Abstract
Despite intense basic research and numerous novel therapies under clinical investigation, cardioprotection as principal pharmacological indication remains as an empty niche. We have rigorously reviewed the investigational development of growth hormone releasing peptides (GHRP) and their clinical niching perspectives. GHRP constitute a group of small synthetic peptides that stimulate the growth hormone secretion and downstream axis activity. Two well-characterized peptidyl secretagogues receptors (GHSR 1a and CD36) are broadly represented including the cardiovascular system. Early studies translated into a new research era that inserted synthetic GHRP as promising cardio and cytoprotective candidates. A broad safety profile and a successful preclinical pharmacology nurtured the enthusiasm before one of these GHRP family members (GHRP-6) as a myocardial reperfusion damages-attenuating candidate. The existence of efficacious although occasional and fragmented clinical interventions in life-threatening conditions, along with the GHRP-6 multifactorial mechanism of action in myocardial infarction, ignite the initiative for a coherent clinical development.
Key words
GHRP, GHS, GH, cardioprotection, cytoprotection
Abbreviations
AKT: Protein kinase B, AMI: acute myocardial infarction, ApoE: Apolipoprotein E, CTGF: Connective tissue growth factor, DCM: dilated cardiomyopathy, dP/dt: indicates the rate of left ventricle maximal pressure rise in early systole. In general is a measure of left ventricle global contractility. DX: doxorubicin, ECM: Extracellular matrix, EGF: epidermal growth factor, ERK1/2: Extracellular signal-regulated kinase 1/2, GH: Growth hormone, GHRH: Growth hormone–releasing hormone, GHRP: Growth hormone releasing peptides, GHS: Growth hormone secretagogues, GHS-R: Growth hormone secretagogue receptor, GHSR-1a: Growth hormone secretagogue receptor type 1a, HIF-1α: hypoxia-inducible factor-1 alpha, I/R: ischemia and reperfusion, IGF-1: insulin-like growth factor-1, IL-1β: Interleukin-1 beta, IL-6: Interleukin 6, LPS: lipopolysacharide, LV: left ventricle, LVEF: left ventricular ejection fraction, MBP: mean blood pressure, MIF: macrophage migration inhibitory factor, MCP-1: Monocyte chemoattractant protein-1, MMP: matrix metalloproteinase, MOD: Multiple Organs Damage, NEP: nitrosylation end-products, NIH: National Institutes of Health, PDGF: Platelet-derived growth factor, PGC1α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha, PI-3K: Phosphatidylinositol-4,5-bisphosphate 3-kinase, PPARγ: Peroxisome proliferator-activated receptor gamma, p-PLB: phosphorylated phospholamban, RAS: Renin-angiotensin system, rhGH: recombinant human growth hormone, ROS: reactive oxygen species, TGF-β: Transforming growth factor beta, TIMP: tissue inhibitor of metalloproteinase, TNF-alpha: Tumor necrosis factor alpha.
Introduction
What represents today a family of peptidyl GH secretagogues with broad cytoprotective properties, came to light by the American endocrinologist Cyril Bowers, who observed that chemical analogs of enkephalin amide showed GH releasing activity upon their incorporation to pituitary cultures. GHRP-6 (His-DTrp-Ala-Trp-DPhe-Lys-NH2) appeared as the first in line synthetic peptide which specifically elicited GH dosage-related release in vitro and in vivo [1]. Afterwards a heptapeptide, GHRP-1, and two other hexapeptides, GHRP-2 and Hexarelin, were synthesized and addressed to basic and sporadic clinical studies. Although at the beginning GHRP seemed to linger as an instrumental tool within the endocrinologist armamentarium, ulterior findings unraveled unexpected pharmacological properties. GHRP family members began to shine by their ability to prevent cardiac cells demise as to induce the restoration of critical cardiac functions upon ischemia/reperfusion episodes [2,3] . A novel generation of promising cardioprotective agents had started to rise and to set a bridge between endocrinology and cardiology.
Although the history of some of the foremost biomedical discoveries is permeated by serendipity [4], we deem that the well-established pivotal role of the GH/insulin-like growth factor-1 (IGF-1) axis for cardiomyocyte physiology, and the subtle alterations of this axis within the pathogenicity of dilated cardiomyopathy and left ventricular dysfunction, ignited the idea of assessing the potentiality of the GHRP to alleviate cardiac pathologies [5]. It was far to be anticipated on those early days, however, that the GHRP-mediated cardiotropic and cytoprotective effects are superior to those shown by the exogenous administration of GH, are not shared by GHRH, and that importantly, GHRP exert their pharmacological actions via GH-independent pathways which obviously represented another turning point in this history [3].
The progresses obtained and the experiences accrued along the years of work with these pioneer synthetic, non-endogenous GHRP gradually led to the discovery of Ghrelin, the 28-aminoacids hormone with GH-releasing action secreted by gastric cells [6,7] with orexigenic, cellular energy and metabolism control; and cardioprotective and cytoprotective abilities for a myriad of cell populations [8]. This endogenous GHS is perhaps the most thoroughly studied GH-secretagogue and a variety of synthetic analogs and receptor inverse agonists have been developed [9-12].
GHRP-6 is a small molecular weight peptide, effective when orally administered, stable, and economically low-priced than others [13]. Significantly important is our observation that GHRP-6 intravenous administration proved to be safe in a doses scale-up clinical trial in healthy human volunteers [14]. Relevant is as well for GHRP-6 pharmacological “positioning” our demonstration that there is no in vivo pharmacological interaction between the peptide and a well-validated cardiovascular drug as the beta blocker agent metroprolol [15]. Since for years GHRP-6 has been the platform of our experimental work, we will address particular attention to its investigational development as for hexarelin and GHRP-2.
In this review, we have attempted to summarize the most relevant achievements of the pharmacological knowledge with synthetic GHRP (GHRP-6, GHRP-2 and Hexarelin) in a historical perspective line. Special focus is spotted on general cyto-and- cardioprotection research fields since all these peptidyl agents, have contributed to the discovery of novel functions and mechanisms involved in cellular survival, senescence and death.
The search strategy was based on the PUBMED/MEDLINE electronic databases including original research publications and reviews. The search was progressively date-escalated from 1980 and included articles in English only. The following search terms were introduced: GHS, GHRP, GHSR, CD36, cardiac ischemia/reperfusion, cardiac stunning, heart failure, cytoprotection, and cardioprotection. We deem cardiologists, clinicians, and basic and clinical pharmacologists would receive some benefit from this text, in correspondence to the futuristic pharmacological opportunities offered by these agents. Cytoprotection remains as an orphan niche in contemporary medical armamentarium.
Repositioning GHRP
Despite their potent and reproducible GH-releasing activity, the clinical use of GHRP as orally active growth-promoting agents, and anabolic anti-aging drugs remains to be confirmed [13]. Thus, the preliminary enthusiasm as an alternative for GH replacement therapy did not experience any significant progress [16]. Nevertheless, it is likely that the myocardial, vascular, and multiorgan expression of the GHRP receptors, may have contributed to reinforce the cardiovascular application stream of these peptides.
A remarkable specific (125)I-Tyr-Ala-hexarelin binding was observed in the human cardiovascular system where the highest binding levels were detected in ventricles, followed by atria, aorta, coronaries, carotid, endocardium and vena cava. In other experiments on H9c2 cardiomyocytes specific GHRP binding was found along with a potent anti-apoptotic activity [3]. The primarily investigated receptor was the growth hormone secretagogue receptor-1a (GHSR1a) which is detected in isolated human cardiomyocytes, myocardium and aorta samples [17]. It has been recently shown that GHSR1a is a sort of “promiscuous receptor” involved in many systems and behavioral patterns as reward, feeding, and memory what makes it an attractive pharmacological target [18]. Years later, the synthetic GHRP hexarelin was acknowledged as a ligand of another protein identified as CD36, a scavenger receptor that is expressed in various tissues, including monocytes/macrophages and the endothelial microvasculature. Activation of CD36 in perfused hearts by hexarelin was shown to increase coronary perfusion pressure in a dose-dependent manner. Contrariwise, this effect was lacking in hearts from CD36-null mice and hearts from spontaneous hypertensive rats genetically deficient in CD36 [19,20]. Thus, it is currently accepted that two cardiac receptor subtypes mediate the pharmacological actions of GHRP-6, GHRP-2, and hexarelin [21,22].
Cardiotropic evidences
The importance of limiting myocardial ischemia/reperfusion injury has been appreciated since Braunwald proposed that the extent and severity of tissue damage were not predetermined at the onset of ischemia, but could be modified by therapeutic manipulations applied during ischemia [23,24]. Few years ago, an NIH expert’s panel concluded that cardioprotection is at a crossroads since approaches to identify cardioprotective therapies have been disappointing during the past 30 years [25]. The fact that the multiple candidates assayed so far are addressed to cut the damage chain by neutralizing one single link, may explain to some extent the mounting failures. Myocardial ischemia/reperfusion damage entails multiple molecular and biochemical mechanisms that each alone is sufficiently injurious to disturb an organ whose mechanical performance is dependent upon the stability of ionic/electrical pumps. Oxidative stress, intracellular calcium overload, pH changes, mitochondrial dysfunction, inflammation, and excessive neurohormones are part of an interactive and self-perpetuating continuum of the myocardial injury cascade (Figure 1). The evidences obtained along the years of experimental screening of the synthetic GHRP, suggest that each single member of this family of peptides is able to simultaneously counteract different injurious operators in the myocardial ischemic event.
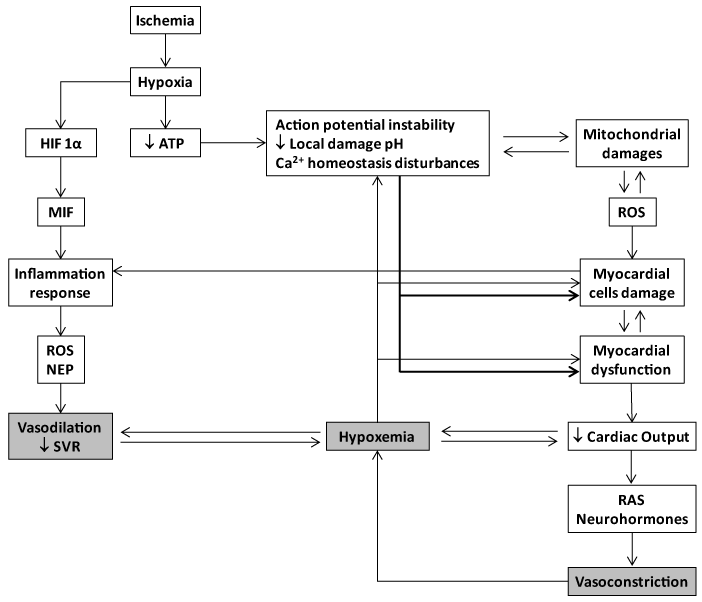
Figure 1. Basic molecular pathophysiological cascade of acute myocardial infarction. Myocardial ischemia results from a demand-supply mismatch between the myocardial energy requirement and the supply of oxygen and other substrates when coronary flow is interrupted. Following ischemia, hypoxia triggers an acute failure in mitochondrial respiratory function when the diffusible oxygen stores become exhausted. Accordingly, adenosine triphosphate (ATP) reserves are rapidly depleted and there is a respiratory shift toward an anaerobic profile. Uner these circumstances, lactate, H+ ions, CO2, and potassium accumulate due to an egress failure during ischemia. Therefore, myocardial ischemia is part of the ischemia/reperfusion injury complex which includes arrhythmias, microendothelial damage, myocardiocytes stunning and cell death. ATP depletion is irrevocably ligated to the inability of maintaining the normal negative resting membrane potential, to an alteration of calcium homeostasis (intracellular Ca2+ ([Ca2+]i) overload) which may eventually lead to different patterns of abonormal cardiac contraction. Mitochondrial morphology and functionality become abonormal when the inner membrane results permeable to solutes (i.e., calcium), thus establishing the so called "open pore" (mitochondrial permeability transition pore-mPTP). The failure of ATP-dependent pumps as the KATP, which is also opened during ischemia/ATP depletion, is a converging causal factor for mitochondrial damage due potassium overload. The involvement of the mPTP during myocardial ischemia/reperfusion promotes local cells death. In this scenario miochondria turn into an active ROS manufacturing plant that increase and perpetuates mitochondrial damages and dysfunction. The failure of myocardial contractility (contractility depression) is a precocious and multifactorial consequence of ischema which may eventually lead to reduced cardiac output and heart failure. A reduced LV srtoke volume with a deficient cardiac output may translate in systemic deleterious consequences for the splanchnic organs system, that if persistent may create a systemic multi-organ hypoxic environmet, as an anticipation to cardiogenic shock and multiple organ failure. Systemic and coronary constriction is an emergency compensatory adrenergic-mediated response before a low coronary pressure and myocardial hypoperfusion. This situation may translate into a self-perpetuative vicious circle thus amplfying the ischemic episode and the myocardial wall stress. The local inflammatory reaction is a useful but critial operator within the myocardial ischemia/reperfusion damage process. Hypoxia itself activates the HIF-α/MIF axis and the consequent downstream inflammatory cascade. HIF-α and NF-κB induce the recruitment of inflammatory cells and enhance the expression of adhesion molecules within the endothelium and the injured zone. Cells debris, scattered sequestered autoantigens, and locally-generated ROS, participate in the orchestration of the inflammatory response. At the end, the ischemia/reperfusion myocardial-targeted area secrete TNF-𝛼, IL-1𝛽, and IL-6. Again these cytokines are involved in a self-perpeatuating process in the ROS chain reaction. The systemic spillover of inflammatory mediators - including cytokines and NEP are potentially harmful for the systemic haemodynamic control. Systemic vascular resistance may drop down under such circumsatnces which may agravate the hypoxemia, the organs perfusion, and the myocardial injury.
Experimental studies by 1997 proved the ability of hexarelin to reverse the cardiac dysfunction in GH-deficient animals immunized by the administration of an anti-GH-releasing hormone (GHRH) serum. Ex vivo and in vivo systems converged to document that hexarelin progressively and globally improved left ventricular function even under post-ischemic scenarios. These experiments had the merit to show that the synthetic secretagogue protective activity was divorced from any further stimulation derived from the somatotropic function [26]. Accordingly in 1998 this group demonstrated that hexarelin protected against postischemic ventricular dysfunction in senescent hearts of aged male rats. Both ex vivo and in vivo the GHRP offered a striking heart protection against reperfusion stunning, improved ventricular pressures and volumes, and reduced CK concentration in perfusate. Again, they sustained the concept that the protection afforded by the peptide is likely due to a direct cardiotropic action which appeared far greater than that induced by GH administration in a concurrent control group [27]. A far more defining protocol was assumed in 1999 as the study included hypophysectomized rats, to ascertain whether hexarelin had non-GH-mediated protective effects on the heart. In this context, the authors showed that hexarelin attenuated the ischemia/reperfusion damage, as prevented elevation of left ventricular end-diastolic pressure, coronary perfusion pressure, reactivity of the coronary vasculature to angiotensin II, and the release of creatine kinase in hypophysectomized animals [28]. These three experiments were pivotal to define GHRP intrinsic cardioprotective ability.
By 1999 seven adult patients with GH deficiency and left ventricular failure received hexarelin administrations (i.v). The GH response to hexarelin was negligible in these patients. Moreover, hexarelin administration increased their LVEF without changing catecholamine levels, mean blood pressure, or cardiac output. For the first time, the acute administration of hexarelin proved to induce a positive inotropic effect in humans, GH-independent and mediated by specific myocardial receptors for a GH secretagogue peptide [29]. A subsequent study involving hexarelin administration to normal adults, severe GH-deficient patients (N=7), and patients with severe ischemic dilated cardiomyopathy (N=12), confirmed that the acute administration of hexarelin exerts a GH-independent positive inotropic effect likely mediated by specific GHRPs myocardial receptors [30]. This pioneering group, subsequently evaluated the cardiac performances of the acute hexarelin administration (2.0 micrograms/kg, i.v.) in patients undergoing by-pass surgery in comparison to patients given GH-releasing hormone, recombinant human GH, or placebo. The study concluded that the acute administration of hexarelin improves cardiac performance without any relevant variation in systemic vascular resistance, induced a reduction of wedge pressure, and significantly, that these cardiotropic effects were not shown by the other concurrent interventions [31].
These human subjects’ studies were paralleled by contemporary experimental progresses in basic science, which demonstrated that hexarelin enhanced H9c2 cardiomyocytes proliferation in a dose-dependent manner. Since these were in vitro experiments, completely excluded a potential intervention of the GH axis and clearly indicated a direct GHRP binding to cardiac cells membranes [32]. Van de Breghe group demonstrated that 14 days of pretreatment with GHRP-2, but not GH, selectively protected against the postischemic diastolic dysfunction and myocardial stunning of excised hearts submitted to ischemia/reperfusion in isolated, perfused rabbit hearts [33].
TO-2 hamster model of dilated cardiomyopathy (DCM) characterized by progressive left ventricular (LV) dilation, LV wall thinning, LV systolic dysfunction, and reduced life span; both GHRP-2 and GHRP-6, ameliorated all the dysfunctional ventricular parameters and reduced the progression of the DCM [34]. We also examined the potential impact of GHRP-6 in a rat model of DCM/heart failure induced by doxorubicin (DX). The concurrent administration of GHRP-6 was undertaken with the purpose to study the potential prophylactic impact before the cardiac function demise. As part of the prolonged treatment with DX, the concurrent administration of GHRP-6 completely prevented failure of cardiac function, evaluated as the percentage of ejection fraction by echocardiography (Figure 2-prevention). This effect significantly increased the animals' survival. Similar results were obtained in the therapeutic administration schedule, with functional recovery of cardiac muscle to physiological levels (Figure 2-regression), also attenuating systemic damages and, consequently, decreasing rats’ mortality rates. In the experimental model of DX-induced cardiac and systemic damage, GHRP-6 additionally attenuated a variety of extracardiac damages observed in the renal tubular and bronchoalveolar epithelial structures as in the hepatic parenchyma [35].
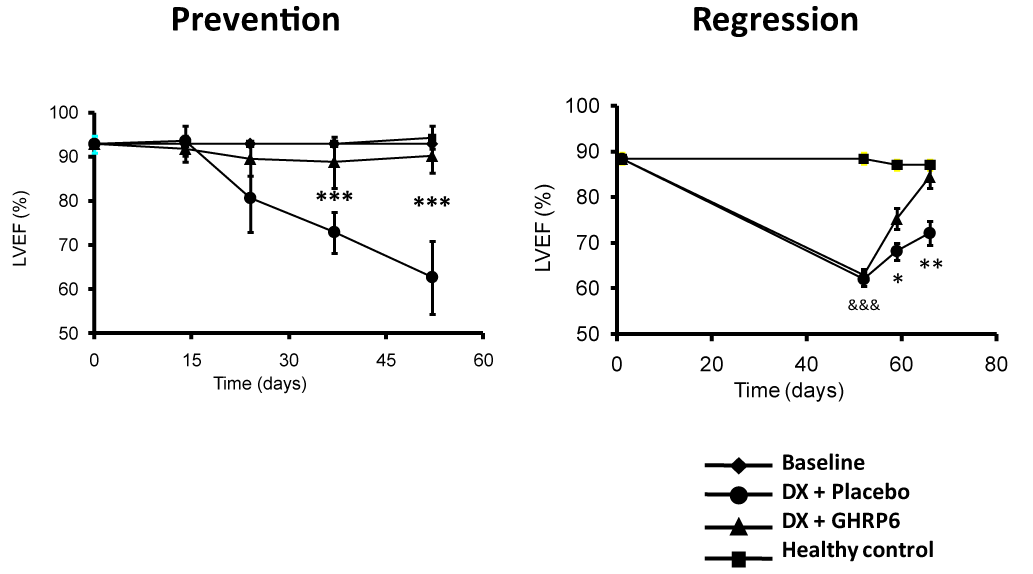
Figure 2. Effect of GHRP-6 on the left ventricle ejection fraction in an experimental model of dilated myocardiopathy (DMC).
Echocardiography data derived from our DMC model including the prevention and the regression study protocols in rats (see reference 35). The prevention protocol conceived the concurrent administration of GHRP-6, as part of a prolonged treatment with doxorubicin. The concomitant GHRP-6 completely prevented cardiac function failure evaluated as the percentage of ejection fraction by echocardiography. The regression approach examined the GHRP-6 intervention once LVEF was already deteriorated. As shown, the therapeutic administration schedule introduced a full functional recovery of cardiac muscle. Data corresponding to percentage of ejection fraction (%EF) are represented as a mean value ± standard error of the mean for each experimental group. (*) represent the statistically significant differences between groups treated either with placebo or GHRP6. Student´s t test.
In more recent years these data were further substantiated using again the TO-2 hamster DCM biomodel in which GHRP-2 reduced the progression of LV remodeling, dysfunction and the ensued myocardial fibrosis by an anti-oxidant mechanism [36]. The above mentioned myocardial fibrotic process amelioration reveals an additional potential use for GHRP in an unmet medical need. In this very context, hexarelin chronic treatment in spontaneously hypertensive rats, in addition to lessen ventricular hypertrophy, diastolic dysfunction, and high blood pressure; significantly reduced cardiac fibrosis by decreasing interstitial and perivascular myocardial collagen deposition and myocardial hydroxyproline content. Mechanistically speaking, hexarelin treatment increased matrix metalloproteinase (MMP)-2 and MMP-9 activities and decreased myocardial mRNA expression of tissue inhibitor of metalloproteinase (TIMP)-1 [37].
Our group has contributed to validate the potential anti-fibrotic abilities of GHRP-6 in animal models of liver cirrhosis [38] and hypertrophic scars (Mendoza-Mari, et al. Already accepted since Wednesday, April 13, 2016. Plastic Surgery International). February 2016), in which via a PPARγ-driven cascade, GHRP-6 intervention reduced TGF-β1 and CTGF expression which translated in a dramatic reduction in the accumulation of collagen and other extracellular matrix proteins.
A seminal report by a Merck Research Laboratories group dated on 2003, demonstrated for the first time that chronic treatment with GHRP-6 (21 days) prevented sudden death in a canine model of dilated cardiomyopathy and subsequently subjected to acute myocardial infarction (AMI). In the meantime the mortality rates for the vehicle and GH-treated groups were about 50%. Although the authors do not precise the mechanism underlying the 100% survival in the GHRP-6 group, an enhanced regional myocardial compensatory function of the nonischemic zone was assumed [39]. This notion could be validated at least in part by the fact that the cardiotropic effects shown by GHRP-1, GHRP-2, GHRP-6, and hexarelin in cardiomyocytes and isolated, denervated, perfused hearts, is mediated by an elevation of Ca2+ influx through the voltage-gated calcium channel, triggering Ca2+ release from thapsigargin-sensitive intracellular stores, which translated in a positive inotropic response without a chronotropic effect [40]. More recent data confirm the ability of hexarelin and other secretagogue peptides that bind and activate the GHS receptor type 1a, to control the cardiac action potential and reduce apoptosis of cardiomyocytes, derived from isolated hearts subjected to ischemia/reperfusion episodes [41].
Consistent with these data, our group observed a transient inotropic effect of about 15 minutes in both healthy and infarcted rabbits following a single GHRP-6 intravenous bolus (400 µg/kg). Echocardiography recordings indicated a 15-20% elevation of the ejection fraction as an increase in shortening fraction (Juan Valiente Mustelier and Jorge Berlanga Acosta. Unpublished observations, 2007). More recent studies based on isolated murine hearts which underwent periods of ischemia and reperfusion (I/R) confirm that pre- or post-treatments with hexarelin for instance prevented the intracellular disturbances in Ca+2 transients through recovery of p-PLB after the I/R insult [42]. Other studies involving adult Wistar rat ventricular myocytes have confirmed the positive inotropic response induce by hexarelin and other secretagogue peptides that bind the GHS-R1a, which activate protein kinase C signaling cascade [43].
In 2005 we undertook a porcine model of AMI via left circumflex artery occlusion for 1 hour followed by a 72-hours reperfusion period. GHRP-6 rescued ischemic myocardium from death for over 70% of the area at risk (Figure 3), and that in addition to enhance survival signaling pathways/gene expression of the PI-3K, AKT, BCL2 pathway; GHRP-6 decreased reactive oxygen species spillover, the inflammatory marker CRP, and preserved the antioxidant defenses [44]. These antioxidant and anti-inflammatory properties have been also attributed to GHRP-2 when its antitherogenic potential was examined in ApoE(-/-) mice so that 12/15-lipoxygenase, interferon gamma, and macrophage migration inhibitory factor gene expression were accounted. Furthermore, in cultured aortic smooth muscle cells GHRP-2 prevented the generation of peroxides, the down-regulation of IGF-1 receptor, and the commitment of apoptosis [45].
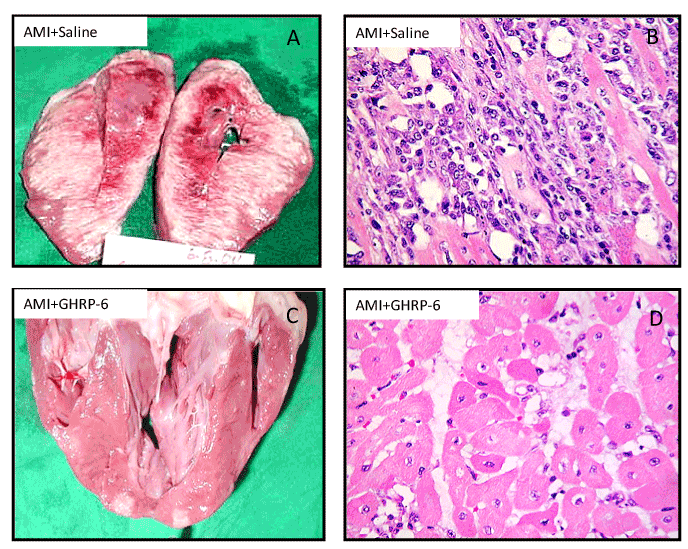
Figure 3. Morphological evidences representative of the GHRP-6 effect in a porcine model of myocardial infarction. Effect of the GHRP-6 on AMI size and severity. A, B – Macroscopic and histological images of AMI damage in animals treated with placebo. C, D- Macroscopic and histological images representative of the GHRP-6 cardioprotective effect. Histological fragments were in every case collected from apparently normal zones, adjacent to the AMI necrotic core. Rats treated with GHRP-6 exhibited mostly preserved or marginally damaged (sarcoplasmic edema) myofibrils. No myofibrolysis was observed although a number of ghost nuclei appeared. (H/E, ×20 magnification).
Hexarelin via CD36 occupation increases the expression of multiple genes involved in fatty acid mobilization in adipocytes toward the mitochondrial oxidative phosphorylation, and many of these up-regulated genes are known targets of peroxisomal proliferator-activated receptor (PPAR)-gamma. Consistent with this, electron microscopy of hexarelin-treated adipocytes reflects highly organized cristae formation that spans the entire width of mitochondria, with a concomitant cytochrome c oxidase activity enhancement. Although this signaling and activation cascade has not been described for myocardial cells so far, the potential existence of these phosphorilative and mitochondriogenic mechanisms in the heart, and its potential amplification by GHRP ligands, may eventually contribute to myocardial salvage during critical ischemia periods [46]. In a more recent study based on a myocardial infarction model, and addressed to examine whether hexarelin treatment can compensate for ghrelin deficiency in ghrelin-knockout mice, the mortality within 2 weeks was significantly lower in the hexarelin (6.7%) and ghrelin groups (14.3%) than in the vehicle group (50%). Furthermore, hexarelin was more effective than ghrelin as judged by the ejection fraction and other left ventricle-dependent physiological constants as dP/dt max and dP/dt min [47].
In extracardiac models of striated muscles atrophy, GHRP-2 exerted a potent myoprotective effect, presumably via the direct agonistic stimulation of the GHS-R1a since no elevation of IGF-1 transcript was observed [48]. Thus, it is likely that GHRP cardioprotective effects in scenarios of dilated myocardiopathy may be somehow mediated by a trophic or anabolic mechanism. Based on the benefits of GHRP-6 on muscle functions, a newly synthesized GHRP-6-biotin conjugate tested on cultured myoblast, showed to induce the expression of myogenic proteins, the IGF-1 levels as the concentrations of energy metabolites and the corresponding enzymes. Practical applications of the GHRP-6-biotin conjugate could include amelioration of sarcopenia and/or cardiac cachexia [49].
Extracardiac cytoprotective action of GHRP
As judged by the Pubmed outcomes, the cytoprotective effects of synthetic peptidyl GHRP appear far less studied in non-cardiac, parenchymal epithelial organs or multiple organ systems than in the cardiovascular system. However, the lessons derived from the studies reviewed are consistent with a broad cytoprotective influence for a variety of splanchnic and non-splanchnic organs by reducing inflammation, and preventing necrosis and/or apoptosis. The constellation of GHRP-mediated pharmacological capabilities is summarized in Figure 4.
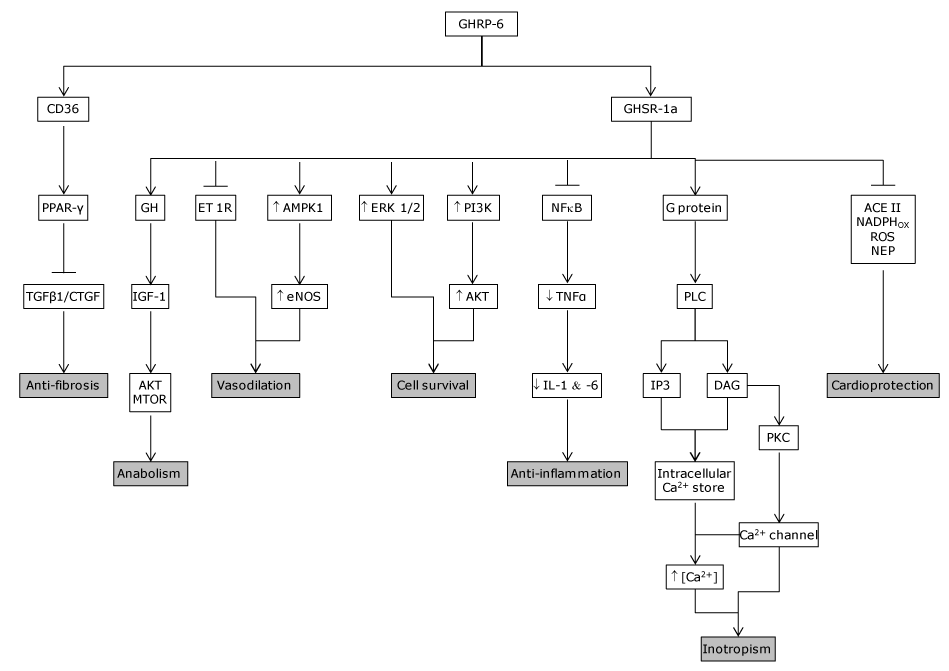
Figure 4. GHRP mechanism of action. GHRP are endowed with the ability to bind two different receptors which seem to mediate its cytoprotective and other pharmacological properties. GHSR1a was the primarily investigated receptor, responsible for the secretagogue and other relevant physiological activities. It has been detected in isolated human cardiomyocytes and endothelial cells among other tissues. Years later, the CD36 was identified in the myocardium and shown to mediate other, non-secretagogue GHRP biological actions. Activation of CD36 in perfused hearts increases coronary perfusion pressure in a dose-dependent manner. CD36 null mice exhibit a significant myocardial vulnerability to ischemia/reperfusion as compared to wild-type controls. The main biological properties/pharmacological actions of GHRP-6 as cyto and cardioprotective candidates are summarized as follows: Inotropic. Its seems to be mediated by an elevation of Ca2+ influx via PLC/DAG/PKC, through the voltage-gated calcium channel, triggering Ca2+ release from thapsigargin-sensitive intracellular stores, which translated in a positive inotropic response without a chronotropic effect. Anti-fibrotic. According to our data, CIGB 500 upregulated PPAR- gamma which is followed by a TGF-beta, CTGF and PDGF downregulation. Anti-inflammatory. Blunts NF.kB expression and activation and the ensued downstream pro-inflammatory cascade. Reduces ROS, NEP and activates SOD expression and activity. Cell survival.
It involves the phosphatidil inositol-3 kinase /protein kinase B (PI-3K/PKB), Akt pathway, as the induction of the hypoxia-inducible factor-1 alpha (HIF-1α) all committed in cellular survival to lethal insults. Cardio-protective. It involves different biological actions which converge to enhance cardiomyocytes survival. I.E., reduction of ROS and NEP cyto-toxicity, reduction of neurohormones, etc.
Vasodilatory. It seems to involve and endothelin activity reduction and an e-NOS up-regulation. Anabolic. It is mediated by the IGF-1/Akt and mTOR pathway activity.
Our group examined the cytoprotective effects exerted by the GHRP-6 administration in a rat model of hepatic tissue subjected to I/R, as in other distal organs from the ischemic site (i.e., lungs, kidneys and small intestine). Histological and biochemical data allowed us to conclude that the pharmacological preconditioning induced with the GHRP-6 treatment attenuated I/R liver damage. Besides, respiratory distress syndrome like pulmonary microscopic changes, intestinal trans-mural infarct and acute tubular necrosis in kidneys were significantly reduced (Figure 5). These results indicated for the first time, a systemic cytoprotective effect for the GHRP-6, suggesting its potential efficacy to control the systemic inflammatory response syndrome sassociated to acute I/R and shock, which eventually originated a Multiple Organs Damage (MOD). We also observed that the cytoprotection induced by GHRP-6 treatment was also related to the attenuation in the generation of reactive oxygen species (ROS) and preservation of the antioxidant defense reserves. Histological analysis as the assessment of myeloperoxidase activity evidenced a clear anti-inflammatory GHRP-6-induced effect in the liver and remote organs. Moreover, the molecular mechanism (Figure 6) mediating the action of GHRP-6 peptide was shown to involve the phosphatidil inositol-3 kinase /protein kinase B (PI-3K/PKB), Akt pathway, as the induction of the hypoxia-inducible factor-1 alpha (HIF-1α) all committed in cellular survival [50]. Subsequent to our study, Granado and co-workers [51] examined the potential anti-inflammatory impact of GHRP-2 in LPS challenged rats. In this context GHRP-2 administration attenuated the effects of LPS on the elevation of circulating levels of transaminases, nitrites/nitrates, and TNF-alpha, via direct interaction with liver nonparenchymal cells. Globally speaking, the exogenous administration of these two synthetic GHRP appeared to exert a potent hepatoprotective role by attenuating the inflammatory response orchestrated by liver resident macrophages. Another line of evidences document the benefits of 15-daily injections of GHRP-2 (100 micrograms/kg) in arthritic rats, so that the treatment ameliorated the external symptoms of arthritis, and decreased the circulating levels of IL-6 as the nitrite/nitrate release from peritoneal macrophages in vitro. This experiment extrapolated the counter-inflammatory properties of GHRP-2 to a nonepithelial organ and suggested again a direct interaction with ghrelin receptor of immune cells [52]. Similarly, effects have been attributed to Ghrelin by inhibiting the inflammatory response via Akt-activated pathway with a concomitant reduction of myeloperoxidase activity, the rate of apoptosis, and oxidative stress [53]. All these data suggests that GHRP exert a mutually inclusive beneficial effect by directly protecting parenchymal organs epithelial cells, and simultaneously by modulating the magnitude of the inflammatory response by direct interaction with the effector immune cells. Supporting the protective effect of GHRP-6 on epithelial organs, a recent study has excellently described and dissected the mechanistic bases on how GHRP-6 prevented gastric mucosal damage induced by water immersion restraint (WRS) and other forms of stress. The data indicated that the protective effect of GHRP-6 on WRS-induced gastric mucosal injury is somehow mediated by peripherally suppressing the vagal efferent effect on the stomach, including gastric acid secretion. Although more studies are clearly demanded, the present findings open the possibility to use GHRP-6 in preventing Curling ulcers [54].
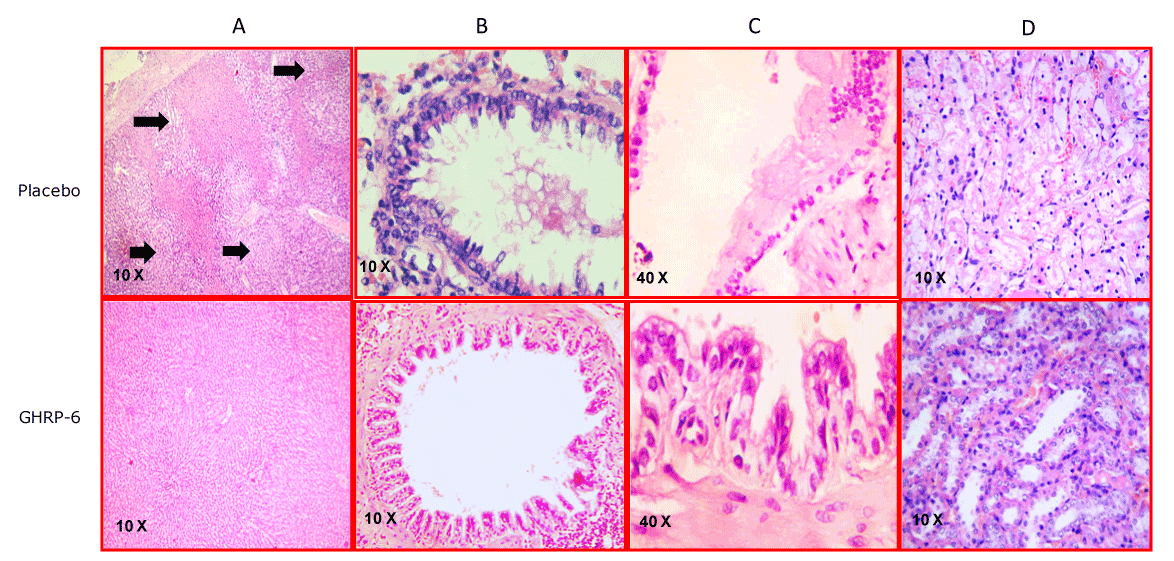
Figure 5. Representative images of the multiorgan protection conferred by GHRP-6. Column A- Liver sections. Black arrows indicate scattered foci of hepatic necrosis in the placebo group. In sharp contrast the animals treated with GHRP-6 exhibited only a mild sinusoidal dilation. Column B- Bronchial sections. The severe damage on the epithelial cells stratum with luminal sloughing in animals receiving placebo is dramatically contrasting with the epithelial integrity detected in the medicated group. Column C-Small intestine images in which the substantial difference is illustrated between the placebo and the GHRP-6 mediated protection. For the former a portion affected by transmural epithelial coagulative necrosis is depicted. Mild edema appeared in the animals treated with GHRP-6. Column D- renal medulla sections. Placebo animals were mostly characterized by tubular epithelial necrosis. For the GHRP-6 medicated group epithelial cells were mostly intact or mildly flattened.
2021 Copyright OAT. All rights reserv
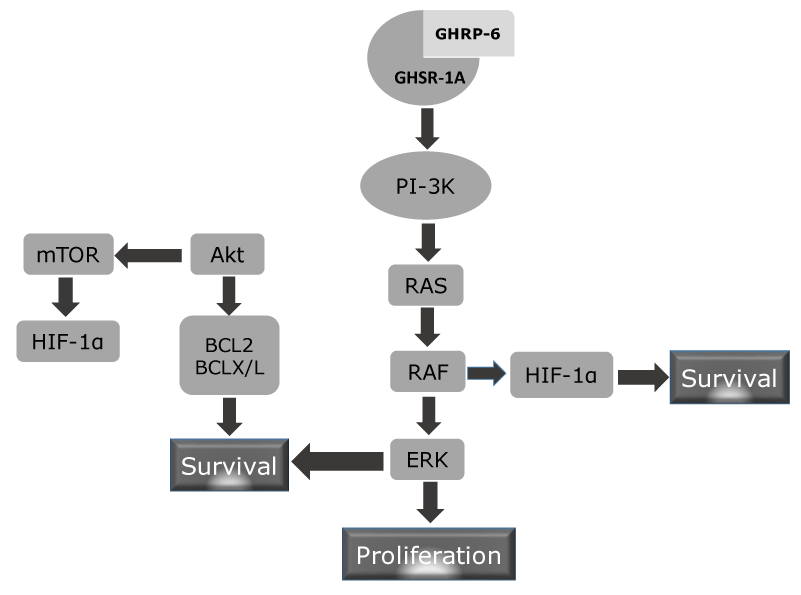
Figure 6. Putative mechanism of GHSR-1A mediated cytoprotection. GHRP-6 agonistic binding to its GHSR elicits two major biological response not related to growth hormone secretion: cell survival and cell proliferation. It is likely that activation of the PI-3K enzyme is a proximal trigger as has been shown for other unrelated ligands and receptors involved in cytoprotection and proliferation. This enzyme activation promotes the posphorylation of Akt and RAS. Akt or PKB is committed in cell survival, metabolim and cell growth. Nevertheless, Akt is a well-reputed anit-apoptotic protein by enhancing Bcl2 and BCLx/L which counteract pro-apoptotic proteins (BAX, BAD). mTOR activation via Akt inducs the expression of HIF-1α which is a prosurvival protein during hypoxia. HIF may be alternatively stimulated by RAF, whih is downstream to RAS in the proliferative cascade via ERK.
Additionally and not less relevant, GHRP-6 appears an excellent partner to combine with other molecules (i.e., EGF) because their exclusive actions seem to achieve a kind of synergism, useful to target the multiples nodes of complex pathophysiological processes, and thus to enhance tissue repair processes [55]. In this respect, Garcia del Barco and co-workers in our group have opened unprecedented avenues, by combining GHRP-6 and EGF as a therapeutic approach to ameliorate the damages of multiple sclerosis [56], peripheral axonal pathology [57] and brain ischemia in animal models [58,59]. They have demonstrated in all these experimental substrates that the combined action of GHRP-6 and EGF is associated to a better outcome in both clinical and pathological fields.
Finally, an exciting medical opportunity could be opened for synthetic GHRP to treat the threatening cancer-associated anorexia-cachexia syndrome in advanced-stage cancer patients. Although the mechanistic bases of this syndrome are not fully understood, it represents a major impediment for the course of chemotherapy. In a rodent model of cancer bearing-chemotherapy, GHRP-2 administration increased appetite/food intake and prolonged median survival time which certainly suggest that GHRP-2 may improve the quality of life of cancer patients by correcting its nutritional and metabolic states [60]. These data may also incite to further studies in the search for a potential niche for GHRP to counteract the catabolic states of prolonged critical illness, invasive surgeries, severe burn traumas, etc.
Concluding remarks
Drug discovery is an uncertain ground in which disappointments and rewards are encountered. Most of those who have been involved in GHRP research have enjoyed clear-cut data which in most of the cases are all-in with very few outs. Exceptionally, a pharmacologically active agent appears to be endowed with such a disparate and useful properties as to make it highly “drugable”. The fact that synthetic GHRP enjoy the faculty to bind at least two different and biologically significant receptors that seem not to be redundant in nature, and that these receptors are largely represented in most organs and tissues; broadens their biological activities and increase their pharmacological potentialities. This suggests that GHRP may stimulate multiple cells and simultaneously trigger different signaling pathways. The information gathered so far in terms of the GHRP molecular cytoprotective mechanism is inconclusive and fragmentary, which has turned difficult to disclose the wizardry behind their biological effects. Nevertheless, it is reasonable that these molecules share the ability to knock life-sensitive pathways and restore critical organelles physiology at very proximal levels.
GHRP members not only enhance GH secretion and upraise the GH/IGF-1 axis bounties. They have also shown to be cardio, neuro and broadly cytoprotective candidates. Just few outspoken examples are: control cardiac electric potentials and ion pumps, myocardial trophism enhancement, cardiomyocytes survival, attenuation of inflammatory soluble messengers and distal cellular effectors, amelioration of the impact of neurohormones in the myocardium, control of peripheral vascular tone, necrosis and apoptosis prevention in a variety of epithelial and non-epithelial structures, tissue remodeling promotion by removing fibrotic material, control of sarcopenia by stimulating skeletal muscle trophism, cachexia and catabolism lessening, and general cytoprotection before chemotherapy regimens with no associated proliferative stimulation. Although GHRP exhibits a broad safety and toxicological profile, its clinical development has been erratic and occasional, although successful, which may have obstructed its positioning within cardiology and intensive medicine fields. In the meantime, novel drugs and therapeutic strategies are demanded to protect organs and tissues exposed to ischemia.
References
- Bowers CY, Momany FA, Reynolds GA, Hong A (1984) On the in vitro and in vivo activity of a new synthetic hexapeptide that acts on the pituitary to specifically release growth hormone. Endocrinology 114: 1537-1545. [Crossref]
- Corneli G, Gasco V, Prodam F, Grottoli S, Aimaretti G, et al. (2007) Growth hormone levels in the diagnosis of growth hormone deficiency in adulthood. Pituitary 10: 141-149. [Crossref]
- Muccioli G, Broglio F, Valetto MR, Ghè C, Catapano F, et al. (2000) Growth hormone-releasing peptides and the cardiovascular system. Ann Endocrinol (Paris) 61: 27-31. [Crossref]
- Szeto HH (2014) First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171: 2029-2050. [Crossref]
- Broglio F, Fubini A, Morello M, Arvat E, Aimaretti G, et al. (1999) Activity of GH/IGF-I axis in patients with dilated cardiomyopathy. Clin Endocrinol (Oxf) 50: 417-430. [Crossref]
- Bowers CY (1998) Growth hormone-releasing peptide (GHRP). Cell Mol Life Sci 54: 1316-1329. [Crossref]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, et al. (1999) Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402: 656-660. [Crossref]
- Root AW, Root MJ (2002) Clinical pharmacology of human growth hormone and its secretagogues. Curr Drug Targets Immune Endocr Metabol Disord 2: 27-52. [Crossref]
- Bhattacharya SK, Andrews K, Beveridge R, Cameron KO, Chen C, et al. (2014) Discovery of PF-5190457, a Potent, Selective, and Orally Bioavailable Ghrelin Receptor Inverse Agonist Clinical Candidate. ACS Med Chem Lett 8;5(5): 474-9. [Crossref]
- Khatib MN, Simkhada P, Gode D (2014) Cardioprotective effects of ghrelin in heart failure: from gut to heart. Heart Views 15: 74-76. [Crossref]
- Kishimoto I, Tokudome T, Hosoda H, Miyazato M, Kangawa K (2012) Ghrelin and cardiovascular diseases. J Cardiol 59: 8-13. [Crossref]
- Pietra C, Takeda Y, Tazawa-Ogata N, Minami M, Yuanfeng X, et al. (2014) Anamorelin HCl (ONO-7643), a novel ghrelin receptor agonist, for the treatment of cancer anorexia-cachexia syndrome: preclinical profile. J Cachexia Sarcopenia Muscle 5: 329-37. [Crossref]
- Broglio F, Arvat E, Gottero C, Benso A, Prodam F, et al. (2003) Natural and synthetic growth hormone secretagogues: do they have therapeutic potential? Treat Endocrinol 2: 153-163. [Crossref]
- Selman-Housein-Bernal KH, Hernández-Bernal F, Abreu-Cruz AA, Valenzuela-Silva C, Berlanga-Acosta J, et al. (2014) Seguridad clínica del péptido liberador de la hormona de crecimiento-6 (GHRP-6) en voluntarios sanos. Investigaciones médico-quirúrgicas 6: 81-91.
- Valiente-Mustelier J, Garcia del Barco D, Guillen-Nieto G, Santana H, Altruda F, et al. (2013) Cardiotropic effect of GHRP-6: in vivo characterization by echocardiography. Biotecnología Aplicada 30: 285-289.
- Marleau S, Mulumba M, Lamontagne D, Ong H (2006) Cardiac and peripheral actions of growth hormone and its releasing peptides: relevance for the treatment of cardiomyopathies. Cardiovasc Res 69: 26-35. [Crossref]
- Zhang G, Yin X, Qi Y, Pendyala L, Chen J, et al. (2010) Ghrelin and cardiovascular diseases. Curr Cardiol Rev 6: 62-70. [Crossref]
- Wellman M, Abizaid A1 (2015) Growth Hormone Secretagogue Receptor Dimers: A New Pharmacological Target(1,2,3). eNeuro 2. [Crossref]
- Demers A, McNicoll N, Febbraio M, Servant M, Marleau S, et al. (2004) Identification of the growth hormone-releasing peptide binding site in CD36: a photoaffinity cross-linking study. Biochem J 382: 417-24. [Crossref]
- Bodart V, Febbraio M, Demers A, McNicoll N, Pohankova P, et al. (2002) CD36 mediates the cardiovascular action of growth hormone-releasing peptides in the heart. Circ Res 90: 844-849. [Crossref]
- Mosa RM, Zhang Z, Shao R, Deng C, Chen J, et al. (2015) Implications of ghrelin and hexarelin in diabetes and diabetes-associated heart diseases. Endocrine 49: 307-323. [Crossref]
- Mao Y, Tokudome T, Kishimoto I (2014) The cardiovascular action of hexarelin. J Geriatr Cardiol 11: 253-258. [Crossref]
- Braunwald E1 (2011) Clinical efforts to reduce myocardial infarct size--the next step. J Cardiovasc Pharmacol Ther 16: 349-353. [Crossref]
- Braunwald E1 (2013) Cardiovascular science: opportunities for translating research into improved care. J Clin Invest 123: 6-10. [Crossref]
- Bolli R, Becker L, Gross G, Mentzer R Jr, Balshaw D, et al. (2004) Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res 95: 125-134. [Crossref]
- De GC, V, Rossoni G, Bernareggi M, Muller EE, Berti F (1997) Cardiac ischemia and impairment of vascular endothelium function in hearts from growth hormone-deficient rats: protection by hexarelin. Eur J Pharmacol 334: 201-207. [Crossref]
- Rossoni G, De Gennaro Colonna V, Bernareggi M, Polvani GL, Müller EE, et al. (1998) Protectant activity of hexarelin or growth hormone against postischemic ventricular dysfunction in hearts from aged rats. J Cardiovasc Pharmacol 32: 260-265. [Crossref]
- Locatelli V, Rossoni G, Schweiger F, Torsello A, De Gennaro Colonna V, et al. (1999) Growth hormone-independent cardioprotective effects of hexarelin in the rat. Endocrinology 140: 4024-4031. [Crossref]
- Bisi G, Podio V, Valetto MR, Broglio F, Bertuccio G, et al. (1999) Cardiac effects of hexarelin in hypopituitary adults. Eur J Pharmacol 381: 31-38. [Crossref]
- Broglio F, Benso A, Valetto MR, Gottero C, Quaranta L, et al. (2001) Growth hormone-independent cardiotropic activities of growth hormone-releasing peptides in normal subjects, in patients with growth hormone deficiency, and in patients with idiopathic or ischemic dilated cardiomyopathy. Endocrine14: 105-108. [Crossref]
- Broglio F, Guarracino F, Benso A, Gottero C, Prodam F, Granata R, et al. (2002) Effects of acute hexarelin administration on cardiac performance in patients with coronary artery disease during by-pass surgery. Eur J Pharmacol 448: 193-200. [Crossref]
- Pettersson I, Muccioli G, Granata R, Deghenghi R, Ghigo E, et al. (2002) Natural (ghrelin) and synthetic (hexarelin) GH secretagogues stimulate H9c2 cardiomyocyte cell proliferation. J Endocrinol 175: 201-209. [Crossref]
- Weekers F, Van HE, Isgaard J, Van den Berghe G (2000) Pretreatment with growth hormone-releasing peptide-2 directly protects against the diastolic dysfunction of myocardial stunning in an isolated, blood-perfused rabbit heart model. Endocrinology 141: 3993-3999. [Crossref]
- Iwase M, Kanazawa H, Kato Y, Nishizawa T, Somura F, et al. (2004) Growth hormone-releasing peptide can improve left ventricular dysfunction and attenuate dilation in dilated cardiomyopathic hamsters. Cardiovasc Res 61: 30-38. [Crossref]
- Cibrian D, Berlanga-Acosta J, Guevara L, Valiente-Mustelier J, Guillen-Nieto G, et al. (2008) Efecto citoprotector cardíaco y extracardíaco del péptido GHRP6. Biotecnología Aplicada 25: 276-278.
- Kato Y, Iwase M, Ichihara S, Kanazawa H, Hashimoto K, et al. (2010) Beneficial effects of growth hormone-releasing peptide on myocardial oxidative stress and left ventricular dysfunction in dilated cardiomyopathic hamsters. Circ J 74: 163-170. [Crossref]
- Xu X, Ding F, Pang J, Gao X, Xu RK, et al. (2012) Chronic administration of hexarelin attenuates cardiac fibrosis in the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 303: H703-711. [Crossref]
- Berlanga-Acosta J, Vázquez D, Cibrian D, Mendoza Y, Ochogavia ME, et al. (2012) Growth Hormone Releasing Peptide 6 (GHRP6) reduces liver fibrosis in CCl4 chronically intoxicated rats. Biotecnología Aplicada 29: 60-72.
- Shen YT, Lynch JJ, Hargreaves RJ, Gould RJ (2003) A growth hormone secretagogue prevents ischemic-induced mortality independently of the growth hormone pathway in dogs with chronic dilated cardiomyopathy. J Pharmacol Exp Ther 306: 815-820.
- Xu XB, Cao JM, Pang JJ, Xu RK, Ni C, et al. (2003) The positive inotropic and calcium-mobilizing effects of growth hormone-releasing peptides on rat heart. Endocrinology 144: 5050-5057. [Crossref]
- Ma Y, Zhang L, Launikonis BS, Chen C (2012) Growth hormone secretagogues preserve the electrophysiological properties of mouse cardiomyocytes isolated from in vitro ischemia/reperfusion heart. Endocrinology 153: 5480-5490. [Crossref]
- Ma Y, Zhang L, Edwards JN, Launikonis BS, Chen C (2012) Growth hormone secretagogues protect mouse cardiomyocytes from in vitro ischemia/reperfusion injury through regulation of intracellular calcium. PLoS One 7: e35265. [Crossref]
- Sun Q, Ma Y, Zhang L, Zhao YF, Zang WJ, et al. (2010) Effects of GH secretagogues on contractility and Ca2+ homeostasis of isolated adult rat ventricular myocytes. Endocrinology 151: 4446-4454. [Crossref]
- Berlanga J, Cibrian D, Guevara L, Dominguez H, Alba JS, et al. (2007) Growth-hormone-releasing peptide 6 (GHRP6) prevents oxidant cytotoxicity and reduces myocardial necrosis in a model of acute myocardial infarction. Clin Sci (Lond ) 112: 241-250. [Crossref]
- Titterington JS, Sukhanov S, Higashi Y, Vaughn C, Bowers C, Delafontaine P (2009) Growth hormone-releasing peptide-2 suppresses vascular oxidative stress in ApoE-/- mice but does not reduce atherosclerosis. Endocrinology 150: 5478-5487. [Crossref]
- Rodrigue-Way A, Demers A, Ong H, Tremblay A (2007) A growth hormone-releasing peptide promotes mitochondrial biogenesis and a fat burning-like phenotype through scavenger receptor CD36 in white adipocytes. Endocrinology 148:1009-1018. [Crossref]
- Mao Y, Tokudome T, Kishimoto I, Otani K, Hosoda H, et al. (2013) Hexarelin treatment in male ghrelin knockout mice after myocardial infarction. Endocrinology 154: 3847-3854. [Crossref]
- Yamamoto D, Ikeshita N, Matsubara T, Tasaki H, Herningtyas EH, et al. (2008) GHRP-2, a GHS-R agonist, directly acts on myocytes to attenuate the dexamethasone-induced expressions of muscle-specific ubiquitin ligases, Atrogin-1 and MuRF1. Life Sci 82: 460-466. [Crossref]
- Lim CJ, Jeon JE, Jeong SK, Yoon SJ, Kwon SD, Lim J, et al. (2015) Growth hormone-releasing peptide-biotin conjugate stimulates myocytes differentiation through insulin-like growth factor-1 and collagen type I. BMB Rep 48: 501-506. [Crossref]
- Cibrián D, Ajamieh H, Berlanga J, León OS, Alba JS, et al. (2006) Use of growth-hormone-releasing peptide-6 (GHRP-6) for the prevention of multiple organ failure. Clin Sci (Lond) 110: 563-573. [Crossref]
- Granado M, Martin AI, Lopez-Menduina M, Lopez-Calderon A, Villanua MA (2008) GH-releasing peptide-2 administration prevents liver inflammatory response in endotoxemia. Am J Physiol Endocrinol Metab 294: E131-E141. [Crossref]
- Granado M, Priego T, Martín AI, Villanúa MA, López-Calderón A (2005) Anti-inflammatory effect of the ghrelin agonist growth hormone-releasing peptide-2 (GHRP-2) in arthritic rats. Am J Physiol Endocrinol Metab 288: E486-492. [Crossref]
- Cao Y, Tang J, Yang T, Ma H, Yi D, et al. (2013) Cardioprotective effect of ghrelin in cardiopulmonary bypass involves a reduction in inflammatory response. PLoS One8: e55021. [Crossref]
- Guo S, Gao Q, Jiao Q, Hao W, Gao X, et al. (2012) Gastric mucosal damage in water immersion stress: mechanism and prevention with GHRP-6. World J Gastroenterol 18: 3145-3155. [Crossref]
- Rodriguez SS, Gonzalez NL, Garcia Del Barco HD, Santos FE, Maza AD, et al. (2014) Role of epidermal growth factor and growth hormone-releasing peptide-6 in acceleration of renal tissue repair after kanamycin overdosing in rats. Iran J Kidney Dis 8: 382-388. [Crossref]
- Del Barco DG, Montero E, Coro-Antich RM, Brown E, Suarez-Alba J, et al. (2011) Coadministration of epidermal growth factor and growth hormone releasing peptide-6 improves clinical recovery in experimental autoimmune encephalitis. Restor Neurol Neurosci. [Crossref]
- Del Barco DG, Perez-Saad H, Rodriguez V, Marin J, Falcon V, et al. (2011) Therapeutic effect of the combined use of growth hormone releasing peptide-6 and epidermal growth factor in an axonopathy model. Neurotox Res 19: 195-209. [Crossref]
- Subiros N, Perez-Saad HM, Berlanga JA, Aldana L, Garcia-Illera G, Gibson CL, et al. (2015) Assessment of dose-effect and therapeutic time window in preclinical studies of rhEGF and GHRP-6 coadministration for stroke therapy. Neurol Res 1743132815Y0000000089. [Crossref]
- Garcia DB-H, Martinez NS, Coro-Antich RM, Machado JM, Alba JS, et al. (2013) Epidermal growth factor and growth hormone-releasing peptide-6: combined therapeutic approach in experimental stroke. Restor Neurol Neurosci 31: 213-223. [Crossref]
- Perboni S, Bowers C, Kojima S, Asakawa A, Inui A (2008) Growth hormone releasing peptide 2 reverses anorexia associated with chemotherapy with 5-fluoruracil in colon cancer cell-bearing mice. World J Gastroenterol 14: 6303-6305. [Crossref]