Despite intensive effort, no effective pharmacological inhibitors of the Ras oncoprotein have been reached the clinic given the impression that Ras proteins are undruggable. However, progresses have been made in several directions to manipulate Ras.
In this manuscript, we describe a novel and promising approach for specifically targeting the Ras/Raf interaction. We have identified the amino acid sequence involved in the binding of Ras to Raf. The amino acids of the binding site were coupled to an optimized penetrating peptide in order to generate a chimeric peptide, Mut3DPT-Ras, able to penetrate the cells and target Ras/Raf interaction. We demonstrate that this protease-resistant peptide has an in vitro apoptotic effect on several cancer cell lines and on primary cells isolated from chronic lymphocytic leukemia (CLL) as well as an antitumoral effect on chronic lymphocytic leukemia-like and lymphoma xenograft model. The new generated peptide allows the modulation of the Ras/Raf interaction and might have a potential as a new therapeutic approach for cancer treatment.
cell penetrating peptides, Ras, chronic lymphocityc leukemia, xenograft models, lymph
Cancer is frequently characterized by mutations that disrupt normal mechanisms controlling cell survival, proliferation, differentiation and apoptosis. Ras family of small GTPases consist of four highly related members: K-Ras 4A, K-Ras 4B, H-Ras and N-Ras. The proteins are normally located at the inner leaflet of the plasma membrane, where they are involved in the signal transduction through interaction with multiple partners/effectors. Ras function switch between GTP-bound active form and GDP-bound inactive form. Active Ras binds to one of their multiple effectors, Raf serine/threonine kinase (c-Raf or B-Raf) and induces their translocation to the plasma membrane where Ras is fully activated [1,2].
The four Ras homologous proteins (H-Ras, N-Ras, K-Ras 4A and K-Ras 4B) are identical in the first 85 amino acids. Ras proteins share 85% identity over the next 80 amino acids and diverge substantially at the C-terminal hypervariable region, which terminates with a CAAX motif, where takes place the posttranslational modifications. Although the role of Ras pathway activation has been described in the development of myeloid disease, growing evidences implicate hyperactive Ras in the development of lymphoid cancers. Lymphoid transformation was observed in primary hematopoietic cell expressing mutant Ras [3]. K-Ras and N-Ras point mutations have been found in several hematological cancers [4], as well as in other solid tumors such as colon, pancreas, lung, ovary and melanoma [1].
Oncogenic Ras is an interesting target for rational drug discovery as the mutant protein is a membrane-associated signalling molecule that is expressed at high level in primary tumor cells. However, targeting Ras is extremely complicated in practice due to the nature of the Ras cycle and the functional consequences of mutations [5,6]. Given these biochemical problems, drug discovery efforts have been primary focused in four areas: 1) direct inhibition of Ras, 2) blocking enzymes that catalyze posttranslational modifications of Ras and that are essential for membrane targeting, 3) developing inhibitors of downstream effectors and 4) searching for synthetic lethal interactors of mutant Ras [1,7].
Cell penetrating peptides (CPP) are molecules that can translocate into cells without causing membrane damage, leading to their proposal use as vectors for intracellular delivering of therapeutic cargos [8]. CPP can cross the membrane and reach the cytoplasm and/or the nucleus [9]. We reported experimental evidence that using a CPP and interfering peptide we were able to dissociate caspase-9/PP2A interaction inducing in vitro and in vivo tumoral cell death without affecting healthy cells [10-13]. Considering the difficulties to reach intracellular or nuclear targets by conventional methods, the use of CPPs in cancer treatment is of great interest [14,15] and peptide-based strategies offer considerable advantages over other delivery systems. Major advantages of peptide therapeutics include reduced immunogenicity as well as rapid delivery of cargoes into cells. Furthermore, they are stable in physiological buffers and are usually non-toxic [8,12].
Chronic lymphocytic leukemia (CLL) is the most common form of leukemia in adults, which is defined by accumulation of monoclonal B cells in hematopoietic organs due to the defect of apoptosis [16]. Clinical treatments are based on the use of purine analogues, often associated to cyclophosphamide [17-19] and the anti-CD20 monoclonal antibody rituximab [19-21]. However, despite a significant improvement of the overall survival of CLL patients, it remains clinical situations for which new therapeutic approaches are needed, especially for relapse or refractory CLL. Hematological malignancies such as CLL provide exceptional platforms for addressing mechanistic questions because established systems for culturing primary cells and robust mouse models.
Mouse models of human B-cells malignancies are important tools for preclinical studies. For human B-cell lymphomas, several mice models have been developed [22]. Here we have used a volumetric model by engrafting Daudi cells into nude mice, which remains of great value for efficiency. Difficulties in establishing permanent B-CLL cell lines have hampered studies on CLL, however few reliable xenografted tumor models are available nowadays. Taking advantage of our previous data [23] we evaluated the efficiency of the chimeric peptide Mut3DPT-Ras in xenograft mouse model obtained by injection of human Jok1 cells and CD5- transfected human Jok1 cells (Jok5.3) into SCID mice [24]. Whereas CD5 is generally lost in long-term cultures of CLL cell lines, Jok5.3 cells derived by stable transfection of the human CD5 gene into Jok-1 cells display a phenotype somewhat close to that of primary leukemic cells. The xenografted mice that we obtained developed a leukemia resembling the CLL type as defined by the French-American-British criteria.
We have taken advantage of our knowledge on the design of peptides for targeting protein/protein interactions in order to identify the amino acids in human K-Ras protein a human B-Raf involved in the complex formation in order to generate new tools for controlled manipulation of Ras/Raf interaction. In this manuscript, we describe the identification of the binding site of Ras to Raf, as well as the generation of chimeric peptides and assessed their potential anti-tumoral effect on human CLL-like and lymphoma xenograft models. This peptide can be used as therapeutic tool to dissociate the complex Ras/Raf in tumoral cells as a potential targeted and selective clinical approach.
In vitro identification of Ras sequence involved in Raf interaction
To determine the residues of human K-Ras protein that interact with the kinase B-Raf, we generated overlapping dodecapeptides from human K-Ras protein, which were immobilized in a cellulose membrane. (Figure 1A) shows the entire human K-Ras amino acid sequence upon hybridization with human Raf protein. One set of spots allowed the identification of a linear interaction area spanning residues from 169 to 188. This sequence corresponds to a solved- exposed structure (data not shown). Chimeric peptide containing Mut3DPT-Sh1, an optimized cell penetrating sequence previously developed in our laboratory, followed by the selected Ras interaction sequence was chemically synthesized and further used for functional assays. (Figure 1B) shows the K-Ras sequence comparison between human and mouse K-Ras and peptide identification, showing the high homology of the Ras/Raf interaction sequence between human and mouse Ras sequences. (Figure 1C) shows the sequence comparison among human H-, K- and N-Ras isoforms. The most important differences in the sequence are located at the C-terminal of the three proteins, strongly suggesting the specificity of our peptide for the K-Ras isoform.

Figure 1. Identification of the binding site of Ras to Raf. A) Raf binding assay on cellulose-bound Ras peptides. The sequence of Ras was developed as series of overlapping dodecapeptides with a shift of two amino acids. Spots were detected using the ECL system. Ras peptides that interact with Raf are boxed and the sequence shown. m, mouse; h, human. B) Comparison of K-Ras sequence between human (h) and mouse (m) species. C) Sequence comparison among human H-, N- and K-Ras isoforms.
We were interested in confirming that the specific target of Mut3DPT-Ras peptide is the complex Ras/Raf. To this end, we analyze whether the peptide was able to target the in vitro interaction Ras/Raf. For the competition assay, lysates from MDA-MB 231 cell line were immunoprecipitated with anti-Raf antibody and the interaction with Raf competed with the peptide Mut3DPT-Ras (Figure 2). Ras was detected in control Raf immunoprecipitates, while it was undetectable after competition with 1.5 mM of Mut3DPT-Ras peptide. Raf was used as internal control showing similar detection in both conditions. This result suggests that Mut3DPT-Ras peptide specifically targets the interaction between human Ras and Raf.
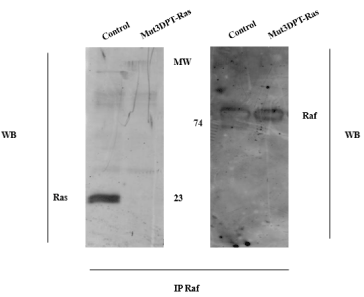
Figure 2. . Mut3DPT-Ras competes in vitro Ras/Raf interaction. The MDA-MB231 cell line was lysed and cytoplasmic extracts immunoprecipitated with anti- Raf antibody. The interaction Ras/Raf was competed in vitro with 1.5 mM of Mut3DPT-Ras peptide for 30 min at room temperature. Immunoprecipitates were washed and immunoblotted with anti-Ras and anti-Raf antibody, the former as internal control of protein loading
The penetrating peptide Mut3DPT-Ras induces apoptosis in cell lines
We first analyzed the effect of Mut3DPT-Ras peptide on the viability of colon, lung and breast cancer cell lines (Table 1). As shown in (Figure 3A), the viability of the colon cancer cell lines HCT1116, SW480 and HT29 was almost not affected by the peptide Mut3DPT-Ras even when used at high dose. On the contrary, the viability of the lung cancer cell lines H1150, HBEC wt and HBEC Ras V12 was significantly affected at high dose of peptide treatment (Figure 3B). Finally, the viability of the lung cancer cell lines H1975, A549 and H1299 was slightly affected by high dose of peptide Mut3DPT-Ras (Figure 3B).
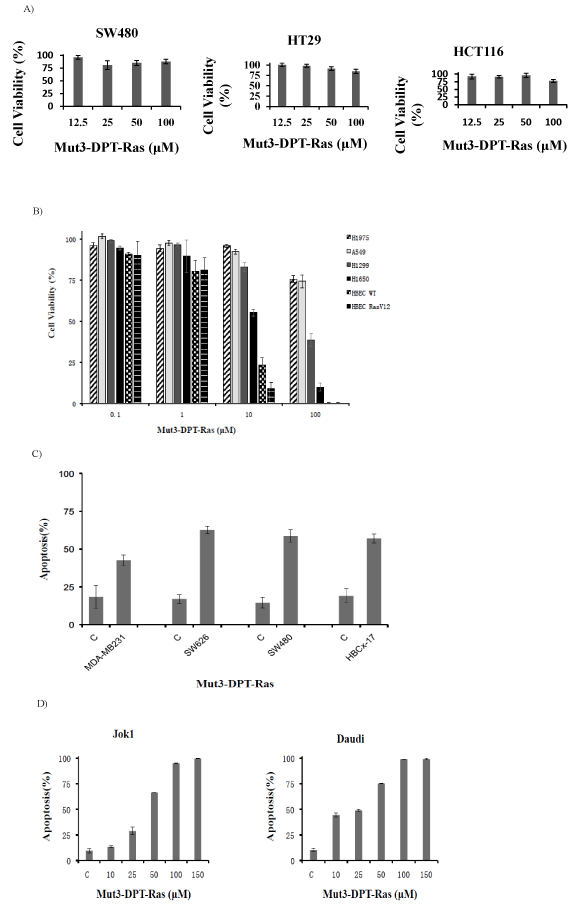
Figure 3. In vitro cell viability and apoptosis assay. A) Colon cancer cell lines HCT116, SW480 and HT29 (1x104/well) were treated with or without different concentrations of peptide Mut3DPT-Ras as indicated. Absorbance was recorded 72h after. The results are presented as percentage of viability relative to control non-treated cells. B) Lung cancer cell lines H1650, HBEC wt H1975, A549, H1299 and HBEC RasV12 (1x104/well) were treated with or without different concentrations of peptide for 72h. The analysis of cell viability was made as above. C) MDA-MB231, BC227 (breast cancer cell lines); SW620 and SW480 (colon cancer cell lines) were treated with 100 µM of Mut3DPT- Ras peptide for 24h and apoptosis was estimated by annexin V-FITC staining. Non treated cells (C) were used as negative control. D) Daudi and Jok1 cell lines were treated for 24h with increasing concentrations of Mut3DPT-Ras peptide and then apoptosis was analyzed by annexin V-FITC staining. C; control.
Table 1. First analyzed effect of Mut3DPT-Ras peptide on the viability of colon, lung and breast cancer cell lines
|
Origin
|
Cell line
|
Mutation
|
|
Non-small cell lung cancer cell line
|
A549
|
K-Ras
|
|
H1299
|
N-Ras
|
|
H1650
|
Ras-wt
|
|
H1975
|
KRas
|
|
HBEC WT
|
Ras wt
|
|
HBEC RasV12
|
K-Ras
|
|
Colon cancer cell line
|
HCT116
|
K-Ras
|
|
HT29
|
K-Ras
|
|
SW480
|
K-Ras
|
|
Ovary cancer cell line
|
SW626
|
K-Ras
|
|
B lymphoma cell line
|
Daudi
|
K-Ras
|
|
CLL-like cell line
|
Jok 1, Jok 5.3
|
-
|
|
Breast cancer cell line
|
MDA-MB-231
|
K-Ras
|
|
BC227
|
Ras-wt
|
We then analyzed the ability of the peptide Mut3DPT-Ras to induce apoptosis on several colon, ovary and breast cancer cell lines, as well as in lymphoma and CLL-like cell lines. As shown in (Figure 3C), the peptide induces apoptosis, as detected by Annexin-V-FITC staining, in MDA-MB 231, BBCx-17 (breast cancer cell lines), SW626 and SW480 (colon and ovary cancer cell lines, respectively) upon 24h of treatment. The basal level of apoptosis in control non-treated cells is shown.
We further analyzed the effect of the peptide in the human Burkitt’s lymphoma B cell lines, Daudi and in the chronic lymphocytic leukemia-like cell line, Jok1 (Figure 3D). In the two cell lines, Mut3DPT-Ras peptide induced apoptosis, which was concentration-dependent and reached the plateau at a concentration of around 100 µM for Daudi and Jok1 cell lines. The non-treated cell lines were used as a negative control. Taken together, these results show an in vitro apoptotic effect of Mut3DPT-Ras peptide in a large panel of human cell lines from different pathologies and relative to control non-treated cells.
Mut3DPT-Ras has apoptotic effect on tumoral primary cell from chronic lymphocytic leukemia
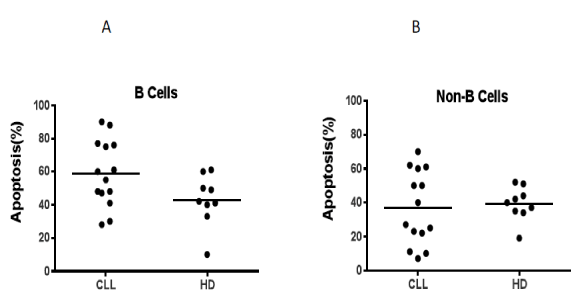
Figure 4. Apoptotic effect of Mut3DPT-Ras peptide on primary cells. A) Peripheral blood mononuclear cells (PBMC) from healthy donors (HD) or chronic lymphocytic leukemia (CLL) patients were isolated by Ficoll gradient and culture in the presence or in the absence of Mut3DPT-Ras peptide (100 μM) for 4h, then washed and apoptosis of B cells analyzed. Selection of B cells was done by anti-CD19 antibody before annexin V-FITC staining. Non-treated cells were used as control. B) PBMC from HD or CLL patients were isolated and treated as above. Apoptosis of non-B cells (T, NK, momocytes) was detected by annexin V-FITC staining. Non-treated cells were used as control.
In order to evaluate the apoptotic effect of the peptide in primary healthy and tumoral cells from the same origin, we used peripheral blood mononuclear cells (PBMC) from healthy donors (HD) and chronic lymphocytic leukemia (CLL) patients. PBMCs from HD and CLL patients were treated 4h with the peptide (50 µΜ) and apoptosis was analyzed by annexin-V staining. The peptide has an important apoptotic effect on B cell isolated from CLL patients and a moderated effect on B cells from HD (Figure 4A) relative to control non treated cells. When non-B cells were analyzed (T, NK, monocytes), we observed similar level of apoptosis between HD and CLL patients relative to control non-treated cells (Figure 4B). The shuttle alone had no effect on apoptosis, neither in HD, nor in CLL patient samples (data not shown). This result might suggest that tumoral B cells could more sensitive to treatment by the peptide than healthy B cells.
Mut3DPT-Ras is stable in human serum: The stability of Mut3DPT-Ras peptide in human serum was assessed by mass spectrometry (MS) before performing the in vivo experiments. When Mut3DPT-Ras (mw 3,732 Da) was incubated with human serum at 37°C and further analyzed by HPLC-MS, we observed that the peptide showed resistance to proteolytic degradation and almost all of the peptide remains intact upon 24h of incubation at 37°C (Figure 5).
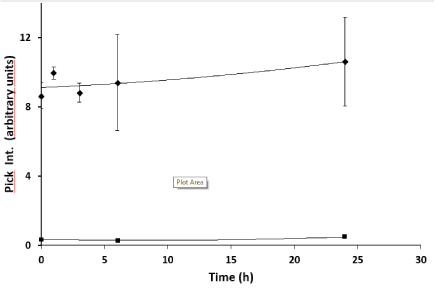
Figure 5. Stability of Mut3DPT-Ras peptide on human serum. Mut3DPT-Ras was incubated at 37oC in human serum for different periods of time and the integrity of the peptide was analyzed by mass spectrometry (MS). Similar results were obtained in two independent experiments. Every measurement was performed in triplicate. Standard deviation is shown.<, Control; ♦Mut3DPT-Ras.
Mut3DPT-Ras has antitumoral effect on human xenograf models of chronic lymphocytic leukemia and lymphoma: The therapeutic efficacy of Mut3DPT-Ras was evaluated in three human xenograft models using the two chronic lymphocytic leukemia Jok1 and Jok5.3 cell lines and the human NHL Daudi cell line.
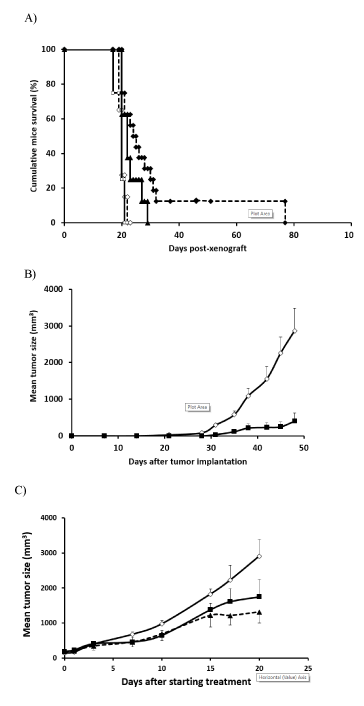
Figure 6. Mut3DPT-Ras peptide has significant in vivo antitumor activity in human chronic lymphocytic leukemia and lymphoma xenograft model. A) SCID mice, inoculated with 2.5 × 106 Jok1 or Jok5.3 cells via tail vein injection, were intra peritoneal (I.P.) treated on days +3 with 5 mg/kg of Mut3DPT-Ras during five consecutive days for 4 weeks. Control mice received NaCl. Survival was monitored for 77 days.¯, Control Jok1; , Control Jok5.3; p, Mut3DPT-Ras Jok5.3; ®, Mut3DPT-Ras Jok1. B) BALB/c nude mice, subcutaneously inoculated with 10 × 106 Daudi cells, were I.P. treated with 5 mg/kg of Mut3DPT-Ras during five consecutive days for 4weeks. Control mice received NaCl. Tumor growth was monitored for 47 days. The average tumor volume of each group (six mice in each group) with SD is shown as a function of time (*, P < 0.05; **, P <0.003). Pooled data from 6-16 mice/group are shown. ◊, Control; <, Mut3DPT-Ras. C) BALB/c nude mice, subcutaneously inoculated with 10 × 106 Daudi cells, were I.P. treated. when palpable tumors were apparent (mean: 150-200 mm3) with 5 mg/kg or 20mg/kg of Mut3DPT-Ras during five consecutive days for 4weeks. Control mice received NaCl. Tumor growth was monitored until tumor burden to reach a maximum ethical-permissible volume. ◊, Control; ■ 5 mg/kg; ▲, 20 mg/kg.
The treatment with Mut3DPT-Ras peptide was initiated three days after injection of the cells in SCID mice xenografted with CLL cell lines. As shown in (Figure 6A), treatment of Jok1 and Jok5.3 tumors with Mut3DPT-Ras at 5 mg/kg yielded a significant survival benefit, providing an increase in median survival. Control mice died of disseminated disease manifested with central nervous system paralysis, with a median survival time of 20 days after Jok1 and jok5.3 tumor inoculation. Median survival in both treated groups was significantly extended to 31 days for Jok1 (p<0.003) and 23 days for Jok5.3 (p<0.01).
As shown in Figure 6B, tumor size (expressed in mm3) in Daudi-inoculated mice treated with Mut3DPT-Ras, was significantly slower, as compared to controls (i.e., mice receiving NaCl) (p<0.05). Treatment with NaCl did not improve the survival of mice (Figure 6B). On the 47th day post-tumor cell inoculation, control mice were euthanized for ethical reasons (medium tumor size, 2866 mm3), whereas the average tumor volume recorded for animals treated with Mut3DPT-Ras was 403 mm3. This tumor reduction of ~81% was statistically significant (p<0.01), compared to controls. In addition, we observed a delay in the appearance of tumor in Mut3DPT-Ras treated mice than in the control group (47 days vs. 25 days).
Subcutaneous Daudi tumors were established in nude mice and allowed to reach a mean tumor volume of 175 mm3 before treatment with saline or Mut3DPT-Ras peptide as described in Materials and Methods. Established, large volume, solid tumors can present a significant challenge to intervention by therapies. Reduction in tumor volume was clearly observed upon Mut3DPT-Ras treatment. At the end of the four weeks of treatment at 20 mg/kg/, peptide- treated mice resulted in a substantial decrease of 58% in the volume of transplanted tumors (1315 ± 308 mm3) when compared with the other two groups, 1750 ± 526 mm3 (44%) and 2904 ± 693 mm3 for the 5 mg/kg/ Mut3DPT-Ras and control groups (p<0.05, Figure 6C). Moreover, 17% of mice treated with 20 mg/kg/ of Mut3DPT-Ras experienced complete tumor remission without relapse (Figure 6C). This result suggests an anti-tumoral effect in both preventive and curative treatment.
Protein-protein interactions are emerging class of molecular targets for many therapeutic areas. However, their relatively large interaction surface has been considered as a factor of risk, since it can be difficult to modulate or to disrupt using small molecules. This obstacle is more prominent when the identification of the interaction amino acids is not obvious. New approaches such as the use of cell penetrating peptides (CPP) are a promising alternative for the modulation of protein-protein interactions.
CPP appear as new class of vectors that cross the membrane overcoming the internalization problem and delivering cargo into cells. They are used for intracellular delivery of cargoes, like peptides [10,11,25,26]. An exciting targeted approach for therapy is the use of chimeric peptides with penetration capability and protein-protein interfering properties. We have published the use of a chimeric peptide consisting of a penetrating sequence associated to the binding site of caspase 9 to PP2A [11,12]. This peptide, as well as the optimized protease- resistant variant [12] induces apoptosis in vitro specifically on primary tumoral cell and has an anti-tumoral effect in vivo.
Ras is mutated at high frequency in various malignancies, including pancreas, colon, lung and melanoma, among others. Ras mutations have been implicated in the development of myeloid pathologies, and increasing evidences implicates hyperactive Ras in the development of lymphoid cancers [3,27-29]. Even if important efforts have been made, no effective pharmacologic inhibitors of Ras proteins have reached the clinic. Today researches are focused in the direct inhibition of Ras, blocking Ras posttranslational modifications, targeting Ras effectors, search for lethal interactors of mutant Ras and, finally, targeting metabolic Ras- induced changes.
We have identified the binding site between the protein Ras and its effector Raf. A blast sequence shows that the identified sequence is highly conserved between human and mouse.
This observation indicates that these sequences should have important functional role. In addition, sequence comparison among human Ras isoforms shows a high homology, differing only in the C-terminal CAAX motif. The peptide derived from the interaction domain of Ras/Raf proteins was associated to an optimized shuttle and evaluated its capacity to interfere with the parental Ras/Raf interaction. This peptide induces apoptosis in all cell lines tested and primary B cells isolated from chronic lymphocytic leukemia patients (CLL). However, the moderated apoptotic effect observed on B cells isolated from healthy donors (HD) might suggest a non-selective (specific) tumoral effect. When analyzed individually the apoptotic effect on B cells from CLL, we observed strong apoptosis on 5 patient samples and a low apoptotic effect on 2 patient samples. When analyzed individually the effect on B cells from HD, we observed a more homogeneous effect except for one HD where the peptide had no effect.
Most of the strategies employed so far did not discriminate between wild type and oncogenic Ras. In this case, the principle of selective action of Ras inhibitors on cancer cells rather than wild-type cells mainly relies on the “oncogenic addition” phenomena whereby cancer cells become more sensitive to the pharmacological inhibitors of Ras function [30]. We cannot exclude that oncogenic Ras and wild type Ras utilize different pathways allowing the specific inhibition of oncogenic Ras. This suggests that this peptide blocks mainly cancer but not or very low the normal cells. When analyzing the effect of the peptide in vivo using xenograft models of lymphoma, we observed a strong anti-tumoral effect even when using a low dose of peptide such as 5 mg/kg and in both, preventive and curative treatment. We observed a moderate effect on CLL like xenograft models, Jok1 and Jok5.3.
Using a computer-based molecular modelling method, Pincus and co-workers have designed two peptides from Ras protein that block proliferation of cancer cells. The rational of their approach is to identify peptide domains from Ras that alter conformation in response to amino acid substitutions. They compare low energy average structures from oncogenic and wild type Ras using conformational energy calculation. The peptides are then synthesized associated to the cell penetrating sequence penetratin. It has been described that region from 38 to 47 amino acids of Ras is the binding site for all of the known targets of Ras, including Raf, GAP, SOS and PI3K [31-37]. The same authors point out that the region comprised between amino acids 55 and 71 of Ras make also contact with GAP and SOS proteins. Pincus and co-workers affirm that their peptides PCN-7 (35-47) and PCN-2 (96-110) block interaction with Raf and have an anti-tumor effect on cell lines. They also affirm that both peptides induce phenotypic reversion of the HT-1080 cell line to non-transformed cells but cause tumor cell necrosis on MIA-PaCa2 cell line. They speculate that PCN-2 might block oncogenic Ras by inhibiting its interaction with JNK. PCN-7 could block the interaction of oncogenic Ras with Raf [38,39]. They suggest that both oncogenic and wild type Ras requires Raf activation, and assume, without demonstration, that each Ras protein interacts with Raf in different ways, resulting in differential activation of downstream kinases. In addition, they also mention, again without experimental demonstration, that their peptides block oncogenic Ras but have not effect on intracellular wild type Ras.
Here, we present a similar chimeric peptide strategy with strong differences. First, we used a proprietary CPP that has been specifically designed to provide stability for in vivo application [12]. Second and more important, the interfering domain of the bifunctional peptide has been designed to specifically target the interaction between Ras and Raf, contrary to the approach used by Pincus. With this direct targeting, we should not affect other Ras or Raf functions, avoiding possible undesired effects. Finally, we have made the proof of concept in vivo using xenograft models of CLL-like and lymphoma and proving the anti-tumoral effect of our peptides.
Despite the involvement of Ras in many types of cancers, there is not clinical effective anti- cancer drugs targeting Ras, which has been considered as “undrugable” target. However, recent studies have identified novel surface packets that could accept small molecules in both the active and inactive form. The discovery of these new Ras inhibitors has been made on the bases of structural information. In the development of Ras inhibitors, the first effort was made in the generation of inhibitors of membrane targeting of Ras such as inhibitors of geranylation, prenylation, farnesylation, myristoilation or palmytoilacion used in monotherapy or in combination with other cytotoxic agents [40-42]. Unfortunately, most of these inhibitors showed no clinical efficacy, in addition to non-specific activities toward other proteins that have the same post-translational modifications.
Efforts have also been made for blocking the activation of Ras. Most of the identified molecules showed no specificity and no selectivity, in addition to the problem that functional Ras wild type is required in normal cells. Concerning the inhibition of the Ras effector interactions, this is the most promising strategy. Several teams have designed compounds that inhibit Ras/Raf binding. However, the lack of evidence for cellular efficacy, low binding affinity, low specificity and weak inhibitory effect may discourage their further optimization [30,43-47].
We have previously used the strategy of using CPP associated to interfering peptides to provide tools that might be useful for targeted treatment of cancer both from efficacy and safety perspectives [10,11], suggesting that Mut3DPT-Ras could also share these favourable properties, as suggested by the results obtained using xenograft model of chronic lymphocytic leukemia and lymphoma. In both cases, the peptide has shown a strong anti tumoral effect, without apparent toxicity. Our data provide further confirmation that Mut3DPT-Ras might be used as new agents for the therapy of CLL and lymphoma.
Cell lines and reagents
H1975, H1299, H1650, HBEC wt, HBEC RasV12 and A549 are human non-small cell lung cancer cell lines [48]. H1975, H1299 and H1650 (NCI-60 collection) were cultured in RPMI 1640 supplemented with 10% FBS. HBEC wt, HBEC RasV12 and A549 were cultured in DMEM medium supplemented with 10% foetal bovin serum (FBS). MDA-MB231 (ATCC) and HBCx-17 (cell line isolated from primary breast cancer tumor in Dr Decaudin Laboratory, LIP) are breast cancer cell lines and were cultured in DMEM supplemented with 10% FBS. HCT116, HT29ans SW480 are colon cancer cell lines obtained from the NCI60 collection from CNIO. They were cultured in DMEM medium supplemented with 10% FBS. SW626 is an ovarian cancer cell line that was maintained in DMEM supplemented with 10% FBS. Human lymphoma cell line Daudi (ATCC CCL-213) and human Chronic Lymphocytic Leukemia Jok1 and Jok5.3 [23] were cultured in RPMI supplemented with 10% FBS, L-glutamine and penicillin/streptomycin. For characteristics of the cell lines, see (Table 1).
Raf protein was from Emzo Life Science. Anti-Ras and anti-Raf antibodies were from Merck Millipore and Abcam respectively. Secondary antibodies were from Dako. Peptides were synthesized by GL Biochem, Shanghai, China. Clarity ECL was from Bio-Rad. MTT viability assay kit was from Bio-Rad. Mice were purchased from Janvier.
2021 Copyright OAT. All rights reserv
Xenograft models were obtained by injection of two human chronic lymphocytic leukemia cell lines. The human cell leukemia Jok1 (CD5-, HLA-DR+, CD71+, CD20+) was stably transfected with pLNCX vector carring human CD5 cDNA. Subclone Jok5.3 stably expressing human CD5+ was selected by limiting dilution and expanded in vitro. The cells stably maintained CD5 expression as determined by standard immunophenotyping and FACS analysis to similar level to that observed in CLL [24]. We verified that Jok5.3 had the following phenotype: CD5-, HLA-DR+, CD71+, CD20+. All cell lines were cultured in RPMI 1640 supplemented with 10% fetal calf serum (FCS) and 2 mM glutamine.
Peptides synthesis and sequences
Peptides were synthesized in an automated multiple peptide synthesizer with solid phase procedure and standard Fmoc chemistry. The purity and composition of the peptides were confirmed by reverse phase HPLC and by mass spectrometry. The sequence of the peptide Mut3DPT-Ras has been protected by a patent (EP13305945).
Raf-binding assays on cellulose-bound peptides containing Ras sequences
Overlapping dodecapeptides scanning the complete human K-Ras sequence were prepared by automated spot synthesis (Abimed, Langerfeld, Germany) onto an amino-derived cellulose membrane, as described [49,50]. The membrane was saturated using 3% non-fat dry milk/3% BSA, incubated with purified Raf protein (4 µg/ml) and after several washing steps, incubated with anti-Raf antibody, followed by PO-conjugated secondary antibody (Dako). Positive spots were visualized using the ECL system (Bio-Rad). Images were acquired using the software Image Quant LAS 4000.
Isolation of cell populations and culture
Fresh blood from healthy donors (HD) was obtained from the Etablissement Français du Sang (EFS). Chronic lymphocytic leukemia (CLL) patient samples were obtained from the Department of Hematology after write consent of the patients. Peripheral blood mononuclear cells (PBMC) from HD and CLL patients were prepared by Ficoll gradient centrifugation. Cells were maintained in RPMI 1640 supplemented with 10% FCS, 1% non-essential amino acids, 1% Hepes, 1% sodium pyruvate and 1% glutamine.
Ficoll gradient
The proportion of Ficoll: blood we used was 1/3 Ficoll and 2/3 blood. Diluted blood in PBS (1:1). Carefully layer diluted blood over Ficoll medium tube. The diluted blood is added to the gradient by gently pipeting onto the separation medium with the tube held at an angle. It is critical that a clear separation be kept between the dense Ficoll medium and the blood layer before centrifugation. Centrifugate at 2,200 rpm for 20 min without brake. Carefully remove the PBMC layer from the tube and transfer to a new tube. Wash PBMC by adding enough PBS. Centrifugate at 1,500 rpm for 5 min. Repeat the washing step, decant the supernatant and resuspend cells in appropriate volume of phosphate buffer saline (PBS).
Detection of apoptosis by Annexin-V-FITC staining on primary cells
Human lymphocytes isolated from HD or CLL patient were stained with anti-hCD19 antibody (APC fluorochrome from BD biosciences) and early apoptotic events were determined using Annexin-V FITC (eBiosciences) as described by the manufacturer. Briefly, cells were washed in 1x binding buffer, centrifuged and resuspended in 100 µl of 1x binding buffer containing Annexin-V FITC (0.1 µg/ml) and propidium iodide (PI) (0.5 µg/ml). After incubation for 15 min, cells were analyzed by flow cytometry. The protocol used for detection of apoptosis in cell lines was the same, omitting the step of staining with the anti-CD19 antibody. Data acquired by FACS Canto (BD biosciences) were analyzed with Diva 60 software.
Cell viability assays
The cell viability was determined using a colorimetric assay based on 3-(4,5-dimethylthiazol- 2-yl)-2,5 diphenyltetrazolium bromide (MTT). Cells were seeded at appropriate concentration in 96-well plates at day 0 and peptide was added to different concentrations at day 1. Cell viability was tested by MTT assay 72h after treatment followed the manufacture’s protocol. Briefly, 15 µl of MTT (5 mg/ml dissolved in PBS) were added to each well. After 4h incubation at 37°C, cell was lysed with 100 µl of 10% SDS in 10 mM HCl. The absorbance at 540 and 620 nn were measured on an Infinite 200 spectrophotometer (Tecan). We performed all experiments in triplicate and the results presented as percentage of cell proliferation normalized to control condition.
Immunoprecipitation and Western blotting
Cells (1 × 107) were lysed for 20 min at 4°C in lysis buffer (50 mM Tris (pH 8), 1% Nonidet P-40, 137 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 10% glycerol, and protease inhibitor mixture). Lysates (800 μg of protein) were immunoprecipitated with the appropriate Ab overnight at 4°C, and protein A-Sepharose was added for 1 h at 4°C. After washing with 1× TBST (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.05% Tween 20), immunoprecipitates were separated by SDS-PAGE, transferred to nitrocellulose, blocked (5% nonfat dry milk in TBST), and incubated with the primary Ab in TBS-0.5% nonfat dry milk. The membrane was washed and incubated with PO-conjugated secondary Ab. Protein detection was performed using the ECL system. Images were acquired using the software Image Quant LAS 4000.
Analysis of peptide stability in mouse serum
Mut3DPT-Ras (10 µM) was incubated at 37ºC in 250 µl mouse serum for different periods of time. Samples were collected and peptide degradation stopped by freezing. Peptides were extracted from collected samples using the ProteoMiner Protein Enrichment System (Bio- Rad). Peptide integrity (percentage of intact peptide) was analyzed by mass spectrometry using a MALDI-TOF (Brücker Autoflex II) following their standard protocols. Every measurement was performed by triplicate. MS data were analyzed using appropriate software (Clinprot tools, Flex analysis, Brücker).
Protein-protein interaction competition
The Ras/Raf interaction was competed using the peptide Mut3DPT-Ras (VKKKKIKAEIKI KMSKDGKKKKKKSKTKCVIM). Lysates from MDA-MB231cells were immunoprecipitated with anti-Ras or anti-Raf antibody, and protein A/G-Sepharose was added for 1h at 4°C. The Ras/Raf interaction was competed with 1.5 mM of the peptide Mut3DPT-Ras for 30 min at room temperature. After several washing steps, immunoprecipitates were transferred to nitrocellulose and blotted with the corresponding Ab.
Animals
6-8 weeks old SCID CB-17 mice (female, 20-23g) and BALB/c nude mice (male, 26-30g) were purchased (Elevage Janvier, Le Genest Saint-Ile, France). All mice were maintained under conditions and protocols in accordance with the Directive 2010/63/UE of the Council of Europe on Animal Welfare. The studies (number authorization 1290 and 5089) were approved by the French Ethics Committee for animal experimentation number 74 and all experiments were conducted following the guidelines of the afore mentioned committee.
In vivo activity
To establish the xenograft lymphoma model, 10 × 106 Daudi cells in 100 µL of PBS were subcutaneously injected into the flank of male BALB/c nude mice. For CLL xenograft model, 2.5 × 106 Jok1 or Jok5.3 cells in 100 µL of PBS were injected into the tail vein of SCID mice. Mice (6-16 mice per group) were then injected with saline or Mut3DPT-Ras (5 mg/kg or 20 mg/kg) intraperitoneally (I.P.) at days 3 after cells transplantation or when palpable tumors were apparent (mean 150-200 mm3) for five consecutive days during four weeks. For intravenous model, mice were monitored daily for the presence of hind-leg paralysis and in that case sacrificed. For subcutaneous model, tumor growth rate was recorded every 2–3 days by measuring the major and minor axes of the tumors formed with a digital caliper. Measurements were transformed into tumor volume using the formula: tumor volume (mm3) = major axis × minor axis2 × 0.5.
Statistical analysis: For in vivo data, statistical comparisons between groups were performed using Factorial analysis of variance ANOVA. Normality and homogeneity were evaluated by Shapiro-Wilk test and found the data valid.
In summary, the approach of cell penetrating and interfering peptides specifically directed against a Ras/Raf interaction is a promising strategy for the development of anti-cancer therapeutic agents. In this article, we have demostrated the proof of principle for the use of the peptide Mut3DPT-Ras for the treatment of lymphoma and CLL, giving a new look as a promising approach for anti-cancer drug development.
This work was supported by SATT- Lutech and Inserm, Paris, France. We thank Valery Frise for help in the collection of some CLL samples. Thanks also to Dr Gerard Zalcman for lung cancer cell lines gift and Jacky Lautridou for statistical in vivo analysis.
The authors declare no conflict of interest.
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ (2014) Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 13: 828-851. [Crossref].
- Adjei AA (2001) Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst 93: 1062-1074. [Crossref]
- Holmes KL, Pierce JH, Davidson WF, Morse HC (1986) Murine hematopoietic cells with pre-B or pre-B/myeloid characteristics are generated by in vitro transformation with retroviruses containing fes, ras, abl, and src oncogenes. J Exp Med 164: 443-457.
- Braun BS, Shannon K (2008) Targeting Ras in myeloid leukemias. Clin Cancer Res 14: 2249-2252. [Crossref]
- Vetter IR, Wittinghofer A (2001) The guanine nucleotide-binding switch in three dimensions. Science 294: 1299-1304. [Crossref]
- Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3: 11-22. [crossref]
- Diaz-Flores E, Shannon K (2007) Targeting oncogenic Ras. Genes Dev 21: 1989-1992.21(16):1989-1992. [Crossref]
- Bitler BG, Schroeder JA (2010) Anti-cancer therapies that utilize cell penetrating peptides. Recent Pat Anticancer Drug Discov 5: 99-108. [Crossref]
- Prochiantz A (2000) Messenger proteins: homeoproteins, TAT and others. Curr Opin Cell Biol 12: 400-406. [Crossref]
- Arrouss I, Decaudin D, Choquet S, Azar N, Parizot C, Zini JM, et al. (2015) Cell penetrating peptides as a therapeutic strategy in chronic lymphocytic leukemia. Protein and Peptide Letters 22: 539-546.
- Arrouss I, Nemati F, Roncal F, Wislez M, Dorgham K, et al. (2013) Specific targeting of caspase-9/PP2A interaction as potential new anti-cancer therapy. PLoS One 8: e60816. [Crossref]
- Fominaya J, Bravo J, Decaudin D, Brossa JY, Nemati F, Rebollo A (2015) Enhanced serum proteolysis resistance of cell-penetrating peptides. Therapeutic Delivery 6: 139-147.
- Guergnon J, Dessauge F, Dominguez V, Viallet J, Bonnefoy S, Yuste VJ, et al. (2006) Use of penetrating peptides interacting with PP1/PP2A proteins as a general approach for a drug phosphatase technology. Molecular Pharmacology 69: 1115-1124.
- Myrberg H, Zhang L, Mae M, Langel U (2008) Design of a tumor-homing cell-penetrating peptide. Bioconjug Chem 19: 70-75. [Crossref]
- Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, et al. (2010) Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci USA 107: 4311-4316. [Crossref]
- Chiorazzi N, Hatzi K, Albesiano E (2005) B-cell chronic lymphocytic leukemia, a clonal disease of B lymphocytes with receptors that vary in specificity for (auto)antigens. Ann N Y Acad Sci 1062: 1-12. [Crossref]
- Dohner K, Schlenk RF, Habdank M, Scholl C, Rucker FG, Corbacioglu A, et al. (2005) Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 106: 3740-3746.
- Austen B, Powell JE, Alvi A, Edwards I, Hooper L, et al. (2005) Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood 106: 3175-3182. [Crossref]
- Hallek M, Pflug N (2010) Chronic lymphocytic leukemia. Annals of oncology: Journal of the European Society for Medical Oncology 21: 154-164.
- Wierda W (2009) New prognostic factors in chronic lymphocytic leukemia. Clinical Advances in Hematology & Oncology 7: 32-33.
- Vega MI, Martinez-Paniagua M, Huerta-Yepez S, Gonzalez-Bonilla C, Uematsu N, Bonavida B (2009). Dysregulation of the cell survival/anti-apoptotic NF-kappaB pathway by the novel humanized BM-ca anti-CD20 mAb: implication in chemosensitization. International Journal of Oncology 35: 1289-1296.
- Donnou S, Galand C, Touitou V, Sautes-Fridman C, Fabry Z, Fisson S (2012). Murine models of B-cell lymphomas: promising tools for designing cancer therapies. Blood 116: 4926-4933
- Loisel S, André PA, Golay J, Buchegger F, Kadouche J, et al. (2011) Antitumour effects of single or combined monoclonal antibodies directed against membrane antigens expressed by human B cells leukaemia. Mol Cancer 10: 42. [Crossref]
- Loisel S, Ster KL, Quintin-Roue I, Pers JO, Bordron A, et al. (2005) Establishment of a novel human B-CLL-like xenograft model in nude mouse. Leuk Res 29: 1347-1352. [Crossref]
- de Figueiredo IR, Freire JM, Flores L, Veiga AS, Castanho MA (2014) Cell-penetrating peptides: A tool for effective delivery in gene-targeted therapies. IUBMB Life. [Crossref]
- Huang Y (2014) Cell-penetrating peptides and drug delivery. Curr Pharm Biotechnol 15: 191. [Crossref]
- Sabnis AJ, Cheung LS, Dail M, Kang HC, Santaguida M, et al. (2009) Oncogenic Kras initiates leukemia in hematopoietic stem cells. PLoS Biol 7: e59. [Crossref]
- Zhang J, Wang J, Liu Y, Sidik H, Young KH, et al. (2009) Oncogenic Kras-induced leukemogeneis: hematopoietic stem cells as the initial target and lineage-specific progenitors as the potential targets for final leukemic transformation. Blood 113: 1304-1314. [Crossref]
- Chiang MY, Xu L, Shestova O, Histen G, L'Heureux S, Romany C, et al. (2008) Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. The Journal of Clinical Investigation. 118: 3181-3194.
- Shima F, Matsumoto S, Yoshikawa Y, Kawamura T, Isa M, et al. (2015) Current status of the development of Ras inhibitors. J Biochem 158: 91-99. [Crossref]
- Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, et al. (1995) The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature 375: 554-560. [Crossref]
- Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, et al. (1997) The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277: 333-338. [Crossref]
- Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, et al. (2003) Structural evidence for feedback activation by Ras GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 112: 685-695.
- Pincus MR (2004) Development of new anti-cancer peptides from conformational energy analysis of the oncogenic ras-p21 protein and its complexes with target proteins. Frontiers in Bioscience 9: 3486-3509.
- Pincus MR, Brandt-Rauf PW, Michl J, Carty RP, Friedman FK (2000) Ras-p21-induced cell transformation: unique signal transduction pathways and implications for the design of new chemotherapeutic agents. Cancer Investigation 18: 39-50.
- Adler V, Pincus MR, Brandt-Rauf PW, Ronai Z (1995) Complexes of p21RAS with JUN N-terminal kinase and JUN proteins. Proc Natl Acad Sci USA 92: 10585-10589. [Crossref]
- Adler V, Pincus MR, Polotskaya A, Montano X, Friedman FK, et al. (1996) Activation of c-Jun-NH2-kinase by UV irradiation is dependent on p21ras. J Biol Chem 271: 23304-23309. [Crossref]
- Bowne WB, Michl J, Bluth MH, Zenilman ME, Pincus MR (2007) Novel peptides from the RAS-p21 and p53 proteins for the treatment of cancer. Cancer Ther 5B: 331-344. [Crossref]
- Adler V, Bowne W, Kamran I, Michl J, Friedman FK, et al. (2008) Two peptides derived from ras-p21 induce either phenotypic reversion or tumor cell necrosis of ras-transformed human cancer cells. Cancer Chemother Pharmacol 62: 491-498. [Crossref]
- Wang, Fang G, Rudolph J (2012) Ras inhibition via direct Ras binding--is there a path forward? Bioorg Med Chem Lett 22: 5766-5776. [Crossref]
- Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, et al. (2013) Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 497: 638-642. [Crossref]
- Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, et al. (2012) The GDI- like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nature Cell Biology 14: 148-158.
- Gonzalez-Perez V, Reiner DJ, Alan JK, Mitchell C, Edwards LJ, Khazak V, et al. (2010) Genetic and functional characterization of putative Ras/Raf interaction inhibitors in C. elegans and mammalian cells. Journal of Molecular Signalling 5: 2.
- Spoerner M, Graf T, Konig B, Kalbitzer HR (2005) A novel mechanism for the modulation of the Ras-effector interaction by small molecules. Biochemical and Biophysical Research Communications 334: 709-713.
- Spoerner M, Nuehs A, Ganser P, Herrmann C, Wittinghofer A, Kalbitzer HR (2005) Conformational states of Ras complexed with the GTP analogue GppNHp or GppCH2p: implications for the interaction with effector proteins. Biochemistry 44: 2225-2236.
- Rosnizeck IC, Spoerner M, Harsch T, Kreitner S, Filchtinski D, et al. (2012) Metal-bis(2-picolyl)amine complexes as state 1(T) inhibitors of activated Ras protein. Angew Chem Int Ed Engl 51: 10647-10651. [Crossref]
- Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, et al (2013) In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci USA 110: 8182-8187.
- Dubois F, Keller M, Calvayrac O, Soncin F, Hoa L, Hergovich A, et al. (2016) RASSF1A Suppresses the Invasion and Metastatic Potential of Human Non-Small Cell Lung Cancer Cells by Inhibiting YAP Activation through the GEF-H1/RhoB Pathway. Cancer Research 76: 1627-1640.
- Frank R, Overwin H (1996) SPOT synthesis. Epitope analysis with arrays of synthetic peptides prepared on cellulose membranes. Methods in Molecular Biology 66: 149-169.
- Gausepohl H, Boulin C, Kraft M, Frank RW (1992) Automated multiple peptide synthesis. Pept Res 5: 315-320. [Crossref]