferative diabetic retinopathy (PDR) that is noticeable by the formation of irregular fragile and friable new blood vessels, which are susceptible to hemorrhage outflow more often as a final point, visual impairment results [44].
With an advent of diabetes and its duration, DR proliferates with various clinical complications, though the initial glycemic control can delay the effect and expansion of DR, it cannot stop the progression of DR [44,47,48]. Thus, the phenomenon of metabolic memory or epigenetic memory has been proposed for the aberrant gene expressions even after normalization of blood glucose once a specific period of hyperglycemic exposure had previously occurred [24-27,49]. To contest this disease therefore candidate gene approach is an essentiality to study the pathogenic mechanisms underlying DR [50-52].
Consistently, numerous genes involved in DR pathways have been targeted as prospective candidate gene segments namely vascular endothelial growth factor, angiotensin-I converting enzyme, glucose transporter 1, angiotensinogen, aldose reductase, transforming growth factor b, angiotensin II type 1 receptor, receptor for advanced glycation end products, inducible and constitutive nitric oxide synthases, endothelial isoforms, and others [53-62]. Various techniques have been used for detection, evaluation and diagnosis of DR using B-scan ultrasonography, fundo-scopic photography, optical coherence tomography and fluorescence angiography [63].
Vascular endothelial growth factor gene (Human chromosome 6p12)
Endothelial growth factors that are involved in the vascular activities function as signaling proteins for both de novo development of the embryonic circulatory system and for the growth of new blood vessels from the already existing vasculature. Secretion of VEGF in the retina of an organism is primarily done from retinal pigmented epithelial cells, Müller glial cells, astrocytes, endothelial cells and pericytes. This growth factor consists of several members such as VEGF-A, VEGF-B, VEGF-C, VEGF-D and PGF (placental growth factor) [64,65]. The numerous polymorphic activities specifically identified and regulated in the promoter regions of the gene also make it a promising gene model to study the vascular complications and the disease associations. These events are indulged in the signaling pathways associated with the metabolic regularities and irregularities of an organism. VEGF being an attractive candidate gene for the study of DR is associated with the development of diabetic macular edema (DME). This event is in association with the polymorphic activity of C-634G which is present in Japanese [66,67] as well as in Indian populations [62,67,68]. Among the referred studies [66], 378 patients with T2D were examined, out of which 203 patients had no retinopathy, 93 had NPDR, and 82 had PDR. The polymorphic study demonstrated that macular edema was present in 16 patients with NPDR and 47 patients with PDR [66]. Other studies had also instigated the role of other VEGF-SNPs in retinopathy initiated with an advent of early diabetes [69,70] leading to the chronic complications. Moreover, at present several clinical trials are exploring and inspecting the effectiveness of anti-VEGF molecules to aid in the treatment of diabetic retinopathy.
History of VEGF molecule: In 1948, Michaelson discussed a vital event in his studies that “in the pathological angiogenesis, there has been observed a secretion and synthesis of a diffusible factor known as “Factor X” by dint of the ischemic retina” [71]. Later in 1971 studies conducted by Folkman, demonstrated the inhibition of angiogenesis for the treatment of cancer, that paved the way to unearthing the anti-angiogenic factors [72]. Outlying studies in 1983 by Senger et al. discovered that a protein mediator is being secreted from the guinea pig tumor cell line that has its active involvement in angiogenesis. This protein has the efficiency to persuade vascular leakage which is why it has been named as Vascular Permeability Factor (VPF) [73].
Similarly in 1989, Ferrara and Henzel acknowledged a molecule existing in bovine pituitary follicular cells and termed it as Vascular Endothelial Growth Factor [74]. Consequently, via cloning of VEGF and VPF confirmed that the two factors have the same tendency and are actually the same proteins [65,75,76].
Substantiations from the previous clinical studies had supported the acute role of VEGF in ophthalmic neovascularization. Further, it was also concluded that the reason behind the elevation of VEGF level in vitreous samples of patients was active proliferative diabetic retinopathies [77] and its associated complications.
VEGF action: Transphosphorylation is a mechanism that activates dimers of tyrosine kinase receptors. These receptors are present on the endothelial cell surface and binds to the VEGF members in turn stimulating the cellular responses. The first member of the VEGF family: VEGF-A is having 2 type of receptors namely, VEGF receptor 1 (VEGFR-1) and 2 (VEGFR-2). VEGFR-1 is a protein present in humans that is encoded by Flt-1 gene [78] and VEGFR-2 is a receptor that has the kinase insert domain which is encoded by KDR [79].
Primary receptor that facilitates the cellular responses to VEGF-A is VEGFR-2. Almost all the receptors are compiled of three parts:
- An extracellular portion, be made up of seven immunoglobulin-like domains (similar)
- A transmembrane hydrophobic spanning region (single)
- An intracellular portion comprising of a tyrosine kinase domain (split) [80].
A cellular signal is transduced when the molecule (VEGF) binds to the extracellular portion of the receptors that is having the immunoglobulin-like domains; this causes the intracellular portion to process phosphorylation of the tyrosine residues, this in turn causes a cascading effect in signaling pathways [81].
VEGF in DR: VEGF plays a dynamic role in the neovascularization in PDR and also in the collapse of blood-retinal barrier, during the emergence of macular edema in diabetic patients [67], in turn altering the permeability of retinal capillaries by enhancing the content of phosphorylation of proteins indulged in the tight-junctions like zonula occludens [82]. Significantly, elevated vitreous levels of VEGF molecules had been a major setback reported in the patients suffering from DR [67,83]. Induction of VEGF molecules activates mitogen-activated proteins, causing the proliferation of endothelial cell. This signaling cascade overlaps with the stimulation of phophatidylinositol 3-kinase pathway after the induction of VEGFR-2 [84].
Another VEGF molecule is VEGF-A which initiates endothelial cells to discharge matrix metallo-proteinases and urokinase-plasminogen activator that results in the degradation of membranes precisely basement membranes which aid in the possible cell migration [85]. Propagation and passage of vascular endothelial cells is tailed by the fabrication of the basement membranes for the capillaries that are formed recently. Stability of these newly formed capillaries is attained by staffing the smooth muscle cells and pericytes that are under control/regulation by platelet derived growth factors [86].
VEGF as a therapeutic target: The mechanisms of VEGF molecules have been well studied in order to reduce neovascularization and vessel leakage by obstructing their effects.
Inhibitors of intracellular Transcripts: The leading drug: Bevasiranib (OPKO, FL, USA) that has been subjected to siRNA technique has been designed to silence the expression of the genes indulged in the production of VEGF. This drug hinders the production of VEGF molecules so as to inhibit the generation of DME. DME is a progressive, vision-limiting barrier of DR that affects almost 30% of patients who are sufferers of diabetes for more than 20 years and is accountable for much of the vision loss due to the proliferation of DR. The standard method of care for DME is macular laser photocoagulation, which stabilizes the vision and reduces the vision rate loss by 50%; however, the frequency of significant vision recovery has been observed in only 15% of treated patients. Further, chronic retinal microvascular impairment results in the elevation of intraocular levels of VEGF-A, which has been a potent, diffusible, endothelial-specific mitogen that mediates various significant physiologic progressions, including the development and penetrability of the vasculatures. Thus this identification of VEGF as a vital mediator of DME recommended that anti-VEGF therapy carried might lead to enhanced and improved visual outcomes as the counter-attack for the disease [87].
Inhibitors of VEGF receptor expression: Aflibercept (Regeneron Pharmaceuticals Inc. and the Sanofi-aventis Inc.) is a complete human fusion protein (recombinant) that muddles with all VEGF-A molecules [88]. This drug has been developed for its utilization in ocular diseases by Regeneron and Bayer Inc. (Leverkusen, Germany) companies. Further, the DA VINCI approach exhibited promising conclusions as compared to laser photocoagulation in cases of DME [89,90].
Anti-VEGF antibodies: Bevacizumab (Avastin, Genentech Inc.) is a humanized monoclonal antibody contrary to all VEGF-A molecules, thus preventing the receptor binding. This monoclonal antibody effectively aid in obstructing neovascularization leading to various retinal diseases such as PDR, DME, macular edema and neovascular glaucoma [91]. Furthermore, Ranibizumab (Lucentis, Genentech Inc.) is also a humanized monoclonal antibody (mab) fragment recovered from the parent molecule of bevacizumab in contrast to VEGF-A [92]. This mab was aimed for improved intra-ocular penetration into the retina [93].
Inhibitors of extracellular VEGF: A RNA aptamer, Pegaptanib sodium (Macugen, Eyetech Pharmaceuticals Inc. and Pfizer Inc.) has been considered as the earliest anti-VEGF drug that has been approved for the treatment of neovascular diseases by binding and blocking the isoform 165 of VEGF family [94].
Aldose reductase gene (ALR/ALR2/ AKR1B1, Human Chromosome 7q35)
Aldose reductase (EC 1.1.1.21) is the leading enzyme in polyol pathway. ALR is a cytosolic, oxidoreductase (monomer) that performs the catalysis of various carbonyl compounds via NADPH-dependent reduction, including its prime target glucose. It has a crystal structure, single domain, 8-stranded comparable and parallel α/β-barrel motifs (folded), having the substrate binding site situated at the carboxy-terminal of the β-barrel [95,96]. The gene encoding for ALR is located on the chromosome 7q35 [97,98], which experiences repeat polymorphism (A-C at 5′end). This repeat polymorphism is associated in the respective studies with DR in Indians [99], Mainland Chinese [59], Chileans [100], Hong Kong Chinese [101,102], Brazilians [103] and in Japanese [104-106]. In non-diabetics, ALR has a truncated affinity for glucose molecules, directing the metabolism of these molecules in a small percentage of the overall glucose utilized. However, a patient is suffering from hyperglycemia, amplified intra-cellular glucose results in enhanced enzymatic conversion to the poly-alcohol sorbitol, with a simultaneous reduction in the level of NADPH [96,107].
ALR action: The major metabolic pathway is polyol pathway connecting hyperglycemia to mellitus tissue complications and aldose reductase enzyme. In this ALR acts as the primary and the rate limiting biocatalyst [50,67,108] in which glucose in the presence of nicotinamide adenine dinucleotide phosphate is reduced to molecules of sorbitol by the action of ALR, which is then converted to fructose by the enzyme sorbitol dehydrogenase and nicotinamice riboside acting as a cofactor [109,110]. It may also be speculated that intracellular fructose can be phosphorylated (Fructose-6-phosphate) and flux through the hexosamine pathway thus experiencing the increase in the UDP-GlcNAc level and protein Ser/Thr-O-GlcNAcylation of histones, kinases and transcription factors [111].
Due to the cellular toxicity of hyperglycemic patients, polyol pathway is held responsible, at least in part, for the development of chronic complications of diabetes. This pathway becomes active primarily when there has been an increment in the level of intra-cellular glucose [110, 112-113]. The biocatalysts of this polyol pathway are present in the tissues of human suffering from diabetes and its complications [114,115].
Discussing the flux mechanism of polyol pathway, the sorbitol present in the organism does not diffuse across the cell membranes easily, which has paved the way for osmotic damage to microvascular cells [96]. During this course of non-diffusive behavior of sorbitol, intra-cellular accumulation of sorbitol initiates, leading to occurrences of osmotic stress [67,116]. Further, with the exhaustive analysis of early studies it was revealed that sorbitol is converted to fructose via sorbitol dehydrogenase, with NAD+ getting reduced to NADH. Detrimental effects of this pathway are: sorbitol-induced osmotic stress, decreased sodium/potassium (ATPase) activity, an upsurge in cytosolic NADH and a drop in cytosolic NADPH [96]. Supplementary to this is the formation of microneurysm in animal models, with pericyte loss and basement membrane thickening [50]. There has been three ALR SNPs that are associated with DR: SNP rs759853, the (CA)n microsatellite polymorphism, and SNP rs9640883 [38,117]. Additionally, considering the facts and results of past studies it can be viewed that the influence of polyol pathway to diabetic hitches may be site specific, tissue and species dependent [96,107].
ALR as a therapeutic target: Inhibition of polyol pathway in in vivo studies brought forth uneven results. In a five-year study conducted on canine species, it was observed that inhibition of ALR gene prevented diabetic symptoms and complications up to an extent specifically in neuropathy, but was unsuccessful in case of proliferative diabetic retinopathy [96,118]. Also, this positive effect of ALR inhibition on neuropathy had given rise to an effective and promising inhibitor Zenarestat against the mechanistic action of ALR [119].
In addition, synthetic ALR inhibitors are carboxylic acid inhibitors, for example Ponalrestat, Tolerestat and Zopolrestat. The former shows the low target permeability and are not effective in in vivo studies and the latter, besides having the enhanced target penetrating capability but, has depicted skin reaction and toxicity in the liver [110, 120-122]. Still the clinical use of ALR inhibitors is yet to be established.
Interpretation and explanation of genetics of DR has been in an infancy stage the reason being an individual’s predisposition to diabetes is not entirely explored and the inheritance of genetic risk variants and their vulnerability to environmental factors are one of the key factors to understand the mechanism of this up-surging disease. A genome wide association study entails high density sampling of common human gene variation. Large-scale GWAS analyses in case of familial inheritance have facilitated for the identification of numerous genetic variants conferring threat to diabetes [18]. Hence, GWAS studies are boundless having ample facts of particular genes and the prospective to ascertain biological effects of genes [123-125].
Besides GWAS, linkage studies tend to focus on the transmission of causative genes in families; while the GWAS identify genetic variants in the diseased population versus healthy individuals. In case of T2D leading to DR, the implications have been insightful as the polymorphism discovery in the HLA region accounts for only 5–10% of disease heritability [126] which states a large constituent of genetic predisposition to T2D, that still requires an identification portfolio. This reflection suggests that environmental or epigenetic factors might influence the disease predisposition [18] and its inclination towards the triggered metabolic and signaling pathways. The initial linkage studies in T2D affected families (further leading to DR) identified CAPN10 and TCF7L2 as risk-conferring genes [127].
Over 50 genetic risk variants (candidate genes) are identified via GWAS namely KCNJ11, PPARG, HNF1B, IRS1, HNF1A, and HNF4A. Most of the disease-causing variants are associated with defective working of pancreatic β-cells, involved as a major factor in the pathology of T2D [128] and its complications. So far, vital genes associated with GWAS of different populations include IGF2BP2, SLC30A8, HHEX, KCNQ11, CDKN2A/B, HMGA2 and NOTCH2-ADAM30 [129]. A directory of all major GWAS studies has been maintained via National Human Genome Research Institute and can be gained access through their website (https://www.genome.gov/) for further exploration of research and development.
Mitochondria are the powerhouse of the cell involved in oxidative phosphorylation and bioenergetics, i.e., the production of adenosine triphosphate (ATP) via its electron transport chain (ETC), which also generates reactive oxygen species. In addition, the mitochondrion also involves in the production epigenetic substrates such as acetyl-coA and betaine as methyl donor in the methionine cycle and S-adenosylmethionine (SAM) biosynthesis. These mitochondrial metabolites such as ATP, acetyl-coA and SAM are known epigenetic substrates. Recently it has been shown that Ser/Thr-O-GlcNAcylation of histones represents potential epigenetic histone codes [111]. Furthermore, mitochondrial tricarboxylic acid (TCA) cycle metabolites and NAD+ have strong influence on histone remodeling via modification of histone acetyltransferases and deacetylases. We and others have shown that under hyperglycemia and diabetes, TXNIP is strongly induced in pancreatic b cells as well as in the retina [20,130]. TXNIP binds to thioredoxin (Trx1 in the cytosol and nucleus and Trx2 in mitochondria) and inhibits its thiol reducing and oxidant scavenging activity thereby causing cellular oxidative/nitrosative stress, NLRP3 inflammasome activation, inflammation, and apoptosis [131]. We also have shown that TXNIP is involved in epigenetic histone modification of pro-inflammatory genes in retinal endothelial cells under hyperglycemia [132]. Furthermore, the TXNIP promoter is under the control histone acetylation (H4K8Ac) and TXNIP expression is highly induced by trichostatin A, a histone deacetylase inhibitor, in retinal endothelial cells , but 5-azacytidine, a DNA methyltransferase inhibitor, was without an effect [130,132].
Epigenetics, as described before, has been distinguished as heritable alterations in gene function that come about without a change in the nucleotide sequence (ref. 24 and Figure 2) by modifying histones (post-translational modification by acetylation, methylation, phosphorylation and others as epigenetic histone marks) and by alterations in DNA methylation patterns [133] and chromatin remodeling [24,26]. These histone and DNA modifications are achieved by different enzyme epigenetic writers (add marks), erasers (remove) and readers (binding proteins). Therefore, these changes are possibly reversible and controlled by the cell environment such as toxins, dietary habits, chronic hyperglycemia (diabetes) or pharmacological medications [134]. Studies also have shown that non-coding RNA sequences, including microRNAs, piwi-RNA and long non-coding RNAs [135], participate in the epigenetic gene expression modulation.
Dysfunctional or depolarized mitochondria produces less ATP but generate more ROS causing mitochondrial protein, mtDNA and lipid damage [26,136]. Under these conditions, defective mitochondrial metabolism may have a greater influence on epigenetic substrate generation and therefore nuclear as well as mitochondrial epigenome modifications in diabetes and its complications (Figure 2). Hence, maintaining a functional mitochondrion may be critical to regulate epigenome and transcriptome profiles in diabetes and prevent or slow down the progression of ocular complications. Mitochondria are dynamic organelles, which undergo its own DNA replication and biogenesis independently of the nuclear DNA replication. Mitochondrial fusion and fission are critical for mixing mitochondrial material (protein, RNA and mtDNA) and removal of damaged mitochondria by autophagy, a process known as mitopahgy [137]. These processes are involved in mitochondrial organelle quality control and efficient bioenergetics to produce ATP and cellular metabolites including epigenetic mechanisms.
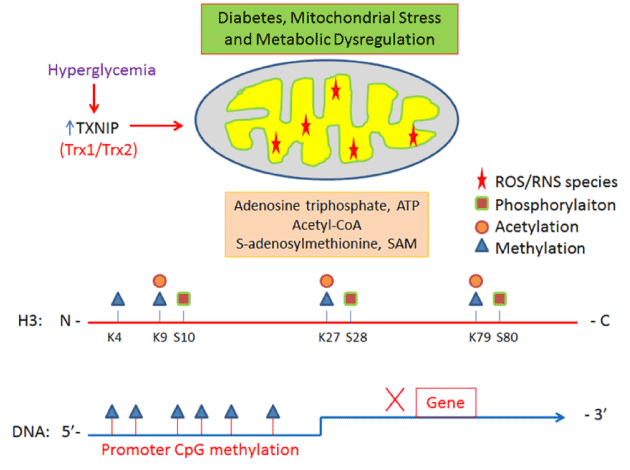
Figure 2. Hyperglycemia-induced TXNIP upregulation, inhibition of Trx1/Trx2 and mitochondrial stress may alter epigenome regulation by changing histone and DNA epigenetic substrates in DR [24]. Some histone H3 and promoter DNA modification examples are shown. In general, histone acetylation and phosphorylation are activation marks while lysine methylations are repressive marks, although H3K4Me is an activation modification. Cytosine methylation at the CpG islands in proximal promoters is a repressive DNA transcriptional mark. Environmental factors such as diet, exercise, and sedentary lifestyle can influence epigenetics and gene expression in aging-related disorders including diabetes and neurodegenerative diseases.
As mentioned above, mitochondrial ETC also generates ROS and causes protein and mtDNA damage. Therefore, mitochondrial quality control also involves a mitochondrion to nuclear retrograde signal known as the mitochondrial unfolded protein response (UPRmt) in which mitochondria-targeted chaperones (mtHSP70, HSP60, HSP10), proteases (ClpXP, LONP1) and anti-oxidants (MnSOD, Trx2) are specifically synthesized and transported to the mitochondrion for maintaining protein functions [138]. However, when the stress level exceeds the quality control, then the damaged mitochondria are fragmented involving dynamin-related protein 1 (Drp1) and fission protein Fis1, and sequestered by autophagosome, which subsequently is removed by lysosomal degradation via mitophagy [139]. Furthermore, mitochondrial oxidative stress may lead to glutathione oxidation and SAM depletion in an attempt to synthesize more glutathione. Recently, it has been shown that SAM levels are reduced in the circulation in diabetes and corelates well with DR [140]. In addition, Trx reductase 2 (TrxR2) mutations and mitochondrial oxidative stress are related to the progression of DR [141]. Furthermore, mtDNA itself undergoes epigenetic cytosine methylation and hydroxymethylation [26]. Hence, mitochondrial oxidative stress can lead to epigenetic alterations both in nuclear-encoded and mitochondrial-encoded genes that are involved in mitochondrial oxidative phosphorylation and metabolic function [142]. It is also known that mtDNA synthesizes only 13 of its own ETC proteins while >1300 proteins are nuclear-encoded and synthesized in free-ribosomes in the cytosol and imported into the mitochondrion. Therefore, a coordinated level of gene expression in the mitochondrion and nucleus is critically important for mitochondrial homeostasis and cellular survival.
In all, the study of epigenetics together with transcriptome and/or proteome (proteogenomics) as an integrated field of study can answer questions in biology and disease that could not be explained by genetics alone. Epigenetic modifications through DNA methylation and histone modifications and the expression of non-coding RNAs such as miRNA and long noncoding RNAs have the ability to manipulate gene expression in nuclear DNA without any mutation or deletion. Thus, the study of epigenetics has been incorporated into biological investigations, such as research in development; stem cell biology and disease mechanisms. We are now scrapping just the surface of epigenetics in disease development and progression of diabetes and its complications, particularly so, in DR. The role of mitochondrial dysfunction and oxidative stress in epigenetic modification and metabolic memory in diabetic complications definitely need further studies [26, 27,143].
The mechanism(s) involved in the pathogenesis of diabetic complications reflects a complex process of chronic hyperglycemia-associated oxidative stress and low-grade inflammation, premature cell death and presumed metabolic memory that provide a framework for the future epigenetic research and development, besides successful clinical trials yet to prove their efficiency on humans [27,96]. Three aspects are of great concern for the complete understanding of the molecular and cellular biology of the diabetic complications. First, the foremost is the phenomenon of metabolic or epigenetic memory, which refers to the remembrance of hyperglycemic episodes inducing microvascular modifications during the normal homeostasis or after glucose normalization [23,26,27]. Second, another aspect is the genetic determinants of susceptibility to macro- and microvascular complications involved in diabetic patients. Thus, to concentrate on this issue, gene mapping studies should be designed to identify predisposition to complications as well as interactions of these genes with the metabolic factors [96,144].
Third, next generation DNA/RNA sequencing (NGS) may provide assistance by identifying exceptional genetic variants having the significant effects on T1D and T2D sufferers that could aid in the early detection of diabetic complications [18]. Epigenomics, transcriptomics, proteomics, metabolomics and systems biology are also rapidly developing techniques that are fitted inside the personalized or precision medicine toolbox. In addition, restricting the overproduction of superoxides by mitochondrial electron-transport pathway, together with activation of NADPH and xanthine oxidases, would put on an equal footing as controlling the polyol pathway, hexosamine flux, PKC activation, AGE formation, and NF-kB activation, inflammation and overall glycemic control [96,145]. Functional studies are also in need to progress at a rapid pace so as to translate these findings into clinical practice. Thus, mitochondrial cell permeable antioxidant therapies may prove to be critical in maintaining mitochondrial bioenergetics, metabolism and epigenetics in diabetes and preventing its complications [146-148]. Finally, the newly acquired CRISPR-Cas9 or dCas9-mediated genome editing approaches [149,150] will also prove to be powerful approaches to correct epigenetic and genetic aberrations in diseases that involve metabolic or epigenetic memory by targeting histone and DNA modifying enzymes and their binding proteins, especially in treating diabetic ocular complications. The retina is a relatively immune privileged and confined organ therefore gene therapy approaches are most suitable via an intravitreal delivery method. Thus, it is an exciting time for epigenetic exploration and ocular gene therapy in DR, which is just beginning to scratch the proverbial tip of the iceberg.
Therapeutic opportunities
The upsurge of diabetic complications demands for novel therapeutic approaches and development of evidence-based and stage-specific drugs. Current drug library available to hit the diabetic complications specifically T2D, involve numerous mechanisms like: blocking the carb digestion; hindering the hepatic glucose production; stimulation in the secretion of pancreatic insulin, etc. Some of the anti-diabetic drugs and their functions are given in Table 2. Nonetheless, the extent to which these drugs work at the epigenetic level is yet to be determined.
Table 2. Anti-diabetic drugs and their functions [18].
Drug |
Drug function |
Metformin |
Reduces hepatic glucose production and insulin resistance |
Sulfonylureas |
Stimulate insulin secretion from pancreatic β-cells |
Thiazolidenediones |
PPAR-γ activators which help decrease triglycerides and free fatty acid levels, and insulin resistance |
Metiglinide |
Stimulate insulin release from Pancreas |
Acarbose |
Inhibition of glycoside hydrolases and hence reduced postprandial glucose |
Prior studies have also pointed out the causal relations between diabetes and epigenetic modifications [26, 151-154]. These include a large variety of molecular inhibitors and/or activators of the enzymatic machinery, signaling factors and involvement of growth factors and their SNPs as well, that could slow down the early or late onset of diabetes and its related chronic complications [155,156].
Personalized medicines
Personalized medicines have vital characteristics so as to tailor the best fit therapies for an individual to treat. Since patients are of diverse subsets, they have varying clinical considerations and features. Factors influencing the treatment goals and strategies include age, gender, diabetes duration, epigenetics, diabetic complications and the presence of comorbidities (cardiovascular diseases, obesity, etc.). For instance, treatment varies from individual to individual, the treatment plan for an individual with an early onset of diabetic complications, who is actually at an increased risk will vary from the patient having late onset of the disease due to prolonged exposure to hyperglycemic conditions [18,157].
Although there has been a limited scope for disease diagnosis, the genetic information of an individual might help to identify the risk involved and aid in differentiating among individuals benefiting with a certain treatment or not. For example, sulfonylureas show enhanced activities over the insulin therapy for the individuals having KCNJ11 mutations (ATP-sensitive potassium channel Kir6.2), that is causing diabetes of neonatal [158], while those individuals with glucokinase (GCK) mutation, persist unresponsiveness with anti-diabetic agents for the control of glycemia [159].
Considering the case of T2D, lifestyle modifications of the individuals at risk can aid in preventing or delaying the growth of T2D [160]. This could also correlate well with the severe diabetic complications and issues such as retinopathy and cardiovascular disease [161]. The degree and extent of epigenetic alterations and marks (histone and/or DNA modifications) may also be different from individual to individual as its marks will be dependent both on genetics and personal life style maintenance – particularly diet and physical activity. Hence, epigenetic studies complementation with GWAS, proteogenomics and metabolomics will add in achieving precision or personalized medicine tailored to suit individual epigenetic and metabolomics profiles.
Authors acknowledge School of Biotechnology, Rajiv Gandhi Technical University and School of Medicine, Wayne State University for providing a platform to prepare and collect the data for the manuscript. Funding to Dr. Lalit Singh Pukhrambam (Lalit P. Singh) from NIH/NEI R01 EY023992 and NIH Core Grant P30EY004068 to the Department of Anatomy and Cell Biology and Research to Prevent Blindness unrestricted grant to the Department of Ophthalmology, Wayne State University are also acknowledged. In addition, we acknowledge and apologize to those researchers whose works, though equally important, have not been cited in this article due to page limitations.
All authors declare no conflict of interest.
- Choudhuri S (2011) From Waddington’s epigenetic landscape to small noncoding RNA: some important milestones in the history of epigenetics research. Toxicol Mech Methods 21: 252-274.
- Waddington CH (2012) The Epigenotpye. Endeavour. 1942. Int J Epidemiol 41: 10-13. [Crossref]
- Egger G, Liang G, Aparicio A, Jones PA (2004) Epigenetics in human disease and prospects for epigenetic therapy. Nature 429: 457-463. [Crossref]
- Holliday R (1987) The inheritance of epigenetic defects. Science 238: 163-170. [Crossref]
- Saban KL, Mathews HL, DeVon HA, Janusek LW (2014) Epigenetics and social context: implications for disparity in cardiovascular disease. Aging Dis 5: 346-355. [Crossref]
- Xu F, Zhou X, Shen F, Pang R, Liu S (2012) Decreased peripheral blood mitochondrial DNA content is related to HbA1c, fasting plasma glucose level and age of onset in type 2 diabetes mellitus. Diabetic Medicine 29: e47-e54. [Crossref]
- Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, et al. (2005) Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54: 8-14. [Crossref]
- Irin AK, Kodamullil AT, Gündel M, Hofmann-Apitius M (2015) Computational Modelling Approaches on Epigenetic Factors in Neurodegenerative and Autoimmune Diseases and Their Mechanistic Analysis. J Immunol Res 2015: 737168.
- Hales CN, Barker DJ (1992) Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35: 595-601. [Crossref]
- Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, et al. (1991) Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303: 1019-1022. [Crossref]
- Barker DJ, Hales CN, Fall C, Osmond C, Phipps K, et al. (1993) Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36: 62-67. [Crossref]
- Lithell HO, McKeigue PM, Berglund L, Mohsen R, Lithell UB, et al. (1996) Relation of size at birth to non-insulin dependent diabetes and insulin concentrations in men aged 50-60 years. BMJ 312: 406-410. [Crossref]
- Rönn T, Ling C (2015) DNA methylation as a diagnostic and therapeutic target in the battle against Type 2 diabetes. Epigenomics 7: 451-460. [Crossref]
- Eriksson J, Forsen T, Tuomilehto J, Jaddoe V, Osmond C, et al. (2002) Effects of size at birth and childhood growth on the insulin resistance syndrome in elderly individuals. Diabetologia 45: 342-348. [Crossref]
- Yamada L, Chong S (2016) Epigenetic studies in Developmental Origins of Health and Disease: pitfalls and key considerations for study design and interpretation. J Dev Orig Health Dis. [Crossref]
- Ling C, Groop L (2009) Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes 58: 2718-2725. [Crossref]
- Cho NH, Whiting D, Guariguata L, Montoya PA, Forouhi N, et al. (2013) IDF diabetes atlas. Brussels, Belgium: International Diabetes Federation.
- Siddiqui K, Tyagi S (2015) Genetics, genomics and personalized medicine in Type 2 diabetes: a perspective on the Arab region. Personalized Medicine 12: 417-431.
- American Diabetes Association (2014) Standards of medical care in diabetes--2014. Diabetes Care 37 Suppl 1: S14-80. [Crossref]
- Shalev A (2014) Minireview: Thioredoxin-interacting protein: regulation and function in the pancreatic β-cell. Mol Endocrinol 28: 1211-1220. [Crossref]
- Singh LP (2013) Thioredoxin Interacting Protein (TXNIP) and Progression of Diabetic Retinopathy. J Clin Exp Ophthalmol 4: 1-29. [Crossref]
- Kreuz S, Fischle W (2016) Oxidative stress signaling to chromatin in health and disease. Epigenomics 8: 843-862. [Crossref]
- Campbell SA, Hoffman BG (2016) Chromatin Regulators in Pancreas Development and Diabetes. Trends Endocrinol Metab 27: 142-152. [Crossref]
- Perrone L, Matrone C, Singh LP (2014) Epigenetic modifications and potential new treatment targets in diabetic retinopathy. J Ophthalmol 2014: 789120.
- Reddy MA, Zhang E, Natarajan R (2015) Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 58: 443-455. [Crossref]
- Mishra M, Kowluru RA (2015) Epigenetic Modification of Mitochondrial DNA in the Development of Diabetic Retinopathy. Invest Ophthalmol Vis Sci 56: 5133-5142. [Crossref]
- Chen Z, Miao F, Paterson AD, Lachin JM, Zhang L, et al. (2016) Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci USA 113: E3002-3011. [Crossref]
- Cheng X, Blumenthal RM (2010) Coordinated chromatin control: structural and functional linkage of DNA and histone methylation. Biochemistry 49: 2999-3008.
- Hashimoto H, Vertino PM, Cheng X (2010) Molecular coupling of DNA methylation and histone methylation. Epigenomics 2: 657-669. [Crossref]
- Muhonen P, Holthofer H (2009) Epigenetic and microRNA-mediated regulation in diabetes. Nephrol Dial Transplant 24: 1088-1096. [Crossref]
- Consitt LA, Bell JA, Koves TR, Muoio DM, Hulver MW, et al. (2010) Peroxisome Proliferator–Activated Receptor-? Coactivator-1a Overexpression Increases Lipid Oxidation in Myocytes From Extremely Obese Individuals. Diabetes 59: 1407-1415.
- Liu MM, Chan CC, Tuo J (2013) Epigenetics in ocular diseases. Curr Genomics 14: 166-172. [Crossref]
- Yang B, Cross DF, Ollerenshaw M, Millward BA, Demaine AG (2003) Polymorphisms of the vascular endothelial growth factor and susceptibility to diabetic microvascular complications in patients with type 1 diabetes mellitus. J Diabetes Complications 17: 1-6.
- Ishida S, Shinoda K, Kawashima S, Oguchi Y, Okada Y, et al. (2000) Coexpression of VEGF receptors VEGF-R2 and neuropilin-1 in proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci 41: 1649-1656. [Crossref]
- Simó R, Carrasco E, García-Ramírez M, Hernández C (2006) Angiogenic and antiangiogenic factors in proliferative diabetic retinopathy. Curr Diabetes Rev 2: 71-98. [Crossref]
2021 Copyright OAT. All rights reserv
- Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, et al. (2008) Vascular endothelial growth factor in eye disease. Prog Retin Eye Res 27: 331-371. [Crossref]
- Wirostko B, Wong TY, Simó R (2008) Vascular endothelial growth factor and diabetic complications. Prog Retin Eye Res 27: 608-621. [Crossref]
- Simó-Servat O, Hernández C, Simó R (2013) Genetics in diabetic retinopathy: current concepts and new insights. Curr Genomics 14: 289-299. [Crossref]
- Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, et al. (2010) GeneCards Version 3: the human gene integrator. Database (Oxford) 2010: baq020. [Crossref]
- Demaine A, Cross D, Millward A (2000) Polymorphisms of the aldose reductase gene and susceptibility to retinopathy in type 1 diabetes mellitus. Invest Ophthalmol Vis Sci 41: 4064-4068. [Crossref]
- Dayeh T, Volkov P, Salö S, Hall E, Nilsson E, et al. (2014) Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet 10: e1004160. [Crossref]
- Rönn T, Ling C (2015) DNA methylation as a diagnostic and therapeutic target in the battle against Type 2 diabetes. Epigenomics 7: 451-460. [Crossref]
- Atlas D (2000) International diabetes federation. Hallado en: http://www.idf. org/diabetesatlas/5e/es/prologo.
- Ng DP (2010) Human genetics of diabetic retinopathy: current perspectives. J Ophthalmol 2010. [Crossref]
- Lai CM, Dunlop SA, May LA, Gorbatov M, Brankov M, et al. (2005) Generation of transgenic mice with mild and severe retinal neovascularisation. Br J Ophthalmol 89: 911-916. [Crossref]
- Robinson R, Barathi VA, Chaurasia SS, Wong TY, et al. (2012) Update on animal models of diabetic retinopathy: from molecular approaches to mice and higher mammals. Dis Model Mech 5: 444-456. [Crossref]
- [No authors listed] (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 329: 977-986. [Crossref]
- Group U.P.D.S. (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). The Lancet 352: 837-853. [Crossref]
- Luna P, Guarner V, Farías JM, Hernández-Pacheco G, Martínez M (2016) Importance of Metabolic Memory in the Development of Vascular Complications in Diabetic Patients. J Cardiothorac Vasc Anesth 30: 1369-1378. [Crossref]
- Engerman RL, Kern TS (1984) Experimental galactosemia produces diabetic-like retinopathy. Diabetes 33: 97-100. [Crossref]
- Kern TS, Engerman RL (1994) Comparison of retinal lesions in alloxan-diabetic rats and galactose-fed rats. Curr Eye Res 13: 863-867. [Crossref]
- Kern TS, Engerman RL (2001) Pharmacological inhibition of diabetic retinopathy: aminoguanidine and aspirin. Diabetes 50: 1636-1642. [Crossref]
- Awata T, Inoue K, Kurihara S, Ohkubo T, Watanabe M, et al. (2002) A common polymorphism in the 5'-untranslated region of the VEGF gene is associated with diabetic retinopathy in type 2 diabetes. Diabetes 51: 1635-1639. [Crossref]
- Beránek M, Kan?ková K, Benes P, Izakovic?ová-Hollá L, Znojil V, et al (2002) Polymorphism R25P in the gene encoding transforming growth factor-beta (TGF-ß1) is a newly identified risk factor for proliferative diabetic retinopathy. Am J Med Genet 109: 278-283.
- Kumaramanickavel G, Ramprasad VL, Sripriya S, Upadyay NK, Paul PG, et al. (2002) Association of Gly82Ser polymorphism in the RAGE gene with diabetic retinopathy in type II diabetic Asian Indian patients. J Diabetes and Complications 16: 391-394. [Crossref]
- Amano S, Yamagishi S, Koda Y, Tsuneoka M, Soejima M, et al. (2003) Polymorphisms of sorbitol dehydrogenase (SDH) gene and susceptibility to diabetic retinopathy. Med Hypotheses 60: 550-551. [Crossref]
- Lam HC, Lee JK, Lu CC, Chu CH, Chuang MJ, et al. (2003) Role of endothelin in diabetic retinopathy. Curr Vasc Pharmacol 1: 243-250. [Crossref]
- Santos KG, Tschiedel B, Schneider J, Souto K, Roisenberg I (2003) Diabetic retinopathy in Euro-Brazilian type 2 diabetic patients: relationship with polymorphisms in the aldose reductase, the plasminogen activator inhibitor-1 and the methylenetetrahydrofolate reductase genes. Diabetes Res Clin Pract 61: 133-136. [Crossref]
- Wang Y, Ng MC, Lee SC, So WY, Tong PC, et al. (2003) Phenotypic heterogeneity and associations of two aldose reductase gene polymorphisms with nephropathy and retinopathy in type 2 diabetes. Diabetes Care 26: 2410-2415. [Crossref]
- Ray D, Mishra M, Ralph S, Read I, Davies R, et al. (2004) Association of the VEGF gene with proliferative diabetic retinopathy but not proteinuria in diabetes. Diabetes 53: 861-864. [Crossref]
- Yoshioka K, Yoshida T, Takakura Y, Umekawa T, Kogure A, et al. (2005) Relation between polymorphisms G1704T and G82S of rage gene and diabetic retinopathy in Japanese type 2 diabetic patients. Intern Med 44: 417-421. [Crossref]
- Suganthalakshmi B, Anand R, Kim R, Mahalakshmi R, Karthikprakash S, et al. (2006) Association of VEGF and eNOS gene polymorphisms in type 2 diabetic retinopathy. Mol Vis 12: 336-341. [Crossref]
- ZvorniÄanin J, ZvorniÄanin E2 (2015) Imaging in Diabetic Retinopathy. Middle East Afr J Ophthalmol 22: 531. [Crossref]
- Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, et al. (1991) The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem 266: 11947-11954. [Crossref]
- Gupta N, Mansoor S, Sharma A, Sapkal A, Sheth J, et al. (2013) Diabetic retinopathy and VEGF. Open Ophthalmol J 7: 4-10. [Crossref]
- Awata T, Kurihara S, Takata N, Neda T, Iizuka H, et al. (2005) Functional VEGF C-634G polymorphism is associated with development of diabetic macular edema and correlated with macular retinal thickness in type 2 diabetes. Biochem Biophys Res Commun 333: 679-685. [Crossref]
- Liew G, Klein R, Wong TY (2009) The role of genetics in susceptibility to diabetic retinopathy. Int Ophthalmol Clin 49: 35-52. [Crossref]
- Uthra S, Raman R, Mukesh BN, Rajkumar SA, Kumari RP, et al. (2008) Association of VEGF gene polymorphisms with diabetic retinopathy in a south Indian cohort. Ophthalmic Genet 29: 11-15. [Crossref]
- Al-Kateb H, Mirea L, Xie X, Sun L, Liu M, et al. (2007) Multiple variants in vascular endothelial growth factor (VEGFA) are risk factors for time to severe retinopathy in type 1 diabetes the DCCT/EDIC genetics study. Diabetes 56: 2161-2168. [Crossref]
- Buraczynska M, Ksiazek P, Baranowicz-Gaszczyk I, Jozwiak L (2007) Association of the VEGF gene polymorphism with diabetic retinopathy in type 2 diabetes patients. Nephrol Dial Transplant 22: 827-832. [Crossref]
- Michaelson I (1948) The mode of development of the vascular system of the retina, with some observations on its significance for certain retinal diseases. Trans Ophthalmol Vis Sci 68: 137-180.
- Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285: 1182-1186. [Crossref]
- Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, et al. (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219: 983-985. [Crossref]
- Ferrara N, Henzel WJ (1989) Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 161: 851-858. [Crossref]
- Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, et al. (1989) Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 246: 1309-1312. [Crossref]
- Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246: 1306-1309. [Crossref]
- Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, et al. (1994) Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Eng J Med 331: 1480-1487. [Crossref]
- Shibuya M, Yamaguchi S, Yamane A, Ikeda T, Tojo A, et al. (1990) Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 5: 519-524. [Crossref]
- Holmes K, Roberts OL, Thomas AM, Cross MJ (2007) Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 19: 2003-2012. [Crossref]
- Veikkola T, Karkkainen M, Claesson-Welsh L, Alitalo K (2000) Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res 60: 203-212. [Crossref]
- Veikkola T, Karkkainen M, Claesson-Welsh L, Alitalo K (2000) Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res 60: 203-212. [Crossref]
- Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW (1999) Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1 A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem 274: 23463-23467. [Crossref]
- Sydorova M, Lee MS (2005) Vascular endothelial growth factor levels in vitreous and serum of patients with either proliferative diabetic retinopathy or proliferative vitreoretinopathy. Ophthalmic Res 37: 188-190. [Crossref]
- Ferrara N (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 25: 581-611. [Crossref]
- Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO (2003) Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res 22: 1-29. [Crossref]
- Wilkinson-Berka JL, Babic S, De Gooyer T, Stitt AW, Jaworski K, et al. (2004) Inhibition of platelet-derived growth factor promotes pericyte loss and angiogenesis in ischemic retinopathy. Am J Pathol 164: 1263-1273. [Crossref]
- Boyer DS, Hopkins JJ, Sorof J, Ehrlich JS (2013) Anti-vascular endothelial growth factor therapy for diabetic macular edema. Ther Adv Endocrinol Metab 4: 151-169. [Crossref]
- [No authors listed] (2008) Aflibercept: AVE 0005, AVE 005, AVE0005, VEGF Trap - Regeneron, VEGF Trap (R1R2), VEGF Trap-Eye. Drugs R D 9: 261-269. [Crossref]
- Do D, Nguyen Q, Shah S, Browning D, Haller J, et al. (2009) An exploratory study of the safety, tolerability and bioactivity of a single intravitreal injection of vascular endothelial growth factor Trap-Eye in patients with diabetic macular oedema. Brit J Ophthalmol 93: 144-149. [Crossref]
- Do DV, Schmidt-Erfurth U, Gonzalez VH, Gordon CM, Tolentino M, et al. (2011) The DA VINCI Study: phase 2 primary results of VEGF Trap-Eye in patients with diabetic macular edema. Ophthalmol 118: 1819-1826. [Crossref]
- Diabetic Retinopathy Clinical Research Network, Scott IU, Edwards AR, Beck RW, Bressler NM, et al. (2007) A phase II randomized clinical trial of intravitreal bevacizumab for diabetic macular edema. Ophthalmology 114: 1860-1867. [Crossref]
- Heier JS, Antoszyk AN, Pavan PR, Leff SR, Rosenfeld PJ, et al. (2006) Ranibizumab for treatment of neovascular age-related macular degeneration: a phase I/II multicenter, controlled, multidose study. Ophthalmol 113: 633-642. [Crossref]
- Rodriguez-Fontal M, Alfaro V, Kerrison JB, Jablon EP (2009) Ranibizumab for diabetic retinopathy. Curr Diabetes Rev 5: 47-51. [Crossref]
- Gragoudas ES, Adamis AP, Cunningham ET Jr, Feinsod M, Guyer DR; VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group (2004) Pegaptanib for neovascular age-related macular degeneration. N Engl J Med 351: 2805-2816. [Crossref]
- Wilson DK, Bohren KM, Gabbay KH, Quiocho FA (1992) An unlikely sugar substrate site in the 1.65 A structure of the human aldose reductase holoenzyme implicated in diabetic complications. Science 257: 81-84. [Crossref]
- Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414: 813-820. [Crossref]
- Uhlmann K, Kovacs P, Boettcher Y, Hammes HP, Paschke R (2006) Genetics of diabetic retinopathy. Exp Clin Endocrinol Diabetes 114: 275-294. [Crossref]
- Abhary S, Hewitt AW, Burdon KP, Craig JE (2009) A systematic meta-analysis of genetic association studies for diabetic retinopathy. Diabetes 58: 2137-2147. [Crossref]
- Kumaramanickavel G, Sripriya S, Ramprasad VL, Upadyay NK, Paul PG, et al. (2003) Z-2 aldose reductase allele and diabetic retinopathy in India. Ophthalmic Genet 24: 41-48. [Crossref]
- Olmos P, Futers S, Acosta AM, Siegel S, Maiz A, et al. (2000) (AC)23 [Z-2] polymorphism of the aldose reductase gene and fast progression of retinopathy in Chilean type 2 diabetics. Diabetes Res Clin Pract 47: 169-176. [Crossref]
- Karen BC, Lam KS, Wat NM, Chung SS (1995) An (AC) n dinucleotide repeat polymorphic marker at the 5' end of the aldose reductase gene is associated with early-onset diabetic retinopathy in NIDDM patients. Diabetes 44: 727-732. [Crossref]
- Lee SC, Wang Y, Ko GT, Critchley JA, Ng MC, et al. (2001) Association of retinopathy with a microsatellite at 5' end of the aldose reductase gene in Chinese patients with late-onset Type 2 diabetes. Ophthalmic Genet 22: 63-67. [Crossref]
- Richeti F, Noronha RM, Waetge RT, de Vasconcellos JP, de Souza OF, et al. (2007) Evaluation of AC(n) and C(-106)T polymorphisms of the aldose reductase gene in Brazilian patients with DM1 and susceptibility to diabetic retinopathy. Mol Vis 13: 740-745. [Crossref]
- Fujisawa T, Ikegami H, Kawaguchi Y, Yamato E, Nakagawa Y, et al. (1999) Length rather than a specific allele of dinucleotide repeat in the 5' upstream region of the aldose reductase gene is associated with diabetic retinopathy. Diabetes Med 16: 1044-1047. [Crossref]
- Ichikawa F, Yamada K, Ishiyama-Shigemoto S, Yuan X, Nonaka K (1999) Association of an (A–C) n dinucleotide repeat polymorphic marker at the 5'-region of the aldose reductase gene with retinopathy but not with nephropathy or neuropathy in Japanese patients with Type 2 diabetes mellitus. Diabetes Med 16: 744-748.
- Ikegishi Y, Tawata M, Aida K, Onaya T (1999) Z-4 allele upstream of the aldose reductase gene is associated with proliferative retinopathy in Japanese patients with NIDDM, and elevated luciferase gene transcription in vitro. Life Sci 65: 2061-2070. [Crossref]
- Obrosova IG, Kador PF (2011) Aldose reductase / polyol inhibitors for diabetic retinopathy. Curr Pharm Biotechnol 12: 373-385. [Crossref]
- Robison W, Nagata M, Laver N, Hohman T, Kinoshita J (1989) Diabetic-like retinopathy in rats prevented with an aldose reductase inhibitor. Invest Ophthalmol Vis Sci 30: 2285-2292. [Crossref]
- Cumbie BC, Hermayer KL (2007) Current concepts in targeted therapies for the pathophysiology of diabetic microvascular complications. Vasc Health Risk Manag 3: 823-832. [Crossref]
- Del-Corso A, Balestri F, Di Bugno E, Moschini R, Cappiello M, et al. (2013) A new approach to control the enigmatic activity of aldose reductase. PLoS One 8: e74076. [Crossref]
- Sakabe K, Wang Z, Hart GW (2010) Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci U S A 107: 19915-19920. [Crossref]
- Gabbay KH (1973) The sorbitol pathway and the complications of diabetes. N Engl J Med 288: 831-836. [Crossref]
- Oates PJ (2002) Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol 50: 325-392. [Crossref]
- Lorenzi M (2007) The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabetes Res 2007: 61038. [Crossref]
- Gallagher EJ, LeRoith D, Stasinopoulos M, Zelenko Z, Shiloach J (2016) Polyol accumulation in muscle and liver in a mouse model of type 2 diabetes. J Diabetes Complications 30: 999-1007. [Crossref]
- Dagher Z, Park YS, Asnaghi V, Hoehn T, Gerhardinger C, et al. (2004) Studies of rat and human retinas predict a role for the polyol pathway in human diabetic retinopathy. Diabetes 53: 2404-2411. [Crossref]
- Abhary S, Burdon KP, Laurie KJ, Thorpe S, Landers J, et al. (2010) Aldose reductase gene polymorphisms and diabetic retinopathy susceptibility. Diabetes Care 33: 1834-1836. [Crossref]
- Engerman RL, Kern TS (1987) Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 36: 808-812. [Crossref]
- Greene DA, Arezzo JC, Brown MB (1999) Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Zenarestat Study Group. Neurology 53: 580-591. [Crossref]
- Kador PF, Robison WG Jr, Kinoshita JH (1985) The pharmacology of aldose reductase inhibitors. Annu Rev Pharmacol Toxicol 25: 691-714. [Crossref]
- Bhatnagar A, Srivastava SK (1992) Aldose reductase: congenial and injurious profiles of an enigmatic enzyme. Biochem Med Metab Biol 48: 91-121. [Crossref]
- Pfeifer MA, Schumer MP, Gelber DA (1997) Aldose reductase inhibitors: the end of an era or the need for different trial designs? Diabetes 46 Suppl 2: S82-89. [Crossref]
- Agardh E, Lundstig A, Perfilyev A, Volkov P, Freiburghaus T, et al. (2015) Genome-wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Medicine 3: 182-188.
- Cho H, Sobrin L (2014) Genetics of diabetic retinopathy. Curr Diab Rep 14: 515. [Crossref]
- Agarwal A, Ingham SA2, Harkins KA3, Do DV1,3, et al. (2016) The role of pharmacogenetics and advances in gene therapy in the treatment of diabetic retinopathy. Pharmacogenomics 17: 309-320. [Crossref]
- da Silva Xavier G, Bellomo EA, McGinty JA, French PM, Rutter GA (2013) Animal models of GWAS-identified type 2 diabetes genes. J Diabetes Res 2013: 906590. [Crossref]
- Ahlqvist E, Ahluwalia TS, Groop L (2011) Genetics of type 2 diabetes. Clin Chem 57: 241-254. [Crossref]
- Billings LK, Florez JC (2010) The genetics of type 2 diabetes: what have we learned from GWAS? Ann N Y Acad Sci 1212: 59-77. [Crossref]
- Ali O (2013) Genetics of type 2 diabetes. World J Diabetes 4: 114-123. [Crossref]
- Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP (2010) Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis 1: e65. [Crossref]
- Devi TS, Lee I, Hüttemann M, Kumar A, Nantwi KC, et al. (2012) Txnip links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: Implications for diabetic retinopathy. Expt Diabetes Res 2012: 1-19.
- Perrone L, Devi TS, Hosoya K, Terasaki T, Singh LP (2009) Thioredoxin-interacting protein (Txnip) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol 221: 262-272. [Crossref]
- Waki H, Yamauchi T, Kadowaki T (2012) The epigenome and its role in diabetes. Curr Diab Rep 12: 673-685. [Crossref]
- Gouri A, Dekaken A, Chefrour M, Guieu R, Benharkat S (2015) Epigenetics and Diabetes: Current and Perspective. Int J Diabetol Vasc Dis Res 3: 94-98.
- Larriba E, del Mazo J2 (2016) Role of Non-Coding RNAs in the Transgenerational Epigenetic Transmission of the Effects of Reprotoxicants. Int J Mol Sci 17: 452. [Crossref]
- Mikhed Y, Daiber A, Steven S (2015) Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int J Mol Sci 16: 15918-15953. [Crossref]
- Mishra P, Chan DC2 (2016) Metabolic regulation of mitochondrial dynamics. J Cell Biol 212: 379-387. [Crossref]
- Pellegrino MW, Nargund AM, Haynes CM (2013) Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta 1833: 410-416. [Crossref]
- Buhlman L, Damiano M, Bertolin G, Ferrando-Miguel R, Lombès A, et al. (2014) Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim Biophys Acta 1843: 2012-2026. [Crossref]
- Van Hecke MV, Dekker JM, Nijpels G, Teerlink T, Jakobs C, et al. (2008) Homocysteine, S-adenosylmethionine and S-adenosylhomocysteine are associated with retinal microvascular abnormalities: the Hoorn Study. Clin Sci (Lond) 114: 479-447. [Crossref]
- Ramus SM, Cilensek I, Petrovic MG, Soucek M, Kruzliak P, et al. (2016) Single nucleotide polymorphisms in the Trx2/TXNIP and TrxR2 genes of the mitochondrial thioredoxin antioxidant system and the risk of diabetic retinopathy in patients with Type 2 diabetes mellitus. J Diabetes Complications 30: 192-198. [Crossref]
- Castegna A, Iacobazzi V, Infantino V (2015) The mitochondrial side of epigenetics. Physiol Genomics 47: 299-307. [Crossref]
- Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, et al. (2014) Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect 122: 1271-1278. [Crossref]
- Engerman RL, Kern TS, Larson ME (1994) Nerve conduction and aldose reductase inhibition during 5 years of diabetes or galactosaemia in dogs. Diabetologia 37: 141-144. [Crossref]
- [No authors listed] (2000) Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. N Engl J Med 342: 381-389. [Crossref]
- Ting HH, Timimi FK, Boles KS, Creager SJ, Ganz P, et al. (1996) Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J Clin Invest 97: 22. [Crossref]
- [No authors listed] (2000) Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 355: 253-259. [Crossref]
- Alam NM, Mills WC 4th, Wong AA, Douglas RM, Szeto HH, et al. (2015) A mitochondrial therapeutic reverses visual decline in mouse models of diabetes. Dis Model Mech 8: 701-710.
- Himeda CL, Jones TI, Jones PL (2016) CRISPR/dCas9-mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-fl in FSH Muscular Dystrophy. Mol Ther 24: 527-535. [Crossref]
- Pham H, Kearns NA, Maehr R (2016) Transcriptional Regulation with CRISPR/Cas9 Effectors in Mammalian Cells. Methods Mol Biol 1358: 43-57. [Crossref]
- El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, et al. (2008) Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 205: 2409-2417. [Crossref]
- Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, et al. (2009) Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58: 1229-1236. [Crossref]
- Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, et al. (2011) Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Research 21(10): 1601-1615. [Crossref]
- Fodor A, Cozma A, Karnieli E (2015) Personalized epigenetic management of diabetes. Personalized Medicine 12: 497-514.
- Cencioni C, Spallotta F, Greco S, Martelli F, Zeiher AM, et al. (2014) Epigenetic mechanisms of hyperglycemic memory. Int J Biochem Cell Biol 51: 155-158. [Crossref]
- Reddy SS (2016) Evolving to Personalized Medicine for Type 2 Diabetes. Endocrinol Metab Clin North Am 45: 1011-1020. [Crossref]
- Wannamethee SG, Shaper AG, Whincup PH, Lennon L, Sattar N (2011) Impact of diabetes on cardiovascular disease risk and all-cause mortality in older men: influence of age at onset, diabetes duration, and established and novel risk factors. Arch Intern Med 171: 404-410. [Crossref]
- Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, et al. (2006) Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 355: 467-477. [Crossref]
- Stride A, Shields B, Gill-Carey O, Chakera AJ, Colclough K, et al. (2014) Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia 57: 54-56. [Crossref]
- Hivert MF, Jablonski KA, Perreault L, Saxena R, McAteer JB, et al. (2011) Updated genetic score based on 34 confirmed type 2 diabetes Loci is associated with diabetes incidence and regression to normoglycemia in the diabetes prevention program. Diabetes 60: 1340-1348. [Crossref]
- Bonora E, Tuomilehto J (2011) The pros and cons of diagnosing diabetes with A1C. Diabetes Care 34 Suppl 2: S184-190. [Crossref]
- Halili MA, Andrews MR, Labzin LI, Schroder K, Matthias G, et al. (2010) Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J Leukoc Biol 87: 1103-1114. [Crossref]
- O'Byrne KJ, Baird AM, Kilmartin L, Leonard J, Sacevich C, et al. (2011) Epigenetic regulation of glucose transporters in non-small cell lung cancer. Cancers (Basel) 3: 1550-1565. [Crossref]
- Yoshikawa M, Hishikawa K, Marumo T, Fujita T (2007) Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-ß1 in human renal epithelial cells. J Am Soc Nephrol 18: 58-65. [Crossref]
- Crosson CE, Mani SK, Husain S, Alsarraf O, Menick DR (2010) Inhibition of histone deacetylase protects the retina from ischemic injury. Invest Ophthalmol Vis Sci 51: 3639-3645. [Crossref]
- Jiang J, Au M, Lu K, Eshpeter A, Korbutt G, et al. (2007) Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells 25: 1940-1953. [Crossref]
- Weems J, Olson AL (2011) Class II histone deacetylases limit GLUT4 gene expression during adipocyte differentiation. J Biol Chem 286: 460-468. [Crossref]
- Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, et al. (2002) The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Nat Acad Sci USA 99: 2995-3000. [Crossref]
- Gilbert RE, Huang Q, Thai K, Advani SL, Lee K, et al. (2011) Histone deacetylase inhibition attenuates diabetes-associated kidney growth: potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int 79: 1312-1321. [Crossref]
- Leoni F, Fossati G, Lewis EC, Lee J, Porro G, et al. (2005) The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med 11: 1-15. [Crossref]
- Lewis EC, Blaabjerg L, Størling J, Ronn SG, Mascagni P, et al. (2011) The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet β cells in vivo and in vitro. Mol Med 17: 369-377. [Crossref]
- Susick L, Senanayake T, Veluthakal R, Woster PM, Kowluru A (2009) A novel histone deacetylase inhibitor prevents IL-1ß induced metabolic dysfunction in pancreatic ß-cells. J Cell Mol Med 13: 1877-1885. [Crossref]
- Takigawa-Imamura H, Sekine T, Murata M, Takayama K, Nakazawa K, et al. (2003) Stimulation of glucose uptake in muscle cells by prolonged treatment with scriptide, a histone deacetylase inhibitor. Biosci Biotechnol Biochem 67: 1499-1506. [Crossref]
- Galmozzi A, Mitro N, Ferrari A, Gers E, Gilardi F, et al. (2013) Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 62: 732-742. [Crossref]
- Tayaramma T, Ma B, Rohde M, Mayer H (2006) Chromatin-Remodeling Factors Allow Differentiation of Bone Marrow Cells into Insulin-Producing Cells. Stem Cells 24: 2858-2867.
- Lenoir O, Flosseau K, Ma FX, Blondeau B, Mai A, et al. (2011) Specific control of pancreatic endocrine β- and δ-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 60: 2861-2871. [Crossref]
- Tedong L, Madiraju P, Martineau LC, Vallerand D, Arnason JT, et al. (2010) Hydro-ethanolic extract of cashew tree (Anacardium occidentale) nut and its principal compound, anacardic acid, stimulate glucose uptake in C2C12 muscle cells. Mol Nutrition & Food Res 54: 1753-1762. [Crossref]
- Kadiyala CSR, Zheng L, Du Y, Yohannes E, Kao HY, et al. (2012) Acetylation of retinal histones in diabetes increases inflammatory proteins effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC). J Biol Chem 287: 25869-25880. [Crossref]
- Chen S, Feng B, George B, Chakrabarti R, Chen M, et al. (2010) Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am J Physiol Endocrinol Metab 298: E127-E137. [Crossref]
- Vecellio M, Spallotta F, Nanni S, Colussi C, Cencioni C, et al. (2014) The histone acetylase activator pentadecylidenemalonate 1b rescues proliferation and differentiation in the human cardiac mesenchymal cells of type 2 diabetic patients. Diabetes 63: 2132-2147. [Crossref]
- Lefebvre B, Belaich S, Longue J, Vandewalle B, Oberholzer J, et al. (2010) 5'-AZA induces Ngn3 expression and endocrine differentiation in the PANC-1 human ductal cell line. Biochem Biophys Res Commun 391: 305-309. [Crossref]
- Chen S, Borowiak M, Fox JL, Maehr R, Osafune K, et al. (2009) A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat Chem Biol 5: 258-265. [Crossref]
- Öström M, Loffler KA, Edfalk S, Selander L, Dahl U, et al. (2008) Retinoic acid promotes the generation of pancreatic endocrine progenitor cells and their further differentiation into ß-cells. PLoS One 3: e2841. [Crossref]
- Yuan Y, Hartland K, Boskovic Z, Wang Y, Walpita D, et al. (2013) A small-molecule inducer of PDX1 expression identified by high-throughput screening. Chem Biol 20: 1513-1522. [Crossref]
- Shen W, Tremblay MS, Deshmukh VA, Wang W, Filippi CM, et al. (2013) Small-molecule inducer of β cell proliferation identified by high-throughput screening. J Am Chem Soc 135: 1669-1672. [Crossref]