Abstract
Excitatory aminoacids (EAAs) are stored in glutamatergic neurons and related into synaptic cleft, where they can activate inotropic or metabotropic receptors. Their action ends due to transport mechanisms performed by EAAT transporters (EAAT1/GLAST, EAAT2/GLT1, EAAT3/EAAC1, and EAAT4 or EAAT5). Glutamate neurotoxicity has been described in several neurodegenerative diseases such as Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD) and amyotropic lateral sclerosis ALS). Some drugs, such as paclitaxel, are able to increase translation of microRNA and could be possible used as regulatory against glutamate neurotoxicity.
Abbreviations
AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; AMPA: α-amino-3-hydroxy-5-methyl-isoxazole-4-propionate; Asp: aspartate; CSF: cerebral spinal fluid; EAA: excitatory amino acid; EAAC1 (EAAT3): excitatory amino acid carrier; EAAT: excitatory amino acid transporter; GABA: gamma-aminobutyric acid; GDH: glutamate dehydrogenase; GLAST (EAAT1): glutamate – aspartate transporter; GLT1 (EAAT2): glutamate transporter; iGluR: ionotropic glutamate receptor; KA: kaninic acid; L-Glu: L-glutamate; mGluR: metabotropic glutamate receptor; miR: micro RNA; NMDA: N-methyl-D-aspartate; PD: Parkinson’s disease
Introduction
Some amino acids act as neurotransmitters in the nervous system, being glutamate and aspartate the common excitatory amino acids and GABA, glycine and taurine the inhibitory ones. Of these amino acids, glutamate and GABA are intimately associated, as their metabolism is associated through glutamic acid decarboxylase (E.C. 4.1.1.15.) (Figure 1). Furthermore, GABA and glutamic acid effects are antagonic and they are related with CO2 fixation (relevant to central ventilation). Glutamic acid metabolism is also related with NH3 detoxification (due to a reduction in a-ketoglutarate and glutamate contents and an increase in glutamine).
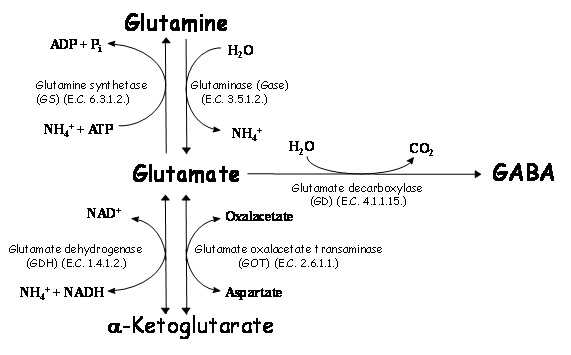
Figure 1. Metabolism of glutamate in brain. Glutamate metabolism is related to formation of glutamine or α-ketoglutarate (which are not neurotransmitters) or GABA (an inhibitory neurotransmitter). Formation of GABA is related with increase of CO2, whereas formation of glutamine is related with a decrease in energy (ATP). Formation of α-ketoglutarate by glutamate dehydrogenase (GDH) is related with an increase of ammonia, whereas reaction by glutamate oxalacetate transaminase (GOT) forms another exitatory aminoacid (aspartate).
The efflux of glutamate from brain across the hemato-encephalic barrier is much higher than the influx [1-3], meaning that metabolism of glutamate must play an important role in regulating the brain glutamate levels. Studies on metabolic generation of glutamate/glutamine by using radioactive substrates in brain shows that two pathways are involved. Glucose, glycerol, lactate, pyruvate, a-ketoglutarate and b-hydroxybutyrate seem to be metabolized to glutamate in neurons [4,5], as a low specific radioactivity of glutamine is obtained. In glial cells [6], where higher glutamine synthase is present [7,8], low radiolabelled glutamate and higher glutamine marked are obtained. This is the case of acetate, propionate, butyrate, citrate, leucine, GABA, aspartate and ammonia [9]. In order to decrease glutamic acid in synaptic cleft, excitatory amino acids transporters have an important effect. Therefore, in this paper we present some aspects of these proteins.
Glutamatergic neurotransmission
The excitatory amino acids (EAAs) are stored in synaptic vesicles in glutamatergic neurons and, upon an action potential, are released via exocytosis into the synaptic cleft where they can activate two different families of receptors: ionotropic (ligand-gated ion channels) and metabotropic (GTP-binding protein coupled) receptors. The ligand-gated ion channels are further divided into three families: a-amino-3-hydroxy-5-methyl-isoxazole-4-propionate (AMPA), kainate (KA) and N–methyl-D-aspartate (NMDA). While AMPA and kainate receptors mediate rapid depolarizing responses at most synapses in the mammalian central nervous system [10], the NMDA receptor participates in synaptic plasticity and synapse formation [11]. The family of metabotropic receptors consists of at least eight subtypes and are involved in the modulation of synaptic signaling by EAAs and other neurotransmitters [12].
The termination of the EAA action takes place by an uptake mechanism that uses the Na+, K+ and pH gradients as a driving force to translocate the neurotransmitters against their concentration gradients, keeping their concentration below the level that activates their receptors (~1 mM) [13-15] (Figure 2). Excessive activation of EAA receptors contributes to brain injury through a process known as excitotoxicity. Therefore, this transport mechanism is not only important for ensuring accurate synaptic signaling but also for limiting the EAA-mediated excitotoxicity
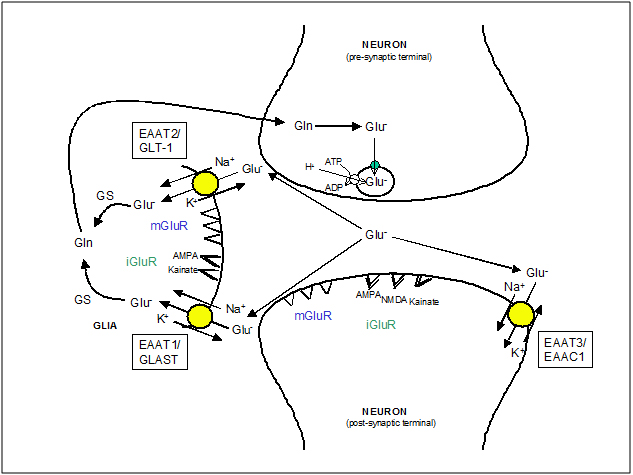
Figure 2. EAA mobilization at glutamatergic synapsis. The diagram illustrates the fate of EAA (i.e. Glutamate, Glu-) released by exocytosis from the pre-synaptic terminal, once they have activated their receptors: ionotropic glutamate receptors (iGluR) and metabotropic glutamate receptors (mGluR). The neuronal glutamate transporter, EAAT3/EAAC1, has a major role in influencing the shape of EPSC but transports only a minor fraction of total glutamate. The glial transporters, EAAT1/GLAST and EAAT2/GLT1, maintain extracellular glutamate at physiological levels. Glutamate in glial cells is metabolized to glutamine (Gln) by glutamine synthase (GS) which is then returned to neurons to replenish the transmitter pool of glutamate. In pre-synaptic neurons glutamate is packed again into synaptic vesicles by a Na+-independent transport driven by the internal positive membrane potential generated by the vesicular ATP-dependent H+ transport.
Excitatory amino acid (EAA) transporters
Three broad subtypes of EAA transport activities have been identified in brain preparations. One type, which is directly coupled to ATP hydrolysis, introduces glutamate into vesicles for release upon depolarization of the synaptic terminal [16]. It indirectly ensures low extracellular concentrations of EAAs by reducing the driving force required to transport an EAA into the cytoplasm. The second activity is a chloride-dependent transport which exchanges amino acids across the plasma membrane [17]. It has a low capacity and cannot concentrate EAA intracellularly. The third type of transport activity is an active Na+-dependent system and it can be differentiated from the others by means of ion selectivity, regional distribution, and sensitivity to inhibition by analogs of EAAs. Since this transport activity is extremely high, it is generally assumed that this activity is primarily responsible for the clearance of extracellular EAAs in brain.
To date, five such Na+-dependent EAA transporters have been characterized in the mammalian central nervous system. In 1992, three groups simultaneously isolated cDNA clones for these transporters. Kanner’s group purified a transport protein from rat brain with biochemical methods [18] and isolated a cDNA clone identified as GLT1 by using sequence information derived from a peptide fragment of this protein [19]. Hediger’s group used expression cloning to isolate a clone called EAAC1 (Excitatory Amino Acid Carrier) from a cDNA library prepared from rabbit intestine [20]. Stoffel’s group while isolating a uridine diphosphate (UDP) galactose: ceramide galactosyltransferase from brain always observed a co-chromatographing protein and after the analysis of its sequence, they identified homology with a bacterial H+-dependent EAA transporter. A cDNA clone encoding a protein called GLAST (glutamate and aspartate transporter) was then isolated [21]. In 1994, Amara’s group isolated the human homologs of all three of these transporters and were called EAAT1-3 for Excitatory Amino Acid Transporter [22]. EAAT1 is the homolog of GLAST, EAAT2 is the homolog of GLT1, and EAAT3 is the homolog of EAAC1. The sequence similarity of these transporters was used to obtain two other EAA transporter clones called EAAT4 and EAAT5 [23,24].
Once the cDNA sequences of these transporters were available, it was possible to study their tissue distribution. The glial transporter EAAT1/GLAST was localized by in situ hybridization and immunochemistry in the oligodendrocyte-related Bergmann glia cells [25] and it was more expressed in cerebellum compared to forebrain regions [26]. Although in one study EAAT1/GLAST immunoreactivity was also detected in neurons [25], this localization was not observed any more by other groups by using several antibodies [26,27]. EAAT1/GLAST is found adjacent to synapses, implicating that the transporter is available for clearance of EAAs after release.
EAAT1 is a 59.5 kDa protein, which shows a saturation kinetics: Km (L-Glu)=77 ± 27 mM [21], Km (D-Asp)=31 ± 7 [28]. Other amino acids, including L-alanine, L-leucine, L-glutamine, L-arginine and L-methionine are not transported in significant amounts. A strong inhibitor of glutamate uptake, D,L-threo-3-hydroxy-aspartate [29] is shown to cause neuronal degeneration [30].
The EAAT2/GLT1 transporter was initially only observed in astrocytes [25,26]. There is now evidence that it is present in neurons during development [31], in neurons in culture [32] and after an ischemic insult [33]. Although these findings provide evidence that neurons can express EAAT2/GLT1, further work is required to reveal how often this occurs and how its expression is regulated. The pattern of EAAT2/GLT1 expression has been discussed controversially. According to Rothstein et al. [25], it is relatively uniformly expressed in different brain regions but Lehre et al. [26] suggested that it is higher expressed in forebrain than in cerebellum. As observed with EAAT1/GLAST, the transporter is found adjacent to synapses, suggesting a similar physiological role.
EAAT2 is a 62.1 kDa protein [19], which shows a saturation kinetic: Km (D-Asp)=28 ± 13 mM [29]. This transporter is a glycoprotein with 2 potential N-linked glycosylation sites and 2 phosphorylation sites [26]. It is inhibited by some receptors agonists: dihydrokainate and L-a-aminoadipate, and by other substances which do not bind to glutamate receptors: L-trans-pyrolidine-2,4-dicarboxylate [34].
In brain, EAAT3/EAAC1 expression is selective for neurons [20,25] and is highly expressed in hippocampus and cortex with lower levels in midbrain, striatum, and cerebellum [25]. EAAC1 immunoreactivity is observed in dendrites and in cell somata, suggesting that it has not a predominant role in regulating EAAs at the synaptic cleft but seems strategic to lower EAAs concentrations during the diffusion process and to restrict transmitter spillover to the nearby areas [35].
EAAT3 has a molecular weight of 57.2 kDa and presents a saturation kinetic: Km (L-Glu)=12.2 ± 1.2 mM [36], Km (D-Asp)=28 ± 13 mM [29, 37]. As EAAT2, it is inhibited by L-a-aminoadipate but not by dihydrokainate.
EAAT4 expression is enriched in the dendritic spines as well as distal dendrites of Purkinje cells in the cerebellum and very low levels of expression are observed in forebrain compared to cerebellum [23]. The amino acid sequence exhibits 65%, 41% and 48% amino acid identity to the human glutamate transporters EAAT1, 2 and 3 respectively. The pharmacological properties of EAAT4 are similar to those described to the other glutamate transporters, with Km (L-Asp)=184 ± 0.46 mM and Km(L-Glu)=3.3 ± 0.4 mM [38].
EAAT5 appears to be retinal based on its mRNA distribution [24]. The lack of antibodies against the mammalian homolog of this transporter unable to study the protein distribution. The amino acid sequence exhibits 46%, 36% and 37% amino acid identity to the human glutamate transporters EAAT1, 2 and 3 respectively. Respect to EAAT4, a 43% identity is observed. The transporter presents a saturation kinetic: Km (L-Glu)=64 ± 6 mM and Km(L-Asp)=13 ± 5 mM, and it is potently inhibited by L-trans-pyrrolidine-2,4-dicarboxylic acid and by threo-b-hydroxyaspartate [24].
The Na+-dependent EAA transporters are not only expressed in the central nervous system and retina but also in the periphery [39,40]. EAAC1 is observed in liver, kidney and intestine [25], GLAST is expressed in heart, lung, skeletal muscle, bone marrow, and placenta [41,42]. GLT1 is thought to be expressed in placenta and bone marrow [41]. In all of these peripheral tissues, these molecules function to transport glutamate and aspartate for other purposes (metabolic) than regulation of synaptic transmission.
The amino acid sequences of the five transporters show between 40 and 65% identity and are likely to form similar structures. Based on the use of hydrophobicity analysis of the amino acid sequences as well as on the identification of molecular tags introduced into the transporters, it has been predicted that there are six a-helical trans membrane segments in the amino-terminal part of these proteins and four trans membrane segments in the highly conserved carboxy-terminal domain which are a-helical, in the model suggested by Slotboom et al. [43], or b-sheets in the model suggested by Wahle and Stoffel [44]. Moreover, It has been demonstrated that EAA transporters form homomultimers [45], but it is still not clear whether each subunit contains a separate pore through which EAAs and the various ions (Na+, H+ and K+) pass or else more subunits associate to form a single pore.
As indicated at the beginning, the energy for active transport of EAAs is derived from the Na+, K+ and pH gradients across the cell membrane. The stoichiometry of uptake is cotransport of three Na+ and one H+ (or possibly counter-transport of a hydroxyl ion) with one glutamate or aspartate and the counter-transport of one K+. This stoichiometry creates a net transfer of positive charge into the cell and, therefore, it is possible to measure it by electrophysiological methods. In addition to the mentioned ion fluxes coupled to EAA transport, the different transporter subtypes allow a varying degree of chloride ion flux which is thermodynamically uncoupled to EAA transport. EAAT4 and EAAT5 allow considerable chloride fluxes; EAAT2/GLT1 and EAAT3/EAAC1 allow very little chloride ion flux, whereas chloride fluxes through EAAT1/GLAST vary depending on substrate: L- and D-aspartate activating larger chloride conductances than L-glutamate [46]. The physiological role of the chloride ions is not clear but it may act as a voltage-clamp [47].
Besides the different degree of the thermodynamically uncoupled chloride flux through the transporters, they also show quite distinct pharmacological profiles. The most important pharmacological difference is the selective sensitivity of EAAT2/GLT1 to competitive inhibition by dihydrokainate, kainate, and threo-3-methylglutamate [22,48]. A number of other pharmacological agents have been reported to inhibit EAA transport by competing with EAA as a substrate for the transporters [22]. The analysis of these competitive inhibitors has been used to delineate the chemical requirements for binding and translocation. Compounds valuable for these efforts were analogues of glutamate that are conformationally constrained by the addition of substituents to the carbon backbone of glutamate or aspartate (b-hydroxyaspartate or methylglutamate derivatives), and the incorporation of ring systems (carboxycyclopropyl)glycines, aminocyclobutane dicarboxylates, or pyrrolidine dicarboxylates. See Table 1 for a summary of different activities of glutamate analogues on the EAA transporters.
| |
Substance
|
System
|
Properties
|
|
Simple aspartate glutamate derivatives and
|
L-glutamate
|
Xenopus oocytes
|
S (T1-5)
|
| |
L-aspartate
|
Xenopus oocytes
|
S (T1-5), I (T2)
|
| |
D-aspartate
|
Xenopus oocytes
|
S (T1-5), I (T2)
|
| |
L-cc- aminoadipate
|
M ammalian cells
|
I (T4), I (T1-3)
|
| |
L-cysteate
|
M ammalian cells
|
I (T1-3)
|
| |
L-serine-O-sulfate
|
M ammalian cells
|
S (T1-2), I (T1-3)
|
|
13-threo-hydroxy-aspartate derivatives
|
3-threo-hydroxy-asp
|
Xenopus oocytes
|
S (T1-4), I (T1-3,5)
|
| |
3-threo-acetoxy-asp
|
Xenopus oocytes
|
S (T1)
|
| |
p-threo-benzoyloxy-asp
|
Xenopus oocytes
|
I (T1-2)
|
| |
3-threo-naphtoyloxy-asp
|
Xenopus oocytes
|
I (T1)
|
| |
3-threo-propionyloxy-asp
|
Xenopus oocytes
|
S (T1)
|
| |
3-threo-benlyloxy-asp
|
Xenopus oocytes
|
I (T1-2)
|
Table 1. Effect of glutamate analogues on EAA transporters activity. Effects were determined in Xenopus oocytes or mammalian cell lines expressing cloned transporters subtypes. T1 denotes EAAT1, T2 denotes EAAT2, T1-T4 denotes EAATs from 1 to 4. S and I denote that glutamate analogues act as substrates or inhibitors, respectively.
|
Substance/condition
|
System
|
Transporter
|
Activity
|
Protein
|
mRNA
|
|
Glutamate
|
Mouse cortical astrocytes
|
GLAST
|
increased
|
increased
|
NE
|
|
Kainate
|
Mouse cortical astrocytes
|
GLAST
|
increased
|
increased
|
NE
|
|
dBcAlv1P
|
Mouse cortical astrocytes
|
GLAST
|
increased
|
increased
|
increased
|
|
Phorbol ester
|
Rat C6 glioma cells
|
EAAC1'
|
increased
|
ND
|
ND
|
|
Arachidonic acid
|
Xenopus oocytes
|
EAAT1
|
decreased
|
ND
|
ND
|
|
Arachidonic acid
|
Xenopus oocytes
|
EAAT2
|
decreased
|
ND
|
ND
|
|
Free oxygen rad
|
Mouse cortical astrocytes
|
ND
|
decreased
|
ND
|
ND
|
|
Nitric oxide
|
Rat synaptosomes
|
ND
|
decreased
|
ND
|
ND
|
|
Arrryloid p peptide
|
Rat cortical astrocytes
|
ND
|
decreased
|
ND
|
ND
|
|
Ischemia
|
Rat hippocampus
|
GLT1
|
ND
|
decreased
|
decreased
|
|
Hypertonic stress
|
Bovine renal NBL-1 cell I.
|
EAAC1
|
increased
|
ND
|
increased
|
|
Aa deprivation
|
Bovine renal NBL-1 cell I.
|
EAAC1
|
increased
|
increased
|
increased
|
|
Electric stimulation
|
Rat frontal cortex
|
ND
|
increased
|
ND
|
ND
|
Table 2. Effects of different substances and experimental conditions on the expression and activity of EAA transporters. ND and NE denote not determined and no effect, respectively.
As indicated previously, the effective removal (uptake) of excessive EAA is crucial for normal glutamatergic neurotransmission and neuronal protection. It has been reported that the dysfunction or loss of EAA transporters is implicated in several neurodegenerative diseases (amyotrophic lateral sclerosis -ALS-, Huntington’s disease) and brain insults (ischemia, hypoxia, status epileptics) [49,50]. Besides, in vivo administration of anti-sense oligonucleotides in rats directed against EAAT1/GLAST and EAAT2/GLT1 reduced the expression levels of the two transporters, increased the extracellular glutamate concentrations and rats had similar pathological characteristics to those seen with glutamate excitotoxicity and also progressive paralysis [51]. In another work, knockout of the EAAT2/GLT1 gene in mice caused epileptic seizures and brain injury [52]. On the other hand, the reduction in EAAT3/EAAC1 expression did not increase the extracellular glutamate concentration but caused low excitotoxicity which resulted in epilepsy [51].
This increasing evidence that EAA transporters are implicated in several neurodegenerative diseases has turned them into a subject of numerous studies regarding their regulation of expression and activity. While expression of EAA transporter genes can be regulated by transcription, mRNA processing and translation, the most known regulatory mechanisms take place at post-translational level. Glutamate, via glutamate receptors, can homeostatically regulate both levels and kinetics of EAA transporters [53]. Other factors that influence EAA transporters include arachidonic acid [54], and cAMP [55]. The regulatory mechanisms can be transporter and cell-type specific: phosphorylation of EAAT2/GLT1 stimulates glutamate transport whereas phosphorylation of EAAT1/GLAST inhibits it; arachidonic acid inhibits EAAT1/GLAST, enhances EAAT2/GLT1 and has little effect on EAAT3/EAAC1; and cAMP highly increases synthesis of EAAT2/GLT1, EAAT1/GLAST and EAAT4 only modestly, and decreases EAAT3/EAAC1. In pathophysiological situations, other compounds, including reactive oxygen species [56] and b–amyloid [57], also regulate EAA transporters. For a summary of the effects of different substances and experimental conditions on the expression and activity of EAA transporters (Table 2).
|
Substance/condition
|
System
|
Transporter
|
Activity
|
Protein
|
mRNA
|
|
Glutamate
|
Mouse cortical astrocytes
|
GLAST
|
increased
|
increased
|
NE
|
|
Kainate
|
Mouse cortical astrocytes
|
GLAST
|
increased
|
increased
|
NE
|
|
dBcAlv1P
|
Mouse cortical astrocytes
|
GLAST
|
increased
|
increased
|
increased
|
|
Phorbol ester
|
Rat C6 glioma cells
|
EAAC1'
|
increased
|
ND
|
ND
|
|
Arachidonic acid
|
Xenopus oocytes
|
EAAT1
|
decreased
|
ND
|
ND
|
|
Arachidonic acid
|
Xenopus oocytes
|
EAAT2
|
decreased
|
ND
|
ND
|
|
Free oxygen rad
|
Mouse cortical astrocytes
|
ND
|
decreased
|
ND
|
ND
|
|
Nitric oxide
|
Rat synaptosomes
|
ND
|
decreased
|
ND
|
ND
|
|
Arrryloid p peptide
|
Rat cortical astrocytes
|
ND
|
decreased
|
ND
|
Ischemia
|
Rat hippocampus
|
GLT1
|
ND
|
decreased
|
decreased
|
|
Hypertonic stress
|
Bovine renal NBL-1 cell I.
|
EAAC1
|
increased
|
ND
|
increased
|
|
Aa deprivation
|
Bovine renal NBL-1 cell I.
|
EAAC1
|
increased
|
increased
|
increased
|
|
Electric stimulation
|
Rat frontal cortex
|
ND
|
increased
|
ND
|
ND
|
Table 2. Effects of different substances and experimental conditions on the expression and activity of EAA transporters. ND and NE denote not determined and no effect, respectively.
Dysfunction of excitatory amino acid neurotransmission
Glutamate appears to be remarkably potent and rapidly acting neurotoxin. Exposure to 100 mM glutamate for 5 min is enough to destroy large numbers of cultured cortical neurons [58]. By the way, glutamate neurotoxicity may be blocked by antagonist compounds and attenuated by antagonists added after glutamate exposure [58].
Many reports have been published on increased levels of glutamate in certain neurodegenerative diseases such as Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) [59-62]. Increased extracellular glutamate has also been implicated in the onset of neurodegeneration associated with hypoxic damage (stroke). The release in glutamate following hypoxia has been suggested to be due, at least in part, to a calcium-independent mechanism following the reversal of the neuronal glutamate uptake carrier [63]. This increase in extracellular glutamate acts afterwards postsynaptically to increase cellular calcium levels with subsequent cell death.
The mechanism by which glutamate is increased in neurodegenerative diseases is unknown. The cause of this increased extracellular glutamate has been ascribed to a decrease in the activity of glutamate dehydrogenase (PD) [64] or a decrease in the number of Na+-glutamate transporters (ALS, HD) [65]. As it is a neurotoxic, it is likely that the high glutamate concentration observed in neurodegenerative diseases are the most likely cause of the neurodegeneration.
Marangos et al. [66] stated the glutamate hypothesis for early Alzheimer’s disease: “Cell death due to neuronal toxicity could result from excessive synthesis or release of glutamate or a glutamate-like substance, faulty glutamate reuptake, decreased glutamate degradation, or decreased inhibition of excitatory neurons. Any of these aberrant processes could, early in the disease, increase local levels of glutamate and so initiate a slow, progressive degeneration and eventual death of neurons”.
Several post-mortem studies have compared brain glutamate levels in Alzheimer’s disease to those in control subjects. Some investigators [67-69] have found lower glutamate levels in the frontal cortex and the temporal cortex of Alzheimer’s patients than in control subjects. Cerebral spinal fluid (CSF) concentration of free glutamate was significantly higher in patients with Alzheimer’s disease than in comparison subjects [59]. It should be noted that measurements of glutamate in CSF are likely to give better approximation of glutamate concentrations at synapses than are plasma glutamate concentrations.
In Huntington’s disease, glutamate and GABA concentrations decrease in striatum and caudate nucleus from brain [60,70]. However, no reduction at all was observed in the frontal cortex of patients. A likely possibility is that the low glutamate content of the caudate and the putamen in HD results from chronic failure of the normal reuptake mechanism for glutamate released at synapses, with or without any excessive rate of release of this neurotransmitter. This possibility is supported by the finding of Cross et al. [71], who observed large reductions in high-affinity glutamate uptake sites in autopsy specimens of caudate and putamen from HD patients.
If either excessive release or decreased reuptake of glutamate occurred in the striatum in HD, concentrations of glutamate might become high at synapses, with resulting damage to neurons. Some of the excess glutamate accumulate in synaptic clefts in HD would be carried away in the extracellular fluid, thus eventually causing a lowered glutamate content in striatal tissue. This fact is supported by an increased glutamate concentration in CSF of living HD patients as it was observed for Alzheimer’s disease patients.
In Parkinson’s disease, Schapire et al. [72] demonstrated a reduced activity of complex I of the mitochondrial respiratory chain in the region of substantia nigra in brain. Deficiencies on complex II and IV have been also observed in muscle biopsia from PD patients [73]. Cedarbaum et al. [64] observed a deficiency of GDH but not on pyruvate dehydrogenase complex. Since complex I is the point of entry for reducing equivalents (as NADH) to the respiratory chain, a decrease in complex I activity might result in feedback end product inhibition of GDH. Decreased levels of GDH might exert an excitotoxic glutamate effect via NMDA receptors on striatal dopamine nerve terminals [74] and contribute to cellular degeneration in PD.
Amyotrophic lateral sclerosis (ALS) is a disease resulting in degeneration of the motor cortex, the brain-stem and the spinal cord. While there are a number of hypotheses underlying this disease, an increase in glutamatergic neurotransmission has been proposed as a key event in the disease onset and recently, this has been described, at least in part, to a defect in the function of the GLT-1 glutamate transporter which is localized on astrocytes [65].
Recent in vitro studies have demonstrated that a blockade of the GLT-1 transporter either by transport inhibitors or by an antisense approach resulted in a slow, selective loss of motor neurons, thus strengthening the case for a critical role for GLT-1 in the etiology of ALS [62,75]. Furthermore, this decrease in uptake does not appear to be associated with a decrease in transporter expression levels [76] and there is no evidence of specific protein mutations associated with the disease. Because of the potential role of free radicals in ALS and the upset in function of the superoxide dismutase enzyme in the familial form of ALS, previous studies have suggested that an increase in free radicals may impair the function of GLT-1 [77].
Glutamate, and particularly the glutamate transporter system, have also been implicated in the ischemic damage associated with anoxia/hypoxia (stroke). For the first few minutes of ischemia, there is a slow acid shift of the cellular pH with a slow rise of extracellular potassium concentration and a subsequent decrease in extracellular sodium and calcium. The rise in potassium depolarizes the cells to around -20mV (anoxic depolarization) and the release of glutamate [78]. The mechanism by which glutamate is released in ischemia has been controversial. Some reports suggest that the release is calcium-dependent, suggesting conventional vesicular release; while others claim that the release is calcium-independent, implying a non-exocytotic mechanism such as the reversed operation of the glutamate uptake carrier.
In the lasts years, microRNAs (miRNAs) have emerged as important regulators of homeostatic mechanisms. Some studies suggest that miRNAs are neuroprotective. Thus, Drosophila microRNA miR-1000 regulates the expression of vesicular glutamate transporter, which loads glutamate into synaptic vesicles [79]. Thus, genetic ablation of this microRNA led to glutamate neurotoxicity, due to an increase of glutamate in synaptic cleft. In mammalian, microRNA miR-137 shows neuroprotection through the vesicular transporter VGluT2. Many other miRNA localized to dendrites could be regulated by synaptic activity. For instance, NMDA receptor activation inhibits microRNA miR-191 expression in dendrites; and increases microRNA miR-501-3p, which controls activation of AMPA receptors [80]. Some drugs, as paclitaxel, can activate transcription of some glutamate receptors, and this might be a therapeutic action against neurotoxicity, that has to be further studied [81].
Conclusions
High glutamate concentrations are related with neurodegenerative diseases [82]. Excitatory amino acid transporters can reduce synaptic concentration of glutamate, therefore diminishing neurotoxic effect in neurons. Furthermore, some drugs (as paclitaxel) or microRNAs could be used therapeutically against neurotoxicity. Their effect could be an increase of glutamate transporters or glutamate receptors.
References
- Oldendorf WH (1971) Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am J Physiol 221: 1629-1639. [Crossref]
- Oldendorf WH, Szabo J (1976) Amino acid assignment to one of three blood-brain barrier amino acid carriers. Am J Physiol 230: 94-98. [Crossref]
- Pratt OE (1976) The transport of metabolizable substances into the living brain. Adv Exp Med Biol 69: 55-75. [Crossref]
- Balázs R, Machiyama Y, Hammond BJ, Julian T, Richter D (1970) The operation of the ?-aminobutyrate bypath of the tricarboxylic acid cycle in brain tissue in vitro. Biochem J 116: 445-467. [Crossref]
- Machiyama Y, Balázs R, Hammond BJ, Julian T, Richter D (1970) The metabolism of gamma-aminobutyrate and glucose in potassium ion-stimulated brain tissue in vitro. Biochem J 116: 469-481. [Crossref]
- Norenberg MD, Martinez-Hernandez A (1979) Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 161: 303-310. [Crossref]
- Berl S, Lajtha A, Waelsch H (1961) Amino acid and protein metabolism. VI. Cerebral compartments of glutamic acid metabolism. J Neurochem 7: 186-197.
- van den Berg CJ, Matheson DF, Ronda G, Reijnierse GLA, Blokhuis GGD, et al. (1975) A model of glutamate metabolism in brain: a biochemical analysis of a heterogenous structure. In Metabolic Compartmentation and Neurotransmission. Plenum Press, New York. 709-723.
- Clarke DD, Ronan EJ, Dicker E, Tirri L (1975) Ethanol and its relation to amino acid metabolism in brain. In Metabolic Compartmentation and Neurotransmission. Plenum Press, New York. 449-460.
- Lerma J, Morales M, Vicente MA, Herreras O (1997) Glutamate receptors of the kainate type and synaptic transmission. Trends Neurosci 20: 9-12. [Crossref]
- Kirkwood A, Lee HK, Bear MF (1995) Co-regulation of long-term potentiation and experience-dependent synaptic plasticity in visual cortex by age and experience. Nature 375: 328-331. [Crossref]
- Conn PJ, Pin JP (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205-237. [Crossref]
- Schousboe A (1981) Transport and metabolism of glutamate and GABA in neurons are glial cells. Int Rev Neurobiol 22: 1-45. [Crossref]
- Fonnum F (1984) Glutamate: a neurotransmitter in mammalian brain. J Neurochem 42: 1-11. [Crossref]
- Nicholls D, Attwell D (1990) The release and uptake of excitatory amino acids. Trends Pharmacol Sci 11: 462-468. [Crossref]
- Maycox PR, Hell JW, Jahn R (1990) Amino acid neurotransmission: spotlight on synaptic vesicles. Trends Neurosci 13: 83-87. [Crossref]
- Zaczek R, Balm M, Arlis S, Drucker H, Coyle JT (1987) Quisqualate-sensitive, chloride-dependent transport of glutamate into rat brain synaptosomes. J Neurosci Res 18: 425-431. [Crossref]
- Danbolt NC, Pines G, Kanner BI (1990) Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glycoprotein from rat brain. Biochemistry 29: 6734-6740. [Crossref]
- Pines G, Danbolt NC, Bjørås M, Zhang Y, Bendahan A, et al. (1992) Cloning and expression of a rat brain L-glutamate transporter. Nature 360: 464-467. [Crossref]
- Kanai Y, Hediger MA (1992) Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 360: 467-471. [Crossref]
- Storck T, Schulte S, Hofmann K, Stoffel W (1992) Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci U S A 89: 10955-10959. [Crossref]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, et al. (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14: 5559-5569. [Crossref]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375: 599-603. [Crossref]
- Arriza JL, Eliasof S, Kavanaugh MP, Amara SG (1997) Excitatory amino acid transporter , a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci U S A 94: 4155-4160. [Crossref]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, et al. (1994) Localization of neuronal and glial glutamate transporters. Neuron 13: 713-725. [Crossref]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC (1995) Differential Expression of Two Glial Glutamate Transporters in the Rat Brain: Quantitative and Immunocytochemical Observations. J Neurosci 15: 1835-1853 . [Crossref]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, et al. (1995) Glutamate transporters in glia plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 1, 711-720. [Crossref]
- Palmada M (2001) Modulation of brain glutamate transport activities by neurotransmitters and benzodiazepines in heterologous expression systems. PhD Thesis in Biochemistry and Molecular Biology.
- Balcar VJ, Johnston GA (1972) The structural specificity of the high affinity uptake of L-glutamate and L-aspartate by rat brain slices. J Neurochem 19: 2657-2666. [Crossref]
- McBean GJ, Roberts PJ (1985) Neurotoxicity of L-glutamate and DL-threo-3-hydroxyaspartate in the rat striatum. J Neurochem 44: 247-254. [Crossref]
- Yamada K, Watanabe M, Shibata T, Nagashima M, Tanaka K, et al. (1998) Glutamate transporter GLT-1 is transiently localized in growing axons of the mouse spinal cord before establishing astrocytic expression. J Neursosci 18: 5706-5713. [Crossref]
- Mennerick S, Dhond RP, Benz A, Xu W, Rothstein JD, et al. (1998) Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J Neurosci 18: 4490-4499. [Crossref]
- Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler R, et al. (1997) Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol 42: 335-348. [Crossref]
- Bridges RJ, Stanley MS, Anderson MW, Cotman CW, Chamberlin AR (1991) Conformationally defined neurotransmitter analogues. Selective inhibition of glutamate uptake by one pyrrolidine-2,4-dicarboxylate diastereomer. J Med Chem 34: 717-725. [Crossref]
- Coco S, Verderio C, Trotti D, Rothstein JD, Volterra A, et al. (1997) Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur J Neurosci 9: 1902-1910. [Crossref]
- Fei YJ, Kanai Y, Nussberger S, Ganapathy V, Leibach FH, et al. (1995) Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature 368: 563-566. [Crossref]
- Palmada M, Böhmer C, Centelles JJ, Kinne RK (1999) Effect of benzodiazepines on the epithelial and neuronal high-affinity glutamate transporter EAAC1. J Neurochem 73: 2389-2396. [Crossref]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375: 599-603. [Crossref]
- McGivan JD, Pastor-Anglada M (1994) Regulatory and molecular aspects of mammalian amino acid transport. Biochem J 299: 321-334. [Crossref]
- Robinson MB, Dowd LA (1997) Heterogeneity and functional properties of subtypes of sodium-dependent glutamate transporters in the mammalian central nervous system. Adv Pharmacol 37: 69-115. [Crossref]
- Mason DJ, Suva LJ, Genever PG, Patton AJ, Steuckle S, et al. (1997) Mechanically regulated expression of a neuronal glutamate transporter in bone: A role for excitatory amino acid as osteotropic agents? Bone 20: 199-205. [Crossref]
- Tanaka K (1993) Expression cloning of a rat glutamate transporter. Neurosci Res 16: 149-153. [Crossref]
- Slotboom DJ, Lolkema JS, Konings WN (1996) Membrane topology of the C-terminal half of the neuronal, glial, and bacterial glutamate transporter family. J Biol Chem 271: 31317-31321. [Crossref]
- Wahle S, Stoffel W (1996) Membrane topology of the high-affinity L-glutamate transporter (GLAST-1) of the central nervous system. J Cell Biol 135: 1867-1877. [Crossref]
- Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honoré T, et al. (1996) Brain glutamate transporter proteins form homomultimers. J Biol Chem 271: 27715-27722. [Crossref]
- Wadiche JI, Amara SG, Kavanaugh MP (1995) Ion fluxes associated with excitatory amino acid transport. Neuron 15: 721-728. [Crossref]
- Eliasof S, Jahr CE (1996) Retinal glial cell glutamate transporter is coupled to an anionic conductance. Proc Natl Acad Sci U S A 93: 4153-4158. [Crossref]
- Vandenberg RJ, Mitrovic AD, Chebib M, Balcar VJ, Johnston GA (1997) Contrasting modes of action of methylglutamate derivatives on the excitatory amino acid transporters, EAAT1 and EAAT2. Mol Pharmacol 51: 809-815. [Crossref]
- Siesjö BK, Zhao Q, Pahlmark K, Siesjö P, Katsura K, et al. (1995) Glutamate, calcium, and free radicals as mediators of ischemic brain damage. Ann Thorac Surg 59: 1316-1320. [Crossref]
- Lazarewicz JW, Salinska E, Wroblewski JT (1992) NMDA receptor-mediated arachidonic acid release in neurons: role in signal transduction and pathological aspects. Adv Exp Med Biol 318: 73-89. [Crossref]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, et al. (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675-686. [Crossref]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, et al. (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699-1702. [Crossref]
- Gegelashvili G, Civenni G, Racagni G, Danbolt NC, Schousboe I, et al. (1996) Glutamate receptor agonists up-regulate glutamate transporter GLAST in astrocytes. Neuroreport 8: 261-265. [Crossref]
- Zerangue N, Arriza JL, Amara SG, Kavanaugh MP (1995) Differential modulation of human glutamate transporter subtypes by arachidonic acid. J Biol Chem 270: 6433-6435. [Crossref]
- Casado M, Bendahan A, Zafra F, Danbolt NC, Aragón C, et al. (1993) Phosphorylation and modulation of brain glutamate transporters by protein kinase C. J Biol Chem 268: 27313-27317. [Crossref]
- Volterra A, Trotti D, Racagni G (1994) Glutamate uptake is inhibited by arachidonic acid and oxygen radicals via two distinct and additive mechanisms. Mol Pharmacol 46: 986-992. [Crossref]
- Harris ME, Carney JM, Cole PS, Hensley K, Howard BJ, et al. (1995) beta-Amyloid peptide-derived, oxygen-dependent free radicals inhibit glutamate uptake in cultured astrocytes: implications for Alzheimer's disease. Neuroreport 6: 1875-1879. [Crossref]
- Choi DW, Maulucci-Gedde M, Kriegstein AR (1987) Glutamate neurotoxicity in cortical cell culture. J Neurosci 7: 357-368. [Crossref]
- Pomara N, Singh R, Deptula D, Chou JC, Schwartz MB, et al. (1992) Glutamate and other CSF amino acids in Alzheimer's disease. Am J Psychiatry 149: 251-254. [Crossref]
- Perry TL, Hansen S (1990) What excitotoxin kills striatal neurons in Huntington's disease? Clues from neurochemical studies. Neurology 40: 20-24. [Crossref]
- Plaitakis A, Constantakakis E, Smith J (1988) The neuroexcitotoxic amino acids glutamate and aspartate are altered in the spinal cord and brain in amyotrophic lateral sclerosis. Ann Neurol 24: 446-449. [Crossref]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, et al. (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675-686. [Crossref]
- Szatkowski M, Attwell D (1994) Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci 17: 359-365. [Crossref]
- Cedarbaum JM, Sheu KF, Harding BJ, Blass JP, Javoy-Agid F, et al. (1990) Deficiency of glutamate dehydrogenase in postmortem brain samples from parkinsonian putamen. Ann Neurol 28: 111-112. [Crossref]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38: 73-84. [Crossref]
- Marangos WF, Greenamyre JT, Penny JB, Young AB (1987) Glutamate dysfunction in Alzheimer’s disease: an hypothesis. Trends Neurosci 10: 65-68.
- Arai H, Kobayashi K, Ichimiya Y, Kosaka K, Iizuka R (1985) Free amino acids in post-mortem cerebral cortices from patients with Alzheimer-type dementia. Neurosci Res 2: 486-490. [Crossref]
- Procter AW, Palmer AM, Francis PT, Lowe SL, Neary D, et al. (1988) Evidence of glutamatergic denervation and possible abnormal metabolism in Alzheimer's disease. J Neurochem 50: 790-802. [Crossref]
- Hyman BT, Van Hoesen GW, Damasio AR (1987) Alzheimer's disease: glutamate depletion in the hippocampal perforant pathway zone. Ann Neurol 22: 37-40. [Crossref]
- Reynolds GP, Pearson SJ (1987) Decreased glutamic acid and increased 5-hydroxytryptamine in Huntington's disease brain. Neurosci Lett 78: 233-238. [Crossref]
- Cross AJ, Slater P, Reynolds GP (1986) Reduced high-affinity glutamate uptake sites in the brains of patients with Huntington's disease. Neurosci Lett 67: 198-202. [Crossref]
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, et al. (1989) Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1: 1269. [Crossref]
- Bindoff LA, Birch-Machin M, Cartlidge NE, Parker WD Jr, Turnbull DM (1989) Mitochondrial function in Parkinson's disease. The Lancet 2:49.
- Clow DW, Jhamandas K (1989) Characterization of L-glutamate action on the release of endogenous dopamine from the rat caudate-putamen. J Pharmacol Exp Ther 248: 722-728. [Crossref]
- Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW (1993) Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci U S A 90: 6591-6595. [Crossref]
- Bristol LA, Rothstein JD (1996) Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol 39: 676-679. [Crossref]
- Nicholls D, Attwell D (1990) The release and uptake of excitatory amino acids. Trends Pharmacol Sci 11: 462-468. [Crossref]
- Attwell D, Barbour B, Szatkowski M (1993) Nonvesicular release of neurotransmitter. Neuron 11: 401-407. [Crossref]
- Verma P, Augustine GJ, Ammar MR, Tashiro A, Cohen SM (2015) A neuroprotective role for microRNA miR-1000 mediated by limiting glutamate excitotoxicity. Nat Neurosci 18: 379-385. [Crossref]
- Hu Z, Zhao J, Hu T, Luo Y, Zhu J, et al. (2015) miR-501-3p mediates the activity-dependent regulation of the expression of AMPA receptor subunit GluA1. J Cell Biol 208: 949-959. [Crossref]
- Masocha W (2015) Astrocytes activation in the anterior cingulated cortex and altered glutamatergic gene expression during paclitaxel-induced neuropathic pain in mice. Peer J 1350: 1-14. [Crossref]
- Lee JE, Jeong da U, Lee J, Chang WS, Chang JW, et al. (2016) The effect of nucleus basalis magnocellularis deep brain stimulation on memory function in a rat model of dementia. BMC Neurol 16: 6. [Crossref]