Abstract
Obesity, which is largely due to energy imbalance, has emerged as one of the most serious health issues in the world. The hypothalamus is the most important organ to regulate feeding behavior and energy expenditure through nutrient sensing and signal integration from central and peripheral pathways. As the main organelle to produce energy, mitochondria play a critical role in energy homeostasis from the organelle level. Besides providing a platform for the oxidation of fuel substrates, mitochondria are also involved in a variety of cell signaling pathways and modulate energy homeostasis through mitochondrial dynamics. Mitochondrial dysfunction may lead to obesity due to inadequate ATP production, oxidative stress, endoplasmic reticulum stress, and inflammation. β, β-carotene-9’,10’-oxygenase2 (BCO2) is a mitochondrial enzyme that catalyzes the asymmetric cleavage of both provitamin A and non-provitamin A carotenoids. This enzyme is localized to the inner mitochondrial membrane, where the electron transport chain is located. Besides the enzymatic function, BCO2 is important for mitochondrial function and is genetically associated with interleukin-18. Moreover, BCO2 protein expression is suppressed in obese and diabetic mice. Given that the important role of BCO2 in mitochondrial structure and function, and the key position of the hypothalamus in energy balance, BCO2 may play a new role in maintaining metabolic homeostasis that has been overlooked before. The mutation of BCO2 might lead to the impairment of whole body energy homeostasis through hypothalamic mitochondrial dysfunction. Here we will be presenting the updates on hypothalamic mitochondria in cellular energy homeostasis and discussing the potential of BCO2 in regulation of hypothalamic mitochondria in health and obesity.
Key words
β, β-carotene-9’,10’-oxygenase2 (BCO2), carotenoid, energy homeostasis, hypothalamus, mitochondria, obesity
Abbreviations
4-PBA: 4-phenylbutyrate; ACS: acetyl-CoA synthetase; AgRP: agouti-related protein; AKT: protein kinase B; AMP: adenosine monophosphate; AMPK: adenosine monophosphate- activated protein kinase; ARC: arcuate nucleus; BAT: brown adipose tissue; BCO2: β, β-carotene-9’,10’-oxygenase2; BMP7: bone morphogenetic protein 7; CART: cocaine- and amphetamine-regulated transcript; CNS: central nervous system; DMH: dorsomedial hypothalamic nucleus; ER: endoplasmic reticulum; ETC: electron transport chain; FFA: free fatty acid; GLP-1: glucagon-like peptide-1; IKK: IκB kinaseβ; IKKβ: inhibitor of nuclear factor kappa-B kinase; InsR: insulin receptor; IRS: insulin receptor substance; JAK: Janus kinase; JNK: c-Jun N-terminal kinase; KATP: ATP-sensitive potassium; LCFA: long-chain fatty acid; LepR: leptin receptor; LHA: lateral hypothalamic area; MAMs: mitochondrial-associated membranes; MAPK: mitogen-activated protein kinase; MC4R: melanocortin 4 receptors; Mfn: mitofusins; mTOR: mammalian target of rapamycin; NF-κβ: nuclear factor kappa–B; NMDA: N-methyl-d-aspartate; NPY: neuropeptide Y; NRFs: nuclear respiratory factors; OXM: oxyntomodulin; OXPHOS: oxidative phosphorylation; PDK1: 3-phosphoinositide-dependent protein kinase 1; PGC: peroxisome proliferator-activated receptor gamma coactivator family; PI3K: phosphoinositide 3-kinase; PIP3: phosphatidylinositol 3,4,5-trisphosphate; PKCτ: protein kinase C iota; POMC: proopiomelanocortin; PRDM16: PR Domain Containing 16; PTP1B: protein tyrosine phosphatase 1B; PVN: paraventricular nucleus; ROS: reactive oxygen species; SOCS3: suppressor of cytokine signaling 3; SOD2: superoxide dismutase; STAT: signal transducers and activators of transcription; TCA: tricarboxylic acid; TGF-β: transforming growth factor beta; TLR4: toll-like receptor 4; TNFα: tumor necrosis factor alpha; TUDCA: tauroursodeoxycholic acid; UCP2: uncoupling protein 2; VMH: ventromedial hypothalamus; α-MSH: α-melanocyte stimulating hormone.
Introduction
Obesity has emerged as one of the most serious health issues in the world, which is closely related to type-2 diabetes [1], neurodegenerative diseases [2], cardiovascular disease [3], and some forms of cancer [4,5]. Obesity is the result of energy intake exceeding energy expenditure. It is a disorder of energy balance [6] commonly caused by a combination of excessive food intake, lack of physical activity , and genetic susceptibility [7].
The hypothalamus is a key region in brain for modulating feeding behavior and energy expenditure through nutrient sensing and signal integration from central and peripheral pathways [8-10].Two populations of neurons in the arcuate nucleus (ARC) of the hypothalamus play an important role in maintaining energy balance. One population of neurons express anorexigenic proopiomelanocortin (POMC) and the other express orexigenic agouti-related protein (AgRP)/neuropeptide Y (NPY). POMC and NPY/AgRP neurons could respond to leptin and insulin and interact with gut hormones such as ghrelin, glucagon-like peptide-1 (GLP-1), peptide YY3–36, cholecystokinin, and pancreatic polypeptide to maintain energy balance [10,11].
Hypothalamic mitochondria are involved in the modulation of energy balance by mitochondrial dynamic to modulate AGRP and POMC neuronal activity [12,13]. Mitochondria could change their number and size by fusion and fission to adapt to the energy environment [12]. It was reported that mitochondria could fuse in AgRP neurons, and fission in POMC neurons during a positive energy balance to enable sustained neuronal activity and maximize the uptake of energy. The morphology of mitochondria also differs between obese and lean mice. Reduced mitochondrial length and loss of mitochondria- endoplasmic reticulum (ER) interactions were observed in diet-induced obese mice compared to lean controls [13].
The mitochondria are responsible for most of the ATP production in the cell through the electron transport chain (ETC)/oxidative phosphorylation (OXPHOS). These processes occur in the inner membrane of mitochondria. The ATP producing capacity of mitochondria is another factor that affects energy homeostasis. A reduced ATP level in the hypothalamus leads to persistent AMPK stimulation. Leptin could function through inhibition of hypothalamic AMPK signaling to increase energy expenditure and lower food intake. Therefore, sustained AMPK stimulation requires a high level of leptin to suppress the effects of AMPK on AgRP and NPY [14]. Such impairment in ATP formation would lead to the tendency of eating more than what the body needs and conserving energy by lowering energy expenditure.
BCO2 (β, β-carotene-9’,10’-oxygenase2) is an enzyme that catalyzes the asymmetric cleavage of carotenoids in the inner membrane of mitochondria [15]. Recent controversial research findings demonstrated that BCO2 may not be activated in the human macula where carotenoids preferentially accumulate [16, 17], suggesting that BCO2 may not only function as a carotenoid cleavage enzyme which warrants further investigation. For example, BCO2 is involved in inflammation by regulating the transcription of the IL-18 gene [18]. It also participates in macular degeneration in humans [16] and in the process of anemia and apoptosis of red blood cells in zebrafish [19]. Furthermore, BCO2 protein expression is reduced in obese [20] and diabetic mice [21]. However, how BCO2 impacts these processes, whether in a direct or an indirect manner, is still unknown.
The expression and activity of BCO2 have been verified in humans [22], mice [23], rats [24], cattle [25], chickens [26], and sheep [27]. BCO2 is expressed in many types of cells, such as liver hepatocytes, epithelial cells of villi and endothelial cells in the duodenum of the small intestine, and other tissues such as kidney and lungs [28-30]. Recently, BCO2 was found to be in the inner mitochondrial membrane [31], where complexes of the ETC/OXPHOS are located. In accordance with this finding, carotenoids were found to be accumulated in the mitochondria of BCO2 knockout mice, which further led to mitochondrial dysfunction, indicated by an increased manganese superoxide dismutase (SOD2) level and decreased respiratory activity [23]. Given that the key position of the hypothalamus in energy balance and the potential role of BCO2 in mitochondrial function, BCO2 may play a novel function in maintaining metabolic homeostasis in the hypothalamus.
The predominant role of the hypothalamus in mediating energy homeostasis
Obesity refers to the metabolic state in which excess fat is accumulated in peripheral tissues, such as white adipose tissue, liver, and muscle [32]. Obesity, which has negative effects on both the quality and length of life, is commonly caused by a combination of excessive food intake, lack of physical activity, and genetic susceptibility [7, 33]. The hypothalamus plays a significant role in controlling energy expenditure and food intake, which are the determinants of metabolic phenotype.
The hypothalamus is organized into anatomically discrete nuclei: paraventricular nucleus (PVN), lateral hypothalamic area (LHA), dorsomedial hypothalamic nucleus (DMH), ventromedial hypothalamus (VMH), and ARC [34]. These neuronal clusters form interconnected neuronal circuits to sense information inputs from the process of eating, ingestion, absorption, and metabolism, as well as the change of energy storage [34]. Early hypothalamic lesioning experiments showed the LHA acts as the “hunger center” and is associated with hypophagia and loss of body weight, while the VMH is the ‘satiety center’ and is linked to hyperphagia and obesity [35]. The ARC is located in the basal part of the hypothalamus and is in close contact with the median eminence. The ARC could sense fluctuations in signals from hormones, nutrients, and other molecules that are transported within the blood [8]. Thus the ARC has been postulated to play a fundamental role in sensing the global energy status of the organism [8,9] and integrating signals from central and peripheral pathways [10].
The POMC and AgRP/NPY are expressed by two key populations of neurons that have opposite effects on appetite in the ARC. POMC neurons exert an anorexigenic effect by co-expressing POMC and cocaine- and amphetamine-regulated transcript (CART) [34]. POMC is the precursor of α-melanocyte stimulating hormone (α-MSH), which could activate melanocortin 4 receptors (MC4R). Activation of MC4R would ultimately inhibit appetite and increase energy expenditure [36]. The other population of neurons co-express orexigenic NPY and AgRP. NPY exerts an orexigenic effect via different subtypes of NPY receptors on downstream neurons, while AgRP could directly block α-MSH action [36]. Both of these populations of neurons act on the PVN, and the ARC also interacts with other hypothalamic nuclei such as the LHA, VMH, and DMH [37].
Hypothalamus regulates energy homeostasis through circulating hormones
NPY/AgRP and POMC/CART neurons respond to a variety of circulating hormones to regulate energy balance. Insulin and leptin are the two most critical anorexigenic hormones that are involved in energy homeostasis [38]. The activation of leptin and insulin are mediated by their receptors in the central nervous system (CNS). Leptin receptor (LepR) and insulin receptor (InsR) are highly expressed in the AgRP and POMC neurons of the ARC [39].
Leptin is a hormone secreted from white adipose tissue and circulates through the body in proportion to body fat mass. It plays a major role in energy homeostasis by inhibiting food intake, enhancing lipolysis, decreasing lipogenesis, and elevating energy expenditure. Leptin acts via LepR in the hypothalamus (predominantly the VMN and ARC, but also the choroid plexus) to regulate energy balance and neuroendocrine function [40-42]. The POMC neurons are activated by leptin, resulting in α-MSH release from POMC axon terminals. The α-MSH could further activate MC4R to increase energy expenditure and suppress food intake [43]. At the same time, the activity of NPY/AgRP neurons is inhibited by leptin [44]. Otherwise, it would antagonize the effect of α-MSH on MC4Rs through the release of AgRP [45]. Besides the actions of leptin via the brain, energy expenditure and food intake could also be modified through the direct action of LepRs on a peripheral target, including the β cells of the pancreas, liver, muscle, and fat [46].
Insulin is secreted by pancreatic β-cell in response to increased plasma glucose levels. It promotes energy utilization and storage through the regulation of glucose uptake and the metabolism of fatty acids. In skeletal muscle, insulin promotes glucose uptake by stimulating the movement of intracellular vesicles containing GLUT4 from cytosol to the plasma membrane, which increases the transport capacity of glucose. In adipose tissue, insulin reduces free fatty acid (FFA) efflux from adipocytes, thus decreasing lipolysis and facilitating fat storage. Furthermore, insulin promotes the storage of glucose as glycogen and/or triacylglycerol in the liver and inhibits hepatic glucose production by decreasing the activity of gluconeogenic enzymes [47].
Hypothalamic leptin and insulin signaling are involved in both short- and long-term energy regulation (Figure 1 (revised from [9])). Janus kinase (JAK) is recruited and activated when leptin binds to the extracellular domain of LepR-b,. The activated JAK in turn helps to phosphorylate LepR-b [48]. During this process, the signal transducer and activator of transcription 3 (STAT3) are activated. The phosphorylated STAT3 could stimulate POMC and inhibit AgRP [49]. The activation of STAT3 is associated with the elevated expression of the suppressor of cytokine signaling 3 (SOCS3). SOCS3 will bind with the LepR-b-JAK-2 complex, thus inhibits leptin signaling [50]. Insulin signaling is initiated by insulin binding to the α subunit of InsR [51]. The conformational change of the α subunit exhibits intrinsic tyrosine kinase activity, which could activate InsR [52]. The insulin receptor substance (IRS) is activated through the phosphorylation process resulting from InsR. The leptin signaling and insulin signaling converge together through the phosphoinositide 3-kinase (PI3K)– Phosphatidylinositol 3,4,5-trisphosphate (PIP3)– 3-phosphoinositide-dependent protein kinase 1(PDK1)– forkhead box protein O1 (FoxO1) signaling pathway [9]. The PI3K, which is activated by leptin or IRS, could promote the synthesis of PIP3 [53]. As the PIP3 accumulates, PDK1 is activated, which leads to the activation of protein kinase B (PKB, also known as AKT). Within the nucleus, FoxO1 could activate AgRP and inhibit POMC expression [54]. Activation of AKT induces the phosphorylation of FoxO1 and further leads to the export of FoxO1 into the cytoplasm. This would facilitate STAT3 to bind with POMC/AgRP. AKT also functions to activate the mammalian target of rapamycin (mTOR), and the activation of mTOR involves phosphorylation and inhibition of adenosine monophosphate (AMP)-activated protein kinase (AMPK). Both mTOR and AMPK act as sensors of the nutrient status that is involved in the regulation of food intake and energy expenditure, which will be discussed below.
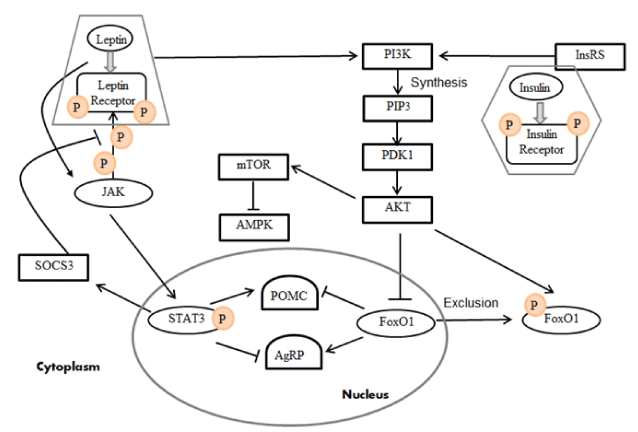
Figure 1. Leptin and insulin signaling pathways in the hypothalamus (Revised from [9]) . Leptin and insulin are secreted from white adipose tissue and the pancreas, respectively. They are activated via their receptors in the hypothalamus. The activation of both LepR‐b and InsR could further activate the PI3K–PIP3–PKD1–FoxO1 signaling pathway. Phosphorylation of LepR-b by JAK2 also leads to the recruitment and phosphorylation of STAT3. SOCS3 would desensitize leptin signaling by suppressing STAT3 activation.
Most obese patients are associated with high serum leptin levels. This situation is called leptin resistance. Similarly, insulin resistance is characterized by a decreased ability to respond to insulin. In most cases, impaired insulin sensitivity of the target organs, rather than the low insulin level in the body, eventually leads to type 2 diabetes. Activation of the IκB kinaseβ (IKK), c-Jun N-terminal kinase (JNK), and protein kinase C iota (PKCτ) signaling pathways and increased expression of SOCS3 and PTP1B are the major molecular mechanisms involved in the induction of hypothalamic leptin resistance and insulin resistance [55-59].
The hypothalamus can also interact with a variety of gut hormones. For example, peptide YY3–36, cholecystokinin, GLP-1, oxyntomodulin (OXM), and pancreatic polypeptide act synergistically to suppress appetite, whereas ghrelin acts to stimulate food intake [10,11]. By relaying signals of nutrition level and energy status from the gut to the CNS, the interaction between NPY/AgRP and POMC/CART neurons and gut hormones plays a critical role in regulating food intake.
Hypothalamus regulates energy homeostasis through sensing nutrient status
Aside from the signals from hormones, the hypothalamus is also sensitive to circulating metabolites. Two types of glucosensing neurons are either inhibited or excited as the glucose level fluctuates [60]. Glucosensing neurons regulate their membrane potential, firing rate, and ion channel function to sense ambient glucose. Glucose sensing is especially important in stimulating appetite when glucose availability drops [61-63].
Circulating lipids such as triglyceride and long-chain fatty acids (LCFAs) function similar to insulin through elevation of the LCFA-CoA level in the hypothalamus [64]. LCFA gets into the brain by passive diffusion or translocation via a carrier protein. Upon entry into the neurons, LCFA is esterified into LCFA-CoA via Acetyl-CoA synthetase (ACS). LCFA-CoA signals nutrient abundance and exhibits insulin-like effects in the brain, including the hypothalamic ARC, to modulate energy homeostasis [65]. Brain fatty acids act as a satiety signal to inhibit appetite[66]. Both the peripheral and central administration of a fatty acid synthase inhibitor significantly suppresses appetite [67,68]. An in vivo study showed food intake and hepatic glucose production were inhibited after three days of intracerebroventricular infusion of oleic acid [69]. The accumulation of LCFA-CoA within specific CNS neurons could activate catabolic neural pathways to inhibit appetite and hepatic glucose production [70]. The anorectic effect of the accumulation of LCFA-CoA may lie downstream of the leptin-induced inhibition of hypothalamic AMPK [71]. LCFA-CoA in the brain activates protein kinase C and further phosphorylates and activates ATP-sensitive potassium (KATP) channels. This lipid-sensing pathway in the hypothalamus activates N-methyl-d-aspartate (NMDA) receptor to trigger the hepatic vagus nerve to regulate glucose production. In this way, neuronal signals are transmitted via the hepatic vagus nerve to the liver.
In contrast to intracellular LCFA-CoA accumulation, which is a sensor of nutrient abundance, the AMPK is a sensor of nutrient insufficiency. At the peripheral level, AMPK activation leads to increased oxidation to maintain cellular energy availability. In the ARC, AMPK activation results in increased food intake and a trend to conserve energy [72]. When cells sense the low fuel availability indicated by an increased AMP/ATP ratio, the activation of AMPK enhances substrate oxidation to replenish the depleted ATP levels [65]. Research conducted in mice showed that within the mediobasal hypothalamus, activation of AMPK increased food intake and body weight, while inhibition of AMPK led to the opposite results [72]. Hypothalamic AMPK activity is inhibited by glucose, insulin, and leptin and stimulated by ghrelin [72,73]. The change of AMPK signaling may also alter the appetite governed by those hormones. A consequence of increased AMPK activity is the oxidation of LCFA-CoA, which might induce orexigenic effects. Furthermore, AMPK could inhibit the activity of mTOR, another nutrient sensor, which will be discussed below.
mTOR is an evolutionarily conserved serine-threonine kinase involved in the regulation of cell-cycle progression and growth via sensing energy status [74]. It was demonstrated that in peripheral tissues, mTOR participates in coupling cellular energy status and facilitated cellular anabolic processes, such as protein synthesis in response to growth factors, amino acids, glucose, and stress [10]. Later, research found that mTOR was also expressed in the hypothalamus and to play an important role in hypothalamic regulation of energy homeostasis in response to nutrient availability. Cota et al. found that administration of leucine in rats led to elevated hypothalamic mTOR signaling and decreased food intake, as well as body weight [74]. They also showed that leptin could activate hypothalamic mTOR activity and that inhibition of mTOR signaling would block the leptin-induced suppression of food intake [74]. Both mTOR and AMPK represent a potential site of convergence for both hormonal and nutrient sensing.
The role of mitochondria in maintaining cellular energy metabolism
We have discussed that the hypothalamus is the most important organ to modulate energy homeostasis in brain. When we take a closer look at the cell organelle level, the mitochondrion is the key organelle that maintains cellular energy metabolism. The mitochondrion is a double-membrane bound organelle in most eukaryotic cells [75]. It is composed of an outer membrane, intermembrane space, inner membrane, and the matrix. A variety of enzymes are involved in different metabolic processes in the mitochondrial matrix. One major role of a mitochondrion is to produce energy through the tricarboxylic acid (TCA) cycle and ETC/OXPHOS. In addition to the utilization of the carbohydrate-derived substrate, the β-oxidation of fatty acids is another resource to generate ATP that mainly takes place in the mitochondria. Besides providing a platform for the oxidation of fuel substrates, mitochondria are also involved in a variety of cell signaling pathways, such as acting as transducers and effectors in the process of cell death, innate immunity, and autophagy [76,77]. Furthermore, mitochondria also serve as calcium buffers and sources of free radicals [78,79].
Hypothalamic mitochondria and energy homeostasis
Hypothalamic mitochondrial dynamic is involved in the energy homeostasis through the regulation of AgRP and POMC neuronal activity [12,13]. Mitochondrial dynamics refer to the dynamic fission and fusion behavior and morphology change of mitochondria [80] in response to the environment, especially to the energy status. Mitofusins (Mfn) namely, Mfn1 and Mfn2, work coordinately to regulate mitochondrial fusion. The depletion of Mfn1 and Mfn2 in AgRP neurons of mice led to decreased activity of the AgRP neuron, which was accompanied by weight loss. [12]. When mice were deprived of food, an increased number and decreased size of mitochondria were observed in AgRP neurons [12]. When fasted mice were exposed to excess food, the number of mitochondria decreased but their size increased in the AgRP neurons. However, the opposite fission-like dynamic phenomenon was observed in the POMC neurons, which exhibited reduced mitochondrial density and coverage [12]. Similarly, mitochondria in the AgRP neurons were fused when mice were fed a high-fat diet. These findings indicate that mitochondria could fuse in AgRP neurons and fissure in POMC neurons during a positive energy balance to enable sustained neuronal activity and maximize the uptake of energy.
The morphology of mitochondria also differs between obese and lean mice. It has been reported that mitochondrial networks and mitochondria- endoplasmic reticulum (ER) contacts in POMC are important for energy homeostasis [13]. Mitochondria form tubular structures or networks in the cell. The branched structure of the mitochondrial network is constituted by separate mitochondria [81]. Decreased mitochondrial length and branching was observed in diet-induced obese mice compared to lean controls. Diet-induced mice also showed a significant reduction of mitochondria-ER contacts in POMC neurons.
Mitochondria and adipocyte transcription factors
Some adipocyte transcription factors play a critical role in modulating mitochondrial function and biogenesis [82]. The peroxisome proliferator-activated receptor gamma coactivator (PGC) family is a group of transcriptional coactivators. A reduced PGC-1α level was observed in the adipose tissues of obese patients [83] and in genetically or diet-induced obese mice [84]. PGC-1α and PGC-1β not only participate in the regulation of glucose metabolism, fatty acid metabolism, and lipid accumulation, but also function to affect the expression of the mitochondrial biogenesis gene and are involved in the development of obesity [85]. PGC-1 stimulates mitochondrial biogenesis and respiration in myotubes of muscle via an induction of uncoupling protein 2 (UCP2), and the regulation of the nuclear respiratory factors (NRFs). Deficiency in either PGC-1α or PGC-1β in preadipocytes of brown adipose tissue (BAT) led to impaired mitochondrial gene expression, density, and respiratory activity [86] .
PR Domain Containing 16 (PRDM16) is the coactivator of PGC-1α and PGC-1β, which is important for mitochondrial biogenesis and uncoupled cellular respiration [87,88]. Decreased PRDM16 expression in BAT leads to the inhibition of mitochondrial gene expression and stimulation of myogenic markers [88, 89]. Another important transcriptional coactivator is bone morphogenetic protein 7 (BMP7), a subgroup of the transforming growth factor beta (TGF-β) superfamily, which increases mitochondrial density and the expression of mitochondrial biogenesis genes through activation of p38 mitogen-activated protein kinases (MAPK) and PGC-1a [90].
Mitochondrial dysfunction in the development of obesity
A renaissance in mitochondrial research has occurred during the past decade. Besides its function in energy production, mitochondria participate in many processes including cell signaling, cell differentiation, and apoptosis [81]. Mitochondria are also involved in the generation of reactive oxygen species (ROS), ER stress, and inflammation. Mitochondrial dysfunction is thought to trigger many chronic diseases, including neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease, liver and kidney disorders, diabetes, and the aging process [91]. By integrating such metabolic information including ATP level, oxidative stress, ER stress, inflammation, and cell signaling, mitochondria play an important role during the development of obesity.
Inadequate ATP production and obesity
A possible source of leptin resistance is impaired mitochondrial ATP production. Research has shown that altered mitochondrial energy production, especially in skeletal muscles, is a major factor to disrupt a chain of metabolic events leading to obesity [14]. In the hypothalamus, the reduced energy producing capacity of mitochondria led to persistently high hypothalamic AMPK stimulation. Leptin could function through inhibition of hypothalamic AMPK signaling to increase energy expenditure and lower food intake. Therefore, sustained AMPK stimulation requires a high level of leptin to suppress the regulation of AMPK on AgRP and NPY [14]. Such impairment in ATP formation would lead to the tendency of eating more than what the body needs and conserving more energy by lowering energy expenditure.
Oxidative stress and obesity
Energy is produced from the TCA cycle, ETC/OXPHOS, and β-oxidation of fatty acids in the mitochondria. Approximately 90% of oxygen in the cell is consumed by the mitochondria [92]. The mitochondrial respiratory chain is one of the major sources of ROS. While mitochondria generate ATP through ETC/OXPHOS, ROS is generated as a byproduct [93]. The electrons are transported from NADH and FADH2 to complex III, and then to complex IV. Eventually, electrons are passed on to oxygen, yielding H2O. However, some electrons might escape from ETC. Once these electrons leak into the mitochondrial matrix, the incomplete electron transfer will lead to the formation of superoxide (O2−⋅). It has been reported that complex I is a major donor of electrons to generate ROS in the brain [94]. Even under normal physiological conditions, approximately 1–5% of the O2 consumed by mitochondria is converted to ROS [81]. ROS refer to a variety of oxygen free radicals, such as superoxide anion radical (O2−⋅), hydroxyl radical (·OH), and non-radical oxidants, such as hydrogen peroxide (H2O2) and singlet oxygen (1O2) [95]. Most of the intracellular ROS are derived from superoxides.
ROS are important for various cell functions and act as a mediator of intracellular signaling cascades. However, excessive production of ROS may cause damage to cells and ultimately lead to the apoptosis or necrosis of the cells [96]. Organisms need to maintain equilibrium between the ROS produced during the energy generating process and the damaging effects of ROS, which is known as oxidative stress [81]. Oxidative stress is usually associated with the etiology of obesity, type 2 diabetes, cancer, and many other chronic diseases [97, 98]. In the cells, there are various anti-oxidative enzymes, such as superoxide dismutase, copper/zinc superoxide dismutase, glutathione peroxidase, and catalase, to deal with the continuous production of ROS [99].
The excess lipid accumulation in obese people represents an excess of energy, but obese individuals fatigue easily and show lower physical endurance, reflecting an energy deficiency [14]. Continuous mitochondrial overload of fuel and incomplete fatty acid oxidation could lead to increased ROS production [100,101]. Oxidative stress also impairs mitochondrial function, resulting in elevated ROS production and mitochondrial dysfunction. This mitochondrial dysfunction may in turn induce insulin resistance, resulting in a vicious cycle that leads to obesity-related pathogenesis. The elevated ROS level due to mitochondrial dysfunction stimulates various downstream pathways including JNK/signal transducers and activators of transcription (STAT) pathway, JNK/ inhibitor of nuclear factor kappa-B kinase (IKKβ), and MAPK, which can disturb leptin and insulin pathways [52,102-104]. A decreased number of mitochondria may also lead to insulin resistance [52]. Petersen and colleagues demonstrated that insulin resistance is caused by dysregulation of cellular fatty acid metabolism, which is a result of an inherited defect in mitochondrial oxidative phosphorylation [105].
ER stress and obesity
ER, an important organelle that forms an interconnected network, is responsible for the synthesis, folding, and maturation of secreted and transmembrane proteins; biosynthesis of lipids; and the storage of Ca2+ [106, 107]. ER stress results from an imbalance between the protein folding capacity and the protein load [55]. The direct result of ER stress is the accumulation of misfolded protein. The ER establishes direct contact with mitochondria through ER domains termed mitochondrial-associated membranes (MAMs). MAMs allow bidirectional communication and trafficking of a signaling molecule [108]. Such association is essential for both mitochondria and ER functions. The interaction between the ER and mitochondria is partially modulated by mitochondrial fusion and fission [108]. The dysfunction of mitochondria is one trigger to ER stress through decreased ATP production and the alteration of the mitochondrial membrane potential and permeability. Both diet-induced and genetic obesity in mice are associated with increased ER stress in the hypothalamus [56,109,110]. Hypothalamic ER stress has emerged as a causative factor in the development of obesity by inducing leptin resistance and insulin resistance.
Enhanced hypothalamic ER stress may play a primary pathogenic role in the development of leptin resistance. Schneeberger et al. put forward the hypothesis that MFN2 in the POMC is the molecular link between hypothalamic ER stress and leptin resistance [13]. To explore whether inhibition of ER stress could reverse the sensitivity of leptin, different approaches aimed at alleviating hypothalamic ER stress were conducted. Diet induced obese mice were treated with chaperones 4-phenylbutyrate (4-PBA) or tauroursodeoxycholic acid (TUDCA). The results showed normalization of the expression of ER stress, accompanied by enhanced leptin sensitivity and reduced food intake and body weight [56,109-112]. Furthermore, ER stress inducing reagents could inhibit leptin signaling by blocking the phosphorylation of STAT3 [113]. Research also found that ER stress leads to leptin resistance via the mediation of protein tyrosine phosphatase 1B (PTP1B) [112].
Besides leptin resistance, ER stress may induce insulin resistance as well. The activation of JNK by ER stress could lead to increased serine phosphorylation of IRS. Phosphorylation of IRS at particular serine residues will disturb the interaction of IRS with InsR [114]. In addition, ER stress in pancreatic β-cells leads to β-cell apoptosis [115]. ER stress could also induce leptin/insulin resistance through elevated inflammation in the hypothalamus [55]. The mechanism of inflammation during the development of obesity will be discussed in the following section.
Hypothalamic inflammation and obesity
Continuous excessive nutrient exposure of the mitochondria could activate inflammatory pathways both in peripheral metabolic organs and in the CNS. Hypothalamic inflammation is characterized by increased interleukin and cytokine levels. For example, increased expression of the proinflammatory cytokines including IL-1, IL-6, and tumor necrosis factor alpha (TNFα), as well as the elevated expression of IKKβ, an important upstream kinase regulator of nuclear factor kappa–B (NF-κβ), were observed in the hypothalamus of obese rats [116,117].
Both lipid infusion and high-fat diet feeding could lead to obesity by activating hypothalamic inflammatory signaling pathways, which results in enhanced food intake and increased nutrient storage. Saturated fatty acids activate neuronal JNK and NF-κB signaling pathways, which directly impact leptin and insulin signaling [56]. The neuronal pro-inflammatory signaling could disturb intracellular signal transduction downstream of insulin receptors through the IRS phosphatidylinositol 3-kinase pathway [118]. Hypothalamic inflammation impairs leptin signaling via the JAK/STAT signaling pathway [119,120] and induces obesity by up-regulating molecules and pathways including an inhibitor of IKKβ, serine kinases, JNK, toll-like receptor 4 (TLR4), the ceramide biosynthesis pathway, and/or the ER stress pathway [121,122]. The induction of hypothalamic inflammation is associated with the activation of toll-like receptor (TLR) signaling [121]. Enhanced TLR expression activates JNK and IKK signaling and stimulates expression of inflammatory genes, such as TNFα and IL1β, which may be the main contributors to leptin resistance and insulin resistance. When TLR4 signaling was blocked, leptin resistance and insulin resistance were reversed [121].
Carotenoids, BCO2, and mitochondrial function
Carotenoids are a group of fat-soluble pigments derived from a 40-carbon basal structure. They contain multiple conjugated double bonds and carry cyclic end-groups that can be substituted with oxygen-containing groups. Based on chemical structure, carotenoids can be categorized into two classes: (1) carotenes, which contain only carbon and hydrogen atoms, and (2) oxocarotenoids (xanthophylls), which carry at least one oxygen atom [123].
Carotenoids are crucial for brain and visual function. Lutein and zeaxanthin are dominant carotenoids in human brain tissue, which account for 66–77% of total carotenoid concentration in the brain [124,125]. They are believed to prevent damage that leads to age-related macular degeneration by quenching free radicals and absorbing blue light [126,127]. Significant correlation between macular pigment density and global cognitive function was found in older adults [128,129]. It was reported that macular lutein and zeaxanthin were significantly correlated with their levels in brain tissue [130]. Therefore, macular pigment can also be used as the biomarker of lutein in brain tissue [131]. The brain is vulnerable to free radical attack because of the high metabolic activity, and high polyunsaturated fatty acid content. Lutein and zeaxanthin are the predominant carotenoids that are selectively taken up by human brain tissue throughout the lifetime that play an important neuro-protective role in brain function [132]. The most well-known benefit of carotenoids in brain function is improvement of cognitive function [131]. These carotenoids have also been suggested to modulate functional properties of synaptic membranes and to be involved in certain changes in the physiochemical and structural features of the membranes [133]. Most of these functions are attributed to their anti-oxidative and anti-inflammatory properties [131].
Considering the critical roles of carotenoids in the brain function, a well-regulated metabolism of carotenoids is necessary for brain, as well as the whole body health. BCO2 is mitochondrial enzyme involves in the cleavage of carotenoids. Compared to β-carotene-15, 15’-monooxygenase (BCO1), BCO2 exhibits broader substrate specificity. It catalyzes asymmetric cleavage of both provitamin A and non-provitamin A carotenoids at the 9’, 10’ double bond to form β-ionone and β-apo-10’-carotenal [15]. The metabolites of carotenoids also exhibit important biological function. The activation of the NF-κB transcription system is associated with harmful effect to bone health, and contributes to cancer development. The 10,10′-diapocarotene-10,10′-dial, which is the cleave products of lycopene by BCO2, was reported to inhibit NF-κB transcription system and to reduce the expression of NF-κB target genes [134]. Such process was mediated by the directly interaction of carotenoid derivatives with key kinases IKKβ and the p65 subunit. The fact that only the carotenoid derivatives, rather than the intact carotenoid exhibited inhibitory effect on NF-κB highlights the critical role of BCO2 among the health benefits of carotenoids.
2021 Copyright OAT. All rights reserv
Previously, most research related to BCO2 was focused on the enzymatic function of BCO2. For example, Amengual et al. reported the deletion of BCO2 led to the disrupted carotenoid homeostasis accompanied by the elevated oxidative and reduced respiratory activity when mice were fed with carotenoids diet [23]. However, our lab’s recent studies found deletion of BCO2 gene in mice leads to increased appetite and higher risk to develop obesity, even when mice were fed with chow diet or high fat diet without containing carotenoids. This observation brings the possibility that BCO2 may play a more complicated role in maintaining mitochondrial function to modulate whole body energy homeostasis, rather than just exhibiting the enzymatic function. The proteomics and metabolomics analysis from our lab further revealed the perturbed energy metabolism, mitochondrial dysfunction, and elevated oxidative stress in hypothalamus caused by deletion of BCO2.
Conclusion and perspectives
The hypothalamus and mitochondria are two important centers for regulation of energy homeostasis at the organ and organelle levels, respectively. As the key organelle that maintains cellular energy metabolism, hypothalamic mitochondria participate in a variety of cell signaling pathways and modulate energy homeostasis through mitochondrial dynamics. Mitochondria is involved in the development of obesity via inadequate ATP production, enhanced inflammation, elevated oxidative stress, and ER stress. Therefore, the mitochondrial integrity is critical for energy homeostasis of the whole body and hypothalamus. Carotenoids in brain have been recently characterized. However the functions of carotenoids and the metabolites in hypothalamus are poorly understood.
Considering the potential role of BCO2 in mitochondrial structure and function and the key position of the hypothalamus in energy balance, BCO2 may play a novel function in maintaining metabolic homeostasis in hypothalamus through regulation of mitochondrial dynamics. However, the exact mechanism by which BCO2 mediates mitochondrial dynamics and hypothalamic function in regulation of energy balance remains unknown. Elucidating how BCO2 and the carotenoid metabolites protect mitochondrial activity and subsequently contribute to maintaining hypothalamic function will provide new perspectives for the prevention and treatment of obesity.
None
References
- Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444: 840-846. [Crossref]
- Sriram K, Benkovic SA, Miller DB, O'Callaghan JP (2002) Obesity exacerbates chemically induced neurodegeneration. Neuroscience 115: 1335-1346. [Crossref]
- Burke GL, Bertoni, AG, Shea, S, Tracy, R, Watson, KE, Blumenthal, RS, Chung, H, Carnethon, MR (2008) The impact of obesity on cardiovascular disease risk factors and subclinical vascular disease: the Multi-Ethnic Study of Atherosclerosis. Arch Intern Med 168: 928-935. [Crossref]
- Hursting SD, Nunez NP, Varticovski L, Vinson C (2007) The obesity-cancer link: lessons learned from a fatless mouse. Cancer Res 67: 2391-2393. [Crossref]
- Vucenik I, Stains JP (2012) Obesity and cancer risk: evidence, mechanisms, and recommendations. Ann N Y Acad Sci 1271: 37-43. [Crossref]
- Grundy SM, Barnett JP (1990) Metabolic and health complications of obesity. Dis Mon 36: 641-731. [Crossref]
- Weinsier RL, Hunter GR, Heini AF, Goran MI, Sell SM (1998) The etiology of obesity: relative contribution of metabolic factors, diet, and physical activity. Am J Med 105: 145-150. [Crossref]
- Cone R, Cowley M, Butler A, Fan W, Marks D, et al. (2001) The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int J Obes Relat Metab Disord 25: S63-67. [Crossref]
- Varela L, Horvath TL (2012) Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep 13: 1079-1086. [Crossref]
- Lenard NR, Berthoud HR (2008) Central and peripheral regulation of food intake and physical activity: pathways and genes. Obesity (Silver Spring) 16 Suppl 3: S11-22. [Crossref]
- Stanley S, Wynne K, McGowan B, Bloom S (2005) Hormonal regulation of food intake. Physiol Rev 85: 1131-1158. [Crossref]
- Dietrich MO, Liu ZW, Horvath TL (2013) Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell 155: 188-199. [Crossref]
- Schneeberger M, Dietrich MO, Sebastián D, Imbernón M, Castaño C, et al. (2013) Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155: 172-187. [Crossref]
- Rogge MM1 (2009) The role of impaired mitochondrial lipid oxidation in obesity. Biol Res Nurs 10: 356-373. [Crossref]
- Lietz G, Oxley A, Boesch-Saadatmandi C, Kobayashi D (2012) Importance of β,β-carotene 15,15'-monooxygenase 1 (BCMO1) and β,β-carotene 9',10'-dioxygenase 2 (BCDO2) in nutrition and health. Mol Nutr Food Res 56: 241-250. [Crossref]
- Li B, Vachali PP, Gorusupudi A, Shen Z, Sharifzadeh H, et al. (2014) Inactivity of human ß, ß-carotene-9', 10'-dioxygenase (BCO2) underlies retinal accumulation of the human macular carotenoid pigment. Proc Natl Acad Sci U S A 111: 10173-10178. [Crossref]
- Babino D, Palczewski G, Widjaja-Adhi MA, Kiser PD2, Golczak M, et al. (2015) Characterization of the Role of β-Carotene 9,10-Dioxygenase in Macular Pigment Metabolism.. J Biol Chem 290: 24844-24857. [Crossref]
- He M, Cornelis MC, Kraft P, van Dam RM, Sun Q, et al. (2010) Genome-wide association study identifies variants at the IL18-BCO2 locus associated with interleukin-18 levels. Arterioscler Thromb Vasc Biol 30: 885-890. [Crossref]
- Lobo GP, Isken A, Hoff S, Babino D, von Lintig J (2012) BCDO2 acts as a carotenoid scavenger and gatekeeper for the mitochondrial apoptotic pathway. Development 139: 2966-2977. [Crossref]
- Lin D, He H, Ji H, Willis J, Willard L, et al. (2014) Wolfberries potentiate mitophagy and enhance mitochondrial biogenesis leading to prevention of hepatic steatosis in obese mice: The role of AMP-activated protein kinase a2 subunit. Mol Nutr Food Res 58: 1005-1015. [Crossref]
- Yu H, Wark L, Ji H, Willard L, Jaing Y, et al. (2013) Dietary wolfberry upregulates carotenoid metabolic genes and enhances mitochondrial biogenesis in the retina of db/db diabetic mice. Mol Nutr Food Res 57: 1158-1169. [Crossref]
- Lindqvist A, Andersson S (2004) Cell type-specific expression of beta-carotene 15,15'-mono-oxygenase in human tissues. J Histochem Cytochem 52: 491-499. [Crossref]
- Amengual J, Lobo GP, Golczak M, Li HN, Klimova T, et al. (2011) A mitochondrial enzyme degrades carotenoids and protects against oxidative stress. FASEB J 25: 948-959. [Crossref]
- Raghuvanshi S, Reed V, Blaner WS2, Harrison EH3 (2015) Cellular localization of β-carotene 15,15' oxygenase-1 (BCO1) and β-carotene 9',10' oxygenase-2 (BCO2) in rat liver and intestine. Arch Biochem Biophys 572: 19-27. [Crossref]
- Berry SD, Davis SR, Beattie EM, Thomas NL, Burrett AK, et al. (2009) Mutation in bovine beta-carotene oxygenase 2 affects milk color. Genetics 182: 923-926. [Crossref]
- Eriksson J, Larson G, Gunnarsson U, Bed'hom B, Tixier-Boichard M, et al. (2008) Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. PLoS Genet 4: e1000010. [Crossref]
- von Lintig J1 (2010) Colors with functions: elucidating the biochemical and molecular basis of carotenoid metabolism. Annu Rev Nutr 30: 35-56. [Crossref]
- Kiefer C, Hessel S, Lampert JM, Vogt K, Lederer MO, et al. (2001) Identification and characterization of a mammalian enzyme catalyzing the asymmetric oxidative cleavage of provitamin A. J Biol Chem 276: 14110-14116. [Crossref]
- Paik J, During A, Harrison EH, Mendelsohn CL, Lai K, et al. (2001) Expression and characterization of a murine enzyme able to cleave beta-carotene. The formation of retinoids. J Biol Chem 276: 32160-32168. [Crossref]
- Raghuvanshi S, Reed V, Blaner WS, Harrison EH (2015) Cellular localization of β-carotene 15,15' oxygenase-1 (BCO1) and β-carotene 9',10' oxygenase-2 (BCO2) in rat liver and intestine.. Arch Biochem Biophys 572: 19-27. [Crossref]
- Palczewski G, Amengual J, Hoppel CL, von Lintig J (2014) Evidence for compartmentalization of mammalian carotenoid metabolism. FASEB J 28: 4457-4469. [Crossref]
- Horvath TL (2005) The hardship of obesity: a soft-wired hypothalamus. Nat Neurosci 8: 561-565. [Crossref]
- Lau DC, Douketis JD, Morrison KM, Hramiak IM, Sharma AM, et al. (2007) 2006 Canadian clinical practice guidelines on the management and prevention of obesity in adults and children [summary]. CMAJ 176: S1-S13. [Crossref]
- López M, Alvarez CV, Nogueiras R, Diéguez C (2013) Energy balance regulation by thyroid hormones at central level. Trends Mol Med 19: 418-427. [Crossref]
- Mayer J, Thomas DW (1967) Regulation of food intake and obesity. Science 156: 328-337. [Crossref]
- Belgardt BF, Okamura T, Brüning JC (2009) Hormone and glucose signalling in POMC and AgRP neurons. J Physiol 587: 5305-5314. [Crossref]
- Suzuki K, Simpson KA, Minnion JS, Shillito JC, Bloom SR (2010) The role of gut hormones and the hypothalamus in appetite regulation. Endocr J 57: 359-372. [Crossref]
- Belgardt BF, Brüning JC (2010) CNS leptin and insulin action in the control of energy homeostasis. Ann N Y Acad Sci 1212: 97-113. [Crossref]
- Lin HV, Plum L, Ono H, Gutiérrez-Juárez R, Shanabrough M, et al. (2010) Divergent regulation of energy expenditure and hepatic glucose production by insulin receptor in agouti-related protein and POMC neurons. Diabetes 59: 337-346. [Crossref]
- Leibowitz SF, Alexander JT (1998) Hypothalamic serotonin in control of eating behavior, meal size, and body weight. Biol Psychiatry 44: 851-864. [Crossref]
- Hâkansson ML, Brown H, Ghilardi N, Skoda RC, Meister B (1998) Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci 18: 559-572. [Crossref]
- Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, et al. (1996) Coexpression of leptin receptor and preproneuropeptide Y mRNA in arcuate nucleus of mouse hypothalamus. J Neuroendocrinol 8: 733-735. [Crossref]
- Diano S (2013) Role of Reactive Oxygen Species in Hypothalamic Regulation of Energy Metabolism. Endocrinol Metab 28: 3. [Crossref]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, et al. (2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411: 480-484. [Crossref]
- Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, et al. (1997) Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 278: 135-138. [Crossref]
- Kahn BB, Flier, JS (2000) Obesity and insulin resistance. J Clin Invest 106: 473-481. [Crossref]
- Sonksen P, Sonksen, J (2000) Insulin: understanding its action in health and disease. Br J Anaesth 85: 69-79. [Crossref]
- Vaisse C, Halaas, JL, Horvath, CM, Darnell, JE, Stoffel, M, Friedman, JM (1996) Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet 14: 95-97. [Crossref]
- Ernst MB, Wunderlich CM, Hess S, Paehler M, Mesaros A, et al. (2009) Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J Neurosci 29: 11582-11593. [Crossref]
- Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, et al. (2000) SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem 275: 40649-40657. [Crossref]
- Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414: 799-806. [Crossref]
- Kim JA, Wei Y, Sowers JR (2008) Role of mitochondrial dysfunction in insulin resistance. Circ Res 102: 401-414. [Crossref]
- Belgardt BF, Husch A, Rother E, Ernst MB, Wunderlich FT, et al. (2008) PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and-independent pathways in control of energy homeostasis and stress response. Cell Metab 7: 291-301. [Crossref]
- Kitamura T, Feng Y, Kitamura YI, Chua SC Jr, Xu AW, et al. (2006) Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med 12: 534-540. [Crossref]
- Cnop M, Foufelle F, Velloso LA (2012) Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med 18: 59-68. [Crossref]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, et al. (2008) Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135: 61-73. [Crossref]
- Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, et al. (2009) Palmitic acid mediates hypothalamic insulin resistance by altering PKC-? subcellular localization in rodents. . J Clin Invest 119: 2577. [Crossref]
- Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjørbaek C, et al. (2004) Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med 10: 734-738. [Crossref]
- Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, et al. (2002) PTP1B regulates leptin signal transduction in vivo. Dev Cell 2: 489-495. [Crossref]
- Levin BE, Kang L, Sanders NM, Dunn-Meynell AA (2006) Role of neuronal glucosensing in the regulation of energy homeostasis. Diabetes 55: S122-S130.
- Blouet C, Schwartz GJ (2010) Hypothalamic nutrient sensing in the control of energy homeostasis. Behav Brain Res 209: 1-12. [Crossref]
- Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI (1995) Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 44: 180-184. [Crossref]
- Levin BE, Magnan C, Dunn-Meynell A, Le Foll C (2011) Metabolic sensing and the brain: who, what, where, and how? Endocrinology 152: 2552-2557. [Crossref]
- He W, Lam TK, Obici S, Rossetti L (2006) Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat Neurosci 9: 227-233. [Crossref]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW (2006) Central nervous system control of food intake and body weight. Nature 443: 289-295. [Crossref]
- Obici S, Feng Z, Arduini A, Conti R, Rossetti L (2003) Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med 9: 756-761. [Crossref]
- Proulx K, Cota D, Woods SC, Seeley RJ (2008) Fatty acid synthase inhibitors modulate energy balance via mammalian target of rapamycin complex 1 signaling in the central nervous system. Diabetes 57: 3231-3238. [Crossref]
- Clegg DJ, Wortman MD, Benoit SC, McOsker CC, Seeley RJ (2002) Comparison of central and peripheral administration of C75 on food intake, body weight, and conditioned taste aversion. Diabetes 51: 3196-3201. [Crossref]
- Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L (2002) Central administration of oleic acid inhibits glucose production and food intake. Diabetes 51: 271-275. [Crossref]
- Sandoval D, Cota D, Seeley RJ (2008) The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regulation. Annu Rev Physiol 70: 513-535. [Crossref]
- Lam TK, Schwartz GJ, Rossetti L (2005) Hypothalamic sensing of fatty acids. Nat Neurosci 8: 579-584. [Crossref]
- Minokoshi Y, Alquier T, Furukawa N, Kim Y-B, Lee A, et al. (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428: 569-574. [Crossref]
- Ruderman NB, Saha AK, Vavvas D, Witters LA (1999) Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol 276: E1-E18. [Crossref]
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, et al. (2006) Hypothalamic mTOR signaling regulates food intake. Science 312: 927-930. [Crossref]
- Henze K, Martin W (2003) Evolutionary biology: essence of mitochondria. Nature 426: 127-128. [Crossref]
- Goldenthal MJ, Marín-García J (2004) Mitochondrial signaling pathways: a receiver/integrator organelle. Mol Cell Biochem 262: 1-16. [Crossref]
- Tait SW, Green DR (2012) Mitochondria and cell signalling. J Cell Sci 125: 807-815. [Crossref]
- Chakraborti T, Das S, Mondal M, Roychoudhury S, Chakraborti S (1999) Oxidant, mitochondria and calcium: an overview. Cell Signal 11: 77-85. [Crossref]
- Papa S, Skulachev VP (1997) Reactive oxygen species, mitochondria, apoptosis and aging. Mol Cell Biochem 174: 305-319. [Crossref]
- Scott I, Logan DC (2011) Mitochondrial dynamics. In Plant mitochondria. (Springer): pp 31-63.
- Bullon P, Newman HN, Battino M (2014) Obesity, diabetes mellitus, atherosclerosis and chronic periodontitis: a shared pathology via oxidative stress and mitochondrial dysfunction? Periodontol 2000 64: 139-153. [Crossref]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, et al. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115-124. [Crossref]
- Semple R, Crowley V, Sewter C, Laudes M, Christodoulides C, et al. (2004) Expression of the thermogenic nuclear hormone receptor coactivator PGC-1a is reduced in the adipose tissue of morbidly obese subjects. Int J Obes Relat Metab Disord 28: 176-179. [Crossref]
- Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, et al. (2007) Peroxisome Proliferator Activator Receptor ? Coactivator-1 Expression Is Reduced in Obesity potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem 282: 15439-15450. [Crossref]
- Bournat JC, Brown CW (2010) Mitochondrial dysfunction in obesity. Curr Opin Endocrinol Diabetes Obes17: 446-452. [Crossref]
- Uldry M, Yang W, St-Pierre J, Lin J, Seale P, et al. (2006) Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell metab 3: 333-341. [Crossref]
- Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, et al. (2008) Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev 22: 1397-1409. [Crossref]
- Seale P, Kajimura S, Yang W, Chin S, Rohas LM, et al. (2007) Transcriptional control of brown fat determination by PRDM16. Cell Metab 6: 38-54. [Crossref]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, et al. (2008) PRDM16 controls a brown fat/skeletal muscle switch. Nature 454: 961-967. [Crossref]
- Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, et al. (2008) New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454: 1000-1004. [Crossref]
- Fernández-Sánchez A, Madrigal-Santillán E, Bautista M, Esquivel-Soto J, Morales-González Á, et al. (2011) Inflammation, oxidative stress, and obesity. Int J Mol Sci 12: 3117-3132. [Crossref]
- Harwood HJ, Petras SF, Shelly LD, Zaccaro LM, Perry DA, et al. (2003) Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J Biol Chem 278: 37099-37111. [Crossref]
- Naik E, Dixit VM (2011) Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med 208: 417-420. [Crossref]
- Kudin AP, Malinska D, Kunz WS (2008) Sites of generation of reactive oxygen species in homogenates of brain tissue determined with the use of respiratory substrates and inhibitors. Biochim Biophys Acta 1777: 689-695. [Crossref]
- Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol rev 94: 909-950. [Crossref]
- Nordberg J, Arnér ES (2001) Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 31: 1287-1312. [Crossref]
- Henriksen EJ, Diamond-Stanic MK, Marchionne EM (2011) Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med 51: 993-999. [Crossref]
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49: 1603-1616. [Crossref]
- Limón-Pacheco J, Gonsebatt ME (2009) The role of antioxidants and antioxidant-related enzymes in protective responses to environmentally induced oxidative stress. Mutat Res 674: 137-147. [Crossref]
- Andreyev AY, Kushnareva YE, Starkov AA (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70: 200-214. [Crossref]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, et al. (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7: 45-56. [Crossref]
- Serra D, Mera P, Malandrino MI, Mir JF, Herrero L (2013) Mitochondrial fatty acid oxidation in obesity. Antioxid Redox Signal 19: 269-284. [Crossref]
- Bashan N, Kovsan J, Kachko I, Ovadia H, Rudich A (2009) Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol Rev 89: 27-71. [Crossref]
- Houstis N, Rosen ED, Lander ES (2006) Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440: 944-948. [Crossref]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350: 664-671. [Crossref]
- van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9: 112-124. [Crossref]
- Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519-529. [Crossref]
- Rowland AA, Voeltz GK (2012) Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13: 607-625. [Crossref]
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, et al. (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9: 35-51. [Crossref]
- Won JC, Jang PG, Namkoong C, Koh EH, Kim SK, et al. (2009) Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity (Silver Spring) 17: 1861-1865. [Crossref]
- Çakir I, Cyr NE, Perello M, Litvinov BP, Romero A, et al. (2013) Obesity induces hypothalamic endoplasmic reticulum stress and impairs proopiomelanocortin (POMC) post-translational processing. J Biol Chem 288: 17675-17688. [Crossref]
- Hosoi T, Sasaki M, Miyahara T, Hashimoto C, Matsuo S, et al. (2008) Endoplasmic reticulum stress induces leptin resistance. Mol pharmacol 74: 1610-1619. [Crossref]
- Hosoi T, Ozawa K (2010) Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clinical Science 118: 19-29. [Crossref]
- Özcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, et al. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457-461. [Crossref]
- Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, et al. (2007) The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 50: 2486-2494. [Crossref]
- De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, et al. (2005) Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 146: 4192-4199. [Crossref]
- Zhang X, Dong F, Ren J, Driscoll MJ, Culver B (2005) High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol 191: 318-325. [Crossref]
- Thaler JP, Schwartz MW (2010) Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology 151: 4109-4115. [Crossref]
- Myers MG, Cowley MA, Münzberg H (2008) Mechanisms of leptin action and leptin resistance. Annu Rev Physiol 70: 537-556. [Crossref]
- Shi X, Wang X, Li Q, Su M, Chew E, et al. (2013) Nuclear factor B (NF-?B) suppresses food intake and energy expenditure in mice by directly activating the Pomc promoter. Diabetologia 56: 925-936. [Crossref]
- Milanski M, Degasperi G, Coope A, Morari J, Denis R, et al. (2009) Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 29: 359-370. [Crossref]
- Williams LM (2012) Hypothalamic dysfunction in obesity. Proc Nutr Soc 71: 521-533. [Crossref]
- Stahl W, Sies H (2003) Antioxidant activity of carotenoids. Mol Aspects Med 24: 345-351. [Crossref]
- Craft N, Haitema TB, Garnett KM, Fitch KA, Dorey CK (2004) Carotenoid, tocopherol, and retinol concentrations in elderly human brain. J Nutr Health Aging 8: 156-62. [Crossref]
- Johnson EJ, Vishwanathan R, Johnson MA, Hausman DB, Davey A, et al. (2013) Relationship between Serum and Brain Carotenoids, α-Tocopherol, and Retinol Concentrations and Cognitive Performance in the Oldest Old from the Georgia Centenarian Study. J Aging Res 2013: 951786. [Crossref]
- Snellen E, Verbeek AL, Van Den Hoogen GW, Cruysberg JR, Hoyng CB (2002) Neovascular age-related macular degeneration and its relationship to antioxidant intake. Acta Ophthalmol Scand 80: 368-371. [Crossref]
- Roberts JE, Dennison J (2015) The Photobiology of Lutein and Zeaxanthin in the Eye. J Ophthalmol 2015: 687173. [Crossref]
- Vishwanathan R, Iannaccone A, Scott TM, Kritchevsky SB, Jennings BJ, et al. (2014) Macular pigment optical density is related to cognitive function in older people. Age Ageing 43: 271-275. [Crossref]
- Feeney J, Finucane C, Savva GM, Cronin H, Beatty S, et al. (2013) Low macular pigment optical density is associated with lower cognitive performance in a large, population-based sample of older adults. Neurobiol Aging 34: 2449-2456. [Crossref]
- Vishwanathan R, Neuringer M, Snodderly DM, Schalch W, Johnson EJ (2013) Macular lutein and zeaxanthin are related to brain lutein and zeaxanthin in primates. Nutr Neurosci 16: 21-29. [Crossref]
- Johnson EJ (2014) Role of lutein and zeaxanthin in visual and cognitive function throughout the lifespan. Nutr Rev 72: 605-612. [Crossref]
- Johnson EJ, Vishwanathan R, Johnson MA, Hausman DB, Davey A, et al. (2013) Relationship between serum and brain carotenoids,-tocopherol, and retinol concentrations and cognitive performance in the oldest old from the Georgia Centenarian Study. J Aging Res 2013:951786. [Crossref]
- Krinsky NI, Mayne ST, Sies H (2004). Carotenoids in health and disease. (CRC Press).
- Linnewiel-Hermoni K, Motro Y, Miller Y, Levy J, Sharoni Y (2014) Carotenoid derivatives inhibit nuclear factor kappa B activity in bone and cancer cells by targeting key thiol groups. Free Radic Biol Med 75: 105-120. [Crossref]