IgE class switch recombination (CSR) is under the tight control to maintain the low concentration of serum IgE. IL-4 and CD40 signal induces IgE CSR. NF-κB classical pathway and STAT6 was indicated in this pathway. In this study, we prepared a M12 cell line expressing TRAF3 in an inducible manner. This cell line showed impaired signal transduction of NF-ΚB alternative pathway and diminished expression of epsilon germ line transcription (ɛGLT), thus indicating NF-ΚB alternative pathway was involved in IgE CSR. CaMKII was activated after IL-4 and anti-CD40 Ab stimulation, and KN-93, a CaMKII inhibitor, inhibited both activation of NF-ΚB alternative pathway and IgE CSR. NF-ΚB alternative pathway is characterized by the NIK activation after TRAF3 degradation through ubiquitination. This ubiquitination of TRAF3 was enhanced by CaMKII and alpha4 in HEK293T cells. Alpha4 was phosphorylated by CaMKII, and alpha4 associated with TRAF3. CaMKII also regulated IgE CSR in normal splenic B cells induced by IL-4 and anti-CD40 Ab stimulation.
IgE, CSR, CaMKII, NF-kB, TRAF3, ubiquitination, alpha4
B cells develop in the bone marrow to produce IgM class antibody by the assembly of V, D and J region of Ig gene independently of the stimulation by antigens [1,2]. When mature B cells encounter with antigens in the peripheral tissue, they may produce different class of antibody with the same antigen specificity by class switch recombination (CSR) to acquire different types of effector functions [3,4]. CSR requires both CD40 signaling and stimulation by cytokines [5]. CD40 ligation is required in all CSR events and induces the activation of AID. AID acts on the single stranded DNA produced by the transcription of switch region [3,6]. Cytokines induce the germ line transcription (GLT) of switch region, thus determining the specificity of the class switch. IgE class antibody is related to the development of allergy and thus is under the control of tight regulation [7]. IL-4/IL-13 stimulation induce the ɛGLT and thereby IgE CSR [8,9]. Iɛ promoter region has STAT6 and NF-kB sites and it was reported that STAT6 and NF-kB classical pathway played an important role in the regulation of IgE CSR [10].
NF-kB is a group of transcription factors including NF-kB1 (p105/p50), NF-kB2 (p100/p52), RelA (p65), RelB and c-Rel [11,12]. They reside in cytoplasm as inactive forms and translocate to nucleus when cells are activated. Various stimulations induce the processing of p105 to p50 and p100 to p52 [13]. This activation of NF-kB is mediated by classical and alternative pathways. Classical pathway is activated in many cells by various stimulations. On the other hand, alternative pathway is mainly activated in B cells by ligation of a limited number of receptors including TNF receptor family members such as CD40 and BAFFR [14,15]. The main transcription factors involved in alternative pathway are RelB and p52 of p100 [11]. The degradation of p100 to p52 is mediated by NIK [11]. NIK phosphorylates and activates IKKα, and then IKKα phosphorylates p100 to induce polyubiquitination and subsequent processing of p100 by proteasome. The amount of NIK protein is low in the non-stimulated cells, because E3 ligases cIAP1/2 ubiquitinate NIK for degradation with the help of TRAF3 [11,15,16]. When receptor is ligated by its ligand, cIAP1/2 ubiquitinate and degrade TRAF3 instead, by assembling to the receptor. TRAF3 belongs to TRAF family and regulates the NF-kB alternative pathway negatively [17,18]. TRAF3 associates with CD40, and the mutant form of CD40 that cannot bind to TRAF3 failed to induce IgE CSR [19].
Alpha4 was originally identified as an associated molecule of B cell receptor (BCR) complex [20]. Later studies demonstrated that alpha4 was ubiquitously expressed and was a regulator of serine/threonine phosphatases such as PP2A and PP6 [21,22]. Alpha4 is an essential molecule for cell survival and the deletion of alpha4 gene even in ES cells was lethal [23,24]. Conditional gene targeting of alpha4 in B cells revealed that alpha4 was involved not only in BCR signal transduction but also in many other signal pathways including LPS, CD40 and IL-4 receptor [25]. B cell specific alpha4 deficient mice showed decreased level of IgG subclasses upon immunization with T dependent Ag. The response of IgM class Ab was intact in the same mice, suggesting that CSR was impaired in alpha4 deficient mice [25]. Neuron specific gene targeting of alpha4 led to the finding that alpha4 regulated learning and memory by associating with CaMKIIα in the cytoplasm of neuron cells [26].
CaMKII is a multisubunit serine/threonine kinase which is activated by Ca2+ influx into cells [27,28]. CaMKII consists of four members (α, β, γ and δ) and each tissue cell in the body expresses at least one type of CaMKII [29]. CaMKIIα is mainly expressed in nervous system but some reports showed that CaMKIIa existed in hematopoietic cells [30]. CaMKIIα plays an important role in learning and memory by phosphorylating AMPA receptor and other substrates in the brain cells [27,28]. CaMKII in lymphocytes regulates the NF-kB pathway by phosphorylating CBM complex in the TCR signal transduction [31].
Here in this report, we demonstrate that IgE CSR is regulated by NF-kB alternative pathway. CaMKII positively regulated NF-kB alternative pathway by enhancing the polyubiquitination of TRAF3 in 293T cells. CaMKII also regulated IgE CSR in normal splenic B cells.
Cells and reagents
The murine B cell line M12 was described previously and the HEK293T (293T) cell line was a kind gift from Dr. Kaisho at Wakayama Medical University in Japan [47]. A CaMKII inhibitor KN-93 was purchased from Wako Pure Chemical Industries (Osaka, Japan). IL-4 was a kind gift from Dr. Nakanishi at Hyogo College of Medicine in Japan. Anti-p52 Ab (Cell signaling technology, Danvers, MA), anti-p65 Ab (Cell signaling), anti-HA Ab (Roche, Manheim, Germany), anti-FLAG Ab (Anti-DYKDDDDK Ab, Wako), anti-phospho-Thr Ab (Invitrogen, CA), anti-histone H3 Ab (Sigma chemical company, MO) and anti-TRAF3 Ab (Santa Cruz, Dallas, Texas) were purchased for Western blot studies. Anti-CD40 Ab [48] and anti-alpha4 Ab were prepared as described previously [20]. Splenic B cells were prepared using B cell isolation kit (Miltenyi Biotech, CA) from BALB/c mice as described previously [48]. Animal experiments were conducted according to guideline of Kumamoto University and were approved by the committee on animal experiments of Kumamoto University (permission number: A 27-071).
M12 cells were collected by centrifugation and loaded with Fluo-4 AM dye (5 mM) in serum-free medium containing Probenecid (1.25 mM) and Pluronic F-127 (0.04%) at 37°C for 60 min in the dark. After washing, the cells were resuspended in medium containing Probenecid (1.25 mM), and the fluorescence was measured in a fluorescence plate reader (MTP-800; Corona Electric, Ibaraki, Japan). In each sample, unstimulated fluorescence baseline was acquired for the first 60 s, and stimulated samples were subsequently analyzed for 10 min.
Preparation of cDNAs and transfection
TRAF6 cDNA was cloned by RT-PCR using the following primers: sense, GAATTCATGAGTCTGCTAAACTGTGA and antisense, GTCGACCTATACCCCTGCATCAGTAC.
TRAF2 cDNA and ubiquitin cDNAs were kind gift from Dr. Kawai at Nara institute of science and technology in Japan. CaMKIIα cDNA was described previously [49]. Kinase-dead mutant of CaMKIIα cDNA in which Lys42 was substituted with Arg was prepared with the PrimeSTARⓇ mutagenesis kit (TaKaRa Bio, Shiga, Japan) using the following primers: sense, GCTGCCAGGATTATCAACACCAAGAA and antisense, GATAATCCTGGCAGCATACTCCTGAC [34]. All the sequences were verified by DNA sequences with an ABI PrismⓇ 310 instrument (Applied Biosystems, CA). The plasmids were transfected with HilyMax transfection reagent (Dojindo, Kumamoto, Japan).
NF-kB luciferase assay
A reporter gene NF-kB luciferase (pNFkB-Luc) was purchased from Clontech (Palo Alto, CA). Luciferase assay was performed as described previously [50]. Briefly, 10 µg of the plasmid was transfected into M12 cells by electroporation at 950 µF, 276 V using gene pulserⓇ II (Bio-Rad, CA). Forty h after transfection, cells were stimulated with 10 U/ml of IL-4 and 10 µg/ml of anti-CD40 Ab. Cells were harvested after 4 h of stimulation and luciferase reaction was performed with PicaGene system (Wako Pure Chemicals).
In vitro phosphorylation assay
The phosphorylation of alpha4 by CaMKII was performed as previously described [49]. The preparation of GST-alpha4 fusion protein was described previously [22]. His-tagged alpha4 was prepared with the pBAD/His vector (Invitrogen) and fusion proteins were purified with Talon metal affinity resins (Clontech) according to the manufacturer’s protocol.
Cell fractionation
Cell fractionation was performed as previously described [49]. M12 cells were lysed in ice-cold 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2 and 0.5% NP-40. The cell lysates were mixed by vortexing at a low speed during 5-min-incubaton on ice. The cell lysates were then centrifuged at 5,000 rpm for 1 min at 4°C. The supernatant was harvested as cytoplasm. The pellet (nuclear fraction) was then resuspended in 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol, and 40% glycerol.
Inducible expression of TRAF3 via Tet-OffⓇsystem
The cDNA of murine TRAF3 in pEF-BOS was a kind gift from Dr. Kaisho at Wakayama Medical University in Japan. The fragment of TRAF3 was recovered by reverse transcription PCR using the following primers: GGATCCATGGACTACAAAGACGATGACGAC and GATATCTCAGGGGTCAGGCAGATCCGAGGT. The recovered cDNA was inserted into pTRE2pur vector. Establishment of M12 cells which inducibly produce TRAF3 (Tet-OffⓇ system, Clontech) was prepared as follows. M12 cells were transfected with firstly with pTet-OffⓇ vector and then with pTRE2pur-TRAF3 vector. M12-TRAF3 which inducibly expressed TRAF3 protein upon withdrawal of doxycycline (MP Biomedicals, CA) was selected after 2 rounds of limiting dilution.
Ubiquitination assay
In vivo ubiquitination assay was performed as previously reported [51]. 293T cells were transfected with 10 µg each of His-tagged ubiquitin and HA-tagged TRAF3 cDNA in 100 mm dish with or without various cDNAs and reagents as indicated in the figure legends. Transfected cells were lysed in 1 ml of buffer A (6 M guanidinium-HCl, 0.1 M Na2HPO4, NaH2PO4, pH 8.0, 10 mM imidazole) and the lysates were sonicated for 30 s on ice. Sonicated lysates were mixed on a rotator with 30 µl (packed volume) of His60 Ni Superflow Resin (Clontech) for 3 h at room temperature. The beads were centrifuged and any non-binding materials were removed from the beads by washing three times with buffer A, and then twice with buffer A diluted in 25 mM Tris-HCl (pH 6.8), 20 mM imidazole at 1:4, and finally twice with 25 mM Tris-HCl (pH 6.8), 20 mM imidazole. His-tagged proteins were recovered from the beads by boiling in SDS-sample buffer supplemented with 200 mM imidazole and they were subjected to Western blot analysis.
Real-Time PCR
Total RNA was extracted using RNA isolation kit (RNAiso plus, TaKaRa, Japan) according to the manufacturer’s recommendations. cDNA was synthesized from 2 µg of total RNA in 20 µl using Prime ScriptTM II 1st strand cDNA Synthesis Kit (TaKaRa) according to the manufacturer’s instructions. Quantitative Real-Time (RT) PCR of ɛGLT was performed using THUNDERBIRDTM SYBER GREENⓇ qPCR MIX (TOYOBO, Japan) with the following primers: sense, CACAGGGGGCAGAAGATG and antisense, AGGGGTAGAGCTGAGGGTTC [52]. The housekeeping gene GAPDH was evaluated as an internal positive control [53]. The primers for GAPDH were as follows: TGAAGCAGGCATCTGAGGG and CGAAGGTGGAAGAGTGGGAG. The RT PCR was performed employing AB 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA). PCRs were performed under following conditions: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C in 96-well optical reaction plates (Applied Biosystems). The specificity of amplicons was verified by melting curve analysis. The comparative cycle of threshold (ΔΔCT) method was used to demonstrate the relative mRNA levels of the ɛGLT.
Statistical analyses
Data in graphs are shown as means + SDs. The Student t-test was used to calculate the p values. The differences were considered to be significant at p value less than 0.05.
IgE class switch recombination (CSR) is positively regulated by NF-kB alternative pathway
To investigate the possible role of NF-kB alternative pathway in the regulation of IgE CSR, a murine B cell line which expressed TRAF3 in an inducible manner (M12-TRAF3) was established as described in the Methods section. The transfectant expressed excess amount of TRAF3 upon withdrawal of doxycycline as indicated in Figure 1A. Parental M12 cells and M12-TRAF3 cells were stimulated with IL-4 and anti-CD40 Ab to induce IgE CSR. IgE CSR was assessed by the induction of ɛGLT which preceded the IgE CSR [5, 6, 10]. Real-Time PCR was performed to detect the ɛGLT as indicated in the Methods section. IL-4 and anti-CD40 Ab stimulation induced ɛGLT in the parental M12 cells after 5 h of stimulation (30.1 + 7.9 fold) (Figure 1B). The same stimulation induced smaller increase of ɛGLT in the M12-TRAF3 cells before doxycycline withdrawal (8.1 + 2.6 fold) (Figure 1B). The response of M12-TRAF3 cells to IL-4 plus anti-CD40 Ab was further reduced after doxycycline withdrawal (6.8 + 1.1 fold) (Figure 1B). Overexpression of TRAF3 was reported to inhibit the NF-kB alternative pathway [32]. In Figure 1C, production and nuclear translocation of p52 was induced by anti-CD40 Ab stimulation to show that NF-kB alterative pathway was activated in M12 cells as previously reported [19]. In M12-TRAF3 cells, activation of NF-kB alternative pathway was reduced compared to parental M12 cells. Therefore, these results suggest that IgE CSR is positively regulated by NF-kB alternative pathway.
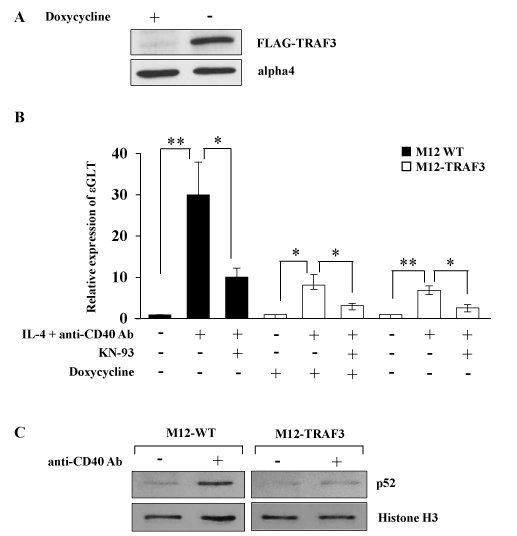
Figure 1. NF-kB alternative pathway is involved in the regulation of IgE CSR. (A) The lysates of M12-TRAF3 cells before and after 24 h-withdrawal of doxycycline were analyzed by Western blot with anti-FLAG Ab. Data are representative of three independent experiments. (B) Real-Time PCR was performed on M12 or M12-TRAF3 cells to detect ɛGLT. Cells were treated with IL-4 and anti-CD40 Ab for 5 h in the presence or absence of 10 µM of KN-93, a CaMKII inhibitor. Doxycycline was withdrawn in M12-TRAF3 cells for 24 h to induce the expression of excess amount of TRAF3. Mean + SD of the four independent experiments. (t-test; *p < 0.05 and **p < 0.01) (C) M12 or M12-TRAF3 cells were stimulated with anti-CD40 Ab for 5 h. Then the lysates were harvested and nuclear fraction was prepared. The samples were analyzed by Western blot with anti-p52 Ab. Membranes were reblotted with anti-histone H3 Ab to show the equal loading. Data are representative of three independent experiments.
CaMKII enhances the NF-kB alternative pathway in the regulation of IgE CSR
To elucidate the regulatory mechanism of NF-kB alternative pathway in IgE CSR, a variety of inhibitors for both kinases and phosphatases were tested whether they had any effect on the IgE CSR by IL-4 plus anti-CD40 Ab stimulation (data not shown). We found that KN-93, a specific inhibitor of CaMKII inhibited the ɛGLT induction in M12-TRAF3 cells as well as in parental M12 cells as shown in Figure 1B. To further confirm the involvement of NF-kB alternative pathway and CaMKII in the regulation of IgE CSR, the following experiments were performed. Firstly, a reporter gene NF-kB luciferase was transiently introduced into M12 cells and the transfected cells were stimulated with anti-CD40 Ab. As shown in Figure 2A, this stimulation induced NF-kB luciferase activity as reported previously [33]. The addition of KN-93 inhibited the luciferase activity induced by CD40 stimulation (Figure 2A). The reduction by KN-93 was modest probably because only alternative pathway was blocked while classical pathway was not affected by KN-93. Nuclear translocation of p65 and p52 was examined after IL-4 and CD40 stimulation to detect the activation of classical and alternative NF-kB pathway respectively. As indicated in Figure 2B, translocation of both p52 and p65 was induced after IL-4 and CD40 stimulation in M12 cells, but only the translocation of p52 was inhibited by KN-93 treatment (Figure 2B). Next, we investigated whether CaMKII was activated by IL-4 and anti-CD40 Ab stimulation in M12 cells. Previously, a signal transduction molecule alpha4 was shown to associate with the CaMKII in the central nervous system [26]. Therefore, we investigated whether alpha4 was a substrate of CaMKII in an in vitro phosphorylation assay. CaMKII phosphorylated both GST-tagged and His-tagged alpha4 produced in E. coli as fusion proteins (Figure 2C). Using this alpha4 as a substrate, the activation of CaMKII was monitored in M12 cells. Thirty minutes of stimulation with IL-4 plus anti-CD40 Ab induced the threonine phosphorylation of alpha4 (Figure 2D). KN-93 treatment inhibited this threonine phosphorylation of alpha4 (Figure 2D). Furthermore, Ca2+ influx was induced in M12 cells by IL-4 and anti-CD40 Ab stimulation (Figure 2E) corroborating the activation of CaMKII in these cells.
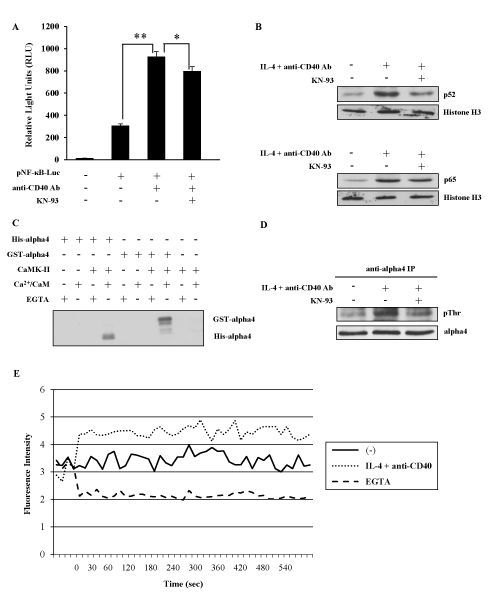
Figure 2. CaMKII positively regulates NF-kB alternative pathway. (A) After transfection of pNF-kB-Luc plasmid, M12 cells were stimulated with anti-CD40 Ab in the presence or absence of KN-93. The light intensity was measured and the intensity of untreated cells was defined as 1. Mean + SD of the three independent experiments. (t-test; *p < 0.05 and **p < 0.01) (B) M12 cells were stimulated with IL-4 and anti-CD40 Ab in the presence or absence of KN-93. Nuclear fraction was analyzed by Western blot with anti-p52 or anti-p65 Ab to detect the activation of NF-kB alternative and classical pathway respectively. Anti-histone H3 Ab was employed to show the equal loading. Data are representative of three independent experiments. (C) In vitro phosphorylation of GST-alpha4 and His-alpha4 fusion proteins by CaMKII in the presence of Ca2+/CaM. Data are representative of two independent experiments. (D) M12 cells were stimulated with IL-4 and anti-CD40 Ab in the presence or absence of KN-93. The lysates were immunoprecipitated by anti-alpha4 Ab and blotted with anti-phosphothreonine Ab. Data are representative of three independent experiments. (E) M12 cells were stimulated with IL-4 and anti-CD40 Ab with or without EGTA. Ca2+ influx was monitored by Fluo-4 AM dye. Data are representative of three independent experiments. GST; glutathione S-transferase. CaM; calmodulin. EGTA; ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid.
CaMKII enhances the polyubiquitination of TRAF3
2021 Copyright OAT. All rights reserv
NF-kB alternative pathway is characterized by the accumulation and activation of NIK, which is induced by the degradation of TRAF3 [11]. To investigate whether CaMKII enhanced the NF-kB alternative pathway by regulating the degradation of TRAF3, polyubiquitination of TRAF3 was analyzed in the transient transfection system of 293T cells as described in the Methods section. Co-transfection of the cDNAs of His-tagged ubiquitin and HA-tagged TRAF3 resulted in the polyubiquitination of TRAF3 as shown in Figure 3A (lane 2 vs. lane 1). Co-transfection of the cDNA of CaMKII enhanced the polyubiquitination of TRAF3 (Figure 3A, lane 4 vs. lane 2). Ten microgram of the cDNA of CaMKII enhanced polyubiquitination maximally and even 1.25 microgram of the cDNA was apparently effective (Figure 3B). To further confirm that CaMKII has a positive regulatory role in the degradation of TRAF3, the effect of a CaMKII inhibitor KN-93 was tested in this system. Treatment of the transfectant with KN-93 indeed inhibited the polyubiquitination of TRAF3 induced in 293T cells (Figure 3C). Kinase dead form of CaMKII cDNA was prepared according to the previous report [34] and was transfected into 293T cells together with the cDNAs of ubiquitin and TRAF3. Kinase dead form of CaMKII had no effect on the polyubiquitination of TRAF3 indicating that enzymatic activity of CaMKII was necessary for the regulation of polyubiquitination of TRAF3 (Figure 3D).
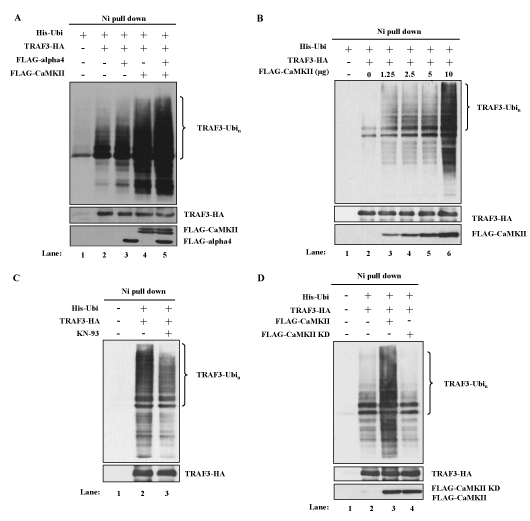
Figure 3. CaMKII and alpha4 enhance the ubiquitination of TRAF3. (A) Effects of overexpression of CaMKII and alpha4 on the ubiquitination of TRAF3. 293T cells were transfected with indicated expression plasmids. MG132 was added for the last 6 h of culture. Cells were lysed under denaturing condition and the lysates were pulled down by Ni Superflow Resin. The purified material was analyzed by Western blot using anti-TRAF3 Ab. Expression level of each transfected protein was monitored by Western blot analysis. Data are representative of four independent experiments. (B) Dose dependent enhancement of ubiquitination of TRAF3 by CaMKII. 293T cells were transfected with fixed amount of ubiquitin and TRAF3 plasmids and increasing amounts of CaMKII plasmid. Ubiquitination was analyzed as in (A). Data are representative of three independent experiments. (C) Effect of KN-93 on the ubiquitination of TRAF3. Cells were transfected with indicated plasmids in the presence or absence of KN-93. Ubiquitination was analyzed as in (A). Data are representative of three independent experiments. (D) Cells were transfected with wild type or mutant CaMKII with indicated plasmids and ubiquitination was analyzed as in (A). Data are representative of three independent experiments.
Alpha4 associates with TRAF3 and regulates its polyubiquitination
B cell specific alpha4 deficient mice showed impaired CSR to IgG1 suggesting that alpha4 might be involved in CSR [25]. We also demonstrated that CaMKII associated with alpha4 in central nervous system (CNS) [26]. Therefore, the effect of the overexpression of alpha4 was tested on the polyubiquitination of TRAF3. Addition of the cDNA of alpha4 further enhanced the polyubiquitination induced by the overexpression of CaMKII (Figure 3A, lane 5 vs. lane 4). However, the effect of transfection of the cDNA of alpha4 alone was much smaller than that of CaMKII (Figure 3A, lane 3 vs. lane 2 and Figure 4A). Because alpha4 is a ubiquitously expressed molecule, endogenous expression of alpha4 may have reduced the effect of overexpression of exogenous alpha4 [25]. We previously described a dominant negative (DN) form of alpha4 which could effectively inhibit the function of endogenous alpha4 [35]. Therefore, we transfected this dominant negative form of alpha4 cDNA together with CaMKII. As demonstrated in Figure 4B, DN form of alpha4 decreased the polyubiquitination of TRAF3 enhanced by CaMKII. We investigated whether alpha4 associated with TRAF3 by co-immunoprecipitation method. Alpha4 associated specifically with TRAF3 when they were co-expressed in 293T cells (Figure 4C). Alpha4 did not associate with other TRAF member molecules such as TRAF2 and 6 (Figure 4C).
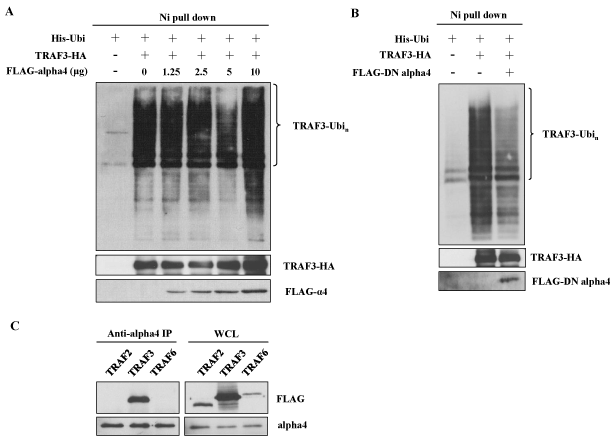
Figure 4. Alpha4 associates with TRAF3 to enhance its ubiquitination. (A) 293T cells were transfected with fixed amount of ubiquitin and TRAF3 plasmids and increasing amounts of alpha4 plasmid. Ubiquitination was analyzed as in Figure 3. Data are representative of three independent experiments. (B) Effect of dominant negative (DN) form of alpha4 on the ubiquitination of TRAF3. 293T cells were transfected with ubiquitin and TRAF3 in the presence or absence of DN-alpha4. Data are representative of three independent experiments. (C) Association of alpha4 with TRAF3. Alpha4 cDNA was transfected into 293T cells together with FLAG-tagged TRAF2, TRAF3 or TRAF6. The cell lysates were immunoprecipitated with anti-alpha4 and subjected to the analysis by Western blot using anti-FLAG Ab. Data are representative of two independent experiments.
IgE CSR is regulated by CaMKII in B cells
Because NF-B pathway is often regulated in a tissue and/or stimulation dependent manner [36], it is important to investigate whether CaMKII regulates NF-kB alternative pathway in a B cell line M12. IL-4 and anti-CD40 Ab stimulation induced TRAF3 degradation in M12 cells as previously reported (Figure 5A) [18]. Real-Time PCR was performed in M12 cells to investigate the effect of transient expression of CaMKII on ɛGLT (Figure 5B). Overexpression of CaMKII cDNA enhanced the ɛGLT. This result suggests that CaMKII enhanced the IgE CSR in M12 cells.
We next investigated whether the phenomena observed in M12 cells were applicable to normal mature B cells. Mature B cells were prepared from spleen of BALB/c mice. IL-4 and anti-CD40 Ab together with BCR crosslinking induced ɛGLT in mature B cells as indicated in Figure 5C. Addition of KN-93 inhibited the ɛGLT induced by these stimulations (Figure 5C).
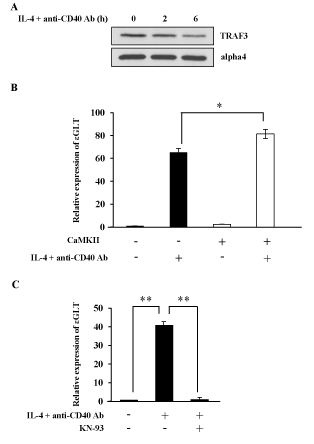
Figure 5. CaMKII regulates NF-kB alternative pathway in normal splenic B cells. (A) M12 cells were stimulated with IL-4 and anti-CD40 Ab for 4 h and the lysates were blotted with anti-TRAF3 Ab or anti-alpha4 Ab. Data are representative of three independent experiments. (B) M12 cells were transfected with CaMKII cDNA. After 40 h of transfection cells were stimulated with IL-4 and anti-CD40 Ab. mRNA was extracted from each sample and Real-Time PCR was performed to detect ɛGLT. The relative mRNA levels of the ɛGLT were expressed as a fold of the value of untreated cells. Mean + SD of the three independent experiments. (t-test; *p < 0.05) (C) Normal B cells were prepared from the spleen of BALB/c mice, and these B cells were stimulated with IL-4 and anti-CD40 Ab in the presence or absence of KN-93. Real-Time PCR was performed to monitor the expression level of ɛGLT. Mean + SD of the three independent experiments. (t-test; **p < 0.01) GLT; germ line transcript. KN-93; a CaMKII inhibitor.
We showed in this report that NF-kB alternative pathway was involved in the regulation of IgE CSR for the first time. CaMKII positively regulated IgE CSR by enhancing the polyubiquitination of TRAF3. Because TRAF3 is a negative regulator of NF-kB alternative pathway, overexpression of TRAF3 resulted in the decreased production of p52 (Figure 1C). In this transfectant, induction of ɛGLT was decreased to demonstrate that NF-kB alternative pathway was functional in IgE CSR. NF-kB pathway is activated by various stimuli in many cells. The classical pathway is activated in most cells while alternative pathway is activated in B cells by ligation of TNF receptor family members. Gene targeting experiments showed that classical pathway was involved in the regulation of CSR, but other papers have suggested that NF-kB alternative pathway was also involved [37]. Aly (alymphoplasia) mutant mice that have no lymph nodes in the body were shown to have a point mutation in the Nik gene which encodes NIK, a key molecule for NF-kB alterative pathway [38]. These mice did not have detectable level of IgA. Another mutant mice (Lym1) which had a point mutation in Nfkb2 also showed impaired IgA CSR [39]. This mutation resulted in the production of non-processible p100 so that NF-kB alternative pathway was compromised. Furthermore, TBK1 deficient B cells showed augmented IgA CSR induced by IL-4 and anti-CD40 Ab [40]. TBK phosphorylated NIK and induced the degradation of NIK to inhibit the NF-kB alternative pathway.
Ca2+ is a well-established second messenger in lymphocytes and CaMKII, in addition to calcineurin and PKC, has been implicated in the signal transduction pathway in lymphocytes [29]. CaMKII have four members (α, β, γ and γ) and it was reported that α and β forms are almost exclusively expressed in brain. However, every tissue expresses at least one form of CaMKII and lymphocytes also express CaMKII [41,42]. In the CNS, CaMKII was reported to function as an upstream signal transducer of NF-kB classical pathway, and in lymphocytes, CaMKII was activated by TCR stimulation to phosphorylate CARMA1, a component of NF-kB classical pathway [31,43]. In this paper, we showed that CaMKII was activated by IL-4 and anti-CD40 Ab to activate NF-kB alternative pathway.
To clearly demonstrate that enzymatic activity of CaMKII is necessary for the induction of polyubiquitination of TRAF3, we prepared the kinase dead (KD) mutant form of CaMKIIα according to a previous report [34]. This kinase dead mutant could interact with Ca2+/calmodulin and also with endogenous CaMKII molecules in the cell. Kinase dead mutant of CaMKIIα had no positive effect on the polyubiquitination of TRAF3 when transfected into 293T cells (Figure 3D). Transfection of KD mutant decreased the ubiquitination slightly, suggesting that this mutant form might function as a dominant negative form by forming a complex composed of wild type and mutant form.
Alpha4 was originally identified as a BCR signal transduction molecule [20]. Later experiments identified alpha4 as a phosphatase regulatory molecule [21,22]. Furthermore, alpha4 was reported to associate with E3 ligases and regulate their enzymatic activity [44-46]. Alpha4 may associate with the substrate, or E3 ligase or both molecules [44-46]. In this study, we showed that alpha4 associated with the substrate for ubiquitination, TRAF3. Only serine phosphorylation of alpha4 was reported by Dr. Brautigan’s group [21]. In our case, threonine was also phosphorylated. The discrepancy may be due to the different stimulation employed by us.
We thank Dr. Takashi Tanaka (RIKEN Center for Integrative Medical Sciences, Japan) for kind advice on ubiquitination assay.
Kano Tanabe was supported by Kumamoto Health Science University special fellowship grant number 2015-C-12.
The authors declare no commercial or financial conflict of interest.
- Bassing CH, Swat W, Alt FW (2002) The mechanism and regulation of chromosomal V(D)J recombination. Cell 109 Suppl: S45-55. [Crossref]
- Schatz DG, Ji Y (2011) Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol 11: 251-263. [Crossref]
- Chaudhuri J, Alt FW (2004) Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol 4: 541-552. [Crossref]
- Stavnezer J, Schrader CE (2014) IgH chain class switch recombination: mechanism and regulation. J Immunol 193: 5370-5378. [Crossref]
- Purkerson J, Isakson P (1992) A two-signal model for regulation of immunoglobulin isotype switching. FASEB J 6: 3245-3252. [Crossref]
- Stavnezer J, Guikema JE, Schrader CE (2008) Mechanism and regulation of class switch recombination. Annu Rev Immunol 26: 261-292. [Crossref]
- Pate MB, Smith JK, Chi DS, Krishnaswamy G (2010) Regulation and dysregulation of immunoglobulin E: a molecular and clinical perspective. Clin Mol Allergy 8: 3. [Crossref]
- Punnonen J, Yssel H, de Vries JE (1997) The relative contribution of IL-4 and IL-13 to human IgE synthesis induced by activated CD4+ or CD8+ T cells. J Allergy Clin Immunol 100: 792-801. [Crossref]
- Geha RS, Jabara HH, Brodeur SR (2003) The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol 3: 721-732. [Crossref]
- Bacharier LB, Geha RS (2000) Molecular mechanisms of IgE regulation. J Allergy Clin Immunol 105: S547-558. [Crossref]
- Vallabhapurapu S, Karin M (2009) Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 27: 693-733. [Crossref]
- Hayden MS, Ghosh S (2012) NF-kB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26: 203-234. [Crossref]