Abstract
Immunomodulatory drugs (IMiDs), today also known as cereblon (CRBN) binding drugs, are therapeutically important anti-cancer and anti-inflammatory drugs. IMiDs are analogs of their prototype compound thalidomide. IMiDs have immune-modulation, anti-angiogenic, anti-inflammatory and anti-proliferative effects. CRBN is a component and substrate receptor of the Cullin 4 Ring E3 Ubiquitin Protein Ligase complex (CRL4). CRL4 consists of Cullin 4, RING finger protein (Roc1), and DNA damage binding protein 1 (DDB1). CRBN binds to its substrate proteins and it leads to ubiquitination of these substrates by the CRL4. CRBN is also involved in IMiDs-mediated T-cell co-stimulation and cytokine production. CRBN is a primary target of thalidomide teratogenicity. The binding of IMiDs to CRBN is associated with cytotoxicity of IMiDs and is used to treat multiple myeloma (MM), myelodysplastic syndromes (MDS), lymphomas and chronic lymphocytic leukemia. CRBN is composed of an N-terminal ATP-dependent serine protease Lon-like domain, which links to the E3 ubiquitin protein ligase complex CRL4, and a C-terminal domain, which binds IMiDs. CRBN binding is mediated by a glutarimide ring in thalidomide, lenalidomide, pomalidomide, CC-122, CC-220, CC-885 and CC-90009. Development of effector molecules mediating targeted ubiquitination of disease related proteins through cereblon is a new important way in pharmacology.
Key words
immunomodulatory drugs, cereblon, cullin 4 E3 ubiquitin protein ligase, Ikaros transcription factors famil, casein kinase 1A1, translation termination factor GSPT1, ubiquitination, proteasome
Introduction
IMiDs include thalidomide, lenalidomide, pomalidomide (Figure 1), CC-122, CC-220, CC-885 and CC-90009 till now. Both, lenalidomide (Revlimid®, initially known as CC-5013), and pomalidomide (Pomalyst®, initially known as CC-4047, Actimid) are a synthetic derivative of thalidomide (Thalomid®, Inmunoprin, Talidex, Talizer).
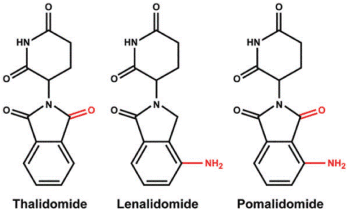
Figure 1. Chemical structures of immunomodulatory drugs (IMiDs) including thalidomide, lenalidomide and pomalidomide. Lenalidomide and pomalidomide are synthetic compounds derived by modifying the chemical structure of thalidomide.
Thalidomide [(RS)-2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione] was synthesized in Germany, in 1954, from α-phtaloylisoglutamine, to be used as a non-barbiturate sedative and antimetic drug (Contergan). In 1957, after a short period of preclinical studies, thalidomide was approved for first trimester gestational sickness in humans. The appearance of malformations such as phocomelia in the newborn banned its use three years later. Thalidomide was responsible for birth defects in more than 10,000 children [1,2]. The US Food and Drug Administration (FDA) approved thalidomide in 1998 for the treatment of erythema nodosum leprosum [3]. Thalidomide exhibits potent antiangiogenic and immunomodulatory effects, and is currently used around the world to treat a range of conditions, mainly multiple myeloma. However, long-term use of thalidomide has detrimental side effects, such as peripheral neuropathy [4,5].
Lenalidomide was developed in order to avoid thalidomide side effects (sedation and neuropathy), and to increase efficacy [6,7]. Lenalidomide shares a number of structural and biological properties with thalidomide but is safer and more potent than thalidomide Lenalidomide [3-(4-amino-1-oxo1,3-dihydro-2H-isoindol-2-yl) piperidine-2,6-dione] is 4-amino-glutarimide analog of thalidomide with potent immunomodulatory, antiangiogenic and direct neoplastic cell inhibitory activity [6-26].
Pomalidomide [4-Amino-2-(2,6-dioxopiperidin-3-yl) isoindole-1,3-dione] is a potent second-generation IMiD [27-34]. Pomalidomide has direct antiproliferative, pro-apoptotic, and antiangiogenic effects, as well as modulatory effects on bone resorption and on the immune system. Chemical structure of the IMiD drugs is shown in Figure 1.
CC-122 hydrochloride is a novel immunomodulatory agent-like thalidomide analog which directly binds to CRBN and promotes ubiquitination and degradation of zinc finger transcription factors Aiolos and Ikaros in difuse large B cell-lymphoma (DLBCL) [35-39]. CC-220 is a further IMiD that binds to CRBN and is currently in phase 1b/2aclinical trials for the treatment of relapsed and refractory multiple myeloma (MM) and in phase 2 clinical trials for systemic lupus erythematosus (SLE) [40]. CC-220 binds tighter to the CRBN in the complex of CRL4 E3 ubiquitin protein ligase than lenalidomide and causes more potent degradation of transcription factors Aiolos and Ikaros. CC-885 binds also to CRL4CRBN but mediates not only degradation of Aiolos but also specifically binds to the eukaryotic translation termination factor 3a (eRF3a), also known as GSPT1 (G1 to S phase transition 1) and mediates degradation of this protein [41,42]. GSPT1 together with the eukaryotic translation termination factor 1 (eRF1) functions in stop codon recognition and nascent protein release from ribosome in the process of mRNA translation. Lenalidomide and pomalidomide do not mediate the degradation of GSPT1. CC-90009 is a further IMiD that binds the E3 protein ubiquitin ligase CRL4CRBN and mediates degradation of certain lymphoid transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. In addition, this downregulates the expression of other proteins, including interferon regulatory factor 4 (IRF4) and c-myc, which plays a key role in the proliferation of certain cancer cell types. CC-90009 is now in phase 1 clinical trials (NCT 02848001) in subjects with relapsed or refractory acute myeloid leukemia (AML) in Canada and USA.
Mechanism of immunomodulatory drugs action in the treatment of multiple myeloma
IMiDs target both MM cells and their microenvironment, while also modulating the immune system. The exact molecular mechanisms of the antitumor effects of IMiDs remain uncertain. IMiDs affect various molecular and cellular elements within the tumor microenvironment. IMiDS change the concentration of various cytokines that support tumor cell growth [8-10,27,43,44]. IMiDs disrupt bone marrow stromal support for malignant MM cells, although the exact mechanisms of these actions remain unclear. IMiDs decrease the expression of adhesion molecules that facilitate the interaction between MM cells and bone marrow stromal cells (BMSCs) [45]. Importantly, the downregulation of MM cells adherence to BMSCs can overcome the cellular adhesion-mediated drug resistence by malignant MM cells. This effect of IMiDs is further increased by their ability to downregulate TNFα [9,43,46,47]. Surprisingly, in contrast to the inhibitory effect of lenalidomide in BMSCs, lenalidomide was totally ineffective in inhibiting TNFα mRNA expression in MM cells [44].
Immunomodulatory activities of IMiDs
IMiDs are a potent co-stimulator of primary human T cells, synergizing with stimulation via T-cell receptor complex to increase IL-2-mediated T-cell proliferation and interferon gamma (IFN-γ) production [12-14,48-50]. Secretion of IL-2 and IFN-γ increases the number of natural killer (NK) cells, improves their function and mediates lysis of MM cells (Figure 2). NKT cells are a heterogeneous group of T-cells that recognize lipids and glycolipids presented by CD1d molecules. NKT cells activated in presence of lenalidomide, have greater ability to secrete IFN-γ. Lenalidomide enhances antigen-specific expansion of NKT cells [51]. Regulatory T-cells (Tregs) are a component of the immune system that suppresses immune response of other cells. Tregs were elevated in MM patients. IMiDs strongly inhibits Tregs proliferation via decreased FOXP3 mRNA expression [52].
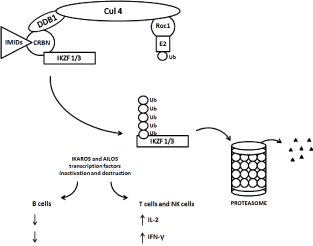
Figure 2. Schematic diagram of ubiquitination and degradation of zinc finger lymphoid transcription factors Ikaros and Aiolos by CRL4cereblon E3 ubiquitin ligase and proteasomes. Binding of cereblon (CRBN) by lenalidomide induces ubiquitination (marking IKZF1 / Ikaros / and IKZF3 / Aiolos /) and degradation of both, Ikaros and Aiolos transcription factors. CRBN functions as a substrate recognition component (substrate receptor) of this E3 ubiquitin ligase enzyme complex. CRL4cereblon complex consists of cullin 4A, RING finger protein regulator of cullins (Roc1), and DNA damage binding protein 1 (DDB1).
Anti-angiogenic properties of IMiDs
All IMiDs have anti-angiogenic activity. Thalidomide has predominant anti-angiogenic activity while lenalidomide and pomalidomide have far greater immune enhancing effects [43]. It appears that anti-angiogenesis occurs via the modulation of chemotactic factors involved in endothelial cells migration including TNFα, vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) from BMSCs rather than a direct inhibition of endothelial cells proliferation [53,54]. Inhibition of VEGF and bFGF production by IMiDs is likely to have multiple other biological effects beyond that of anti-angiogenesis, including inhibition of IL-6 production by BMSC. VEGF and bFGF up-regulate IL-6 and other pro-inflammatory cytokines [55].
Inhibition of cell cycle and induction of apoptosis by IMiDs
IMiDs induce cell cycle arrest via upregulation of tumor suppressor genes (cyclin dependent kinase inhibitors p15INK4b, p16INK4a, p21Cip1,Waf1, p27Kip1; early response transcription factors / Erg1, Erg2 and Erg3/) and apoptosis by caspase activation [56,57]. The activity of the transcription factor NF-κB inhibition by IMiDs resulted in reduced expression of anti-apoptotic proteins including cellular inhibitor of apoptosis protein 2 (cIAP2) [58] and FLIP [Fas-associated protein with death protein (FADD)-like interleukin-1β-converting enzyme (FLICE) inhibitor protein] [59]. These anti-apoptotic proteins inhibit caspase-8, that is on the other hand stimulated by IMiDs [57].
Disruption of bone marrow stromal support for malignant MM cells
IMiDs down-regulate the expression of adhesion molecules [leukocyte function-associated antigen 1 (LFA-1, CD11a), intercellular adhesion molecule 1 (ICAM-1, CD54), vascular cell adhesion molecule 1 (VCAM-1, CD106) and very late antigen 4 (VLA-4)] that facilitate the interaction between MM cells and BMSC. As we described, IMiDs inhibit NF-κB, a transription factor that has important growth and anti-apoptotic roles and which is connected with the upregulation of intracellular adhesion molecules and many cytokines [60].
IMiDs effect on myeloma cell proliferation
The direct anti-MM effect of IMiDs was shown to occur through the induction of a G1 phase of cell cycle growth arrest of MM cells [54] and was associated with a decrease in interferon regulatory factor 4 (IRF4), a transcription factor that is critical for MM cell growth and survival, (Figure 2), [61,62].
Raje, et al. [63] showed strong synergism of anti-MM activity of rapamycin (Rapamune), a specific mTOR inhibitor, combined with CC-5013. Importantly, this combination was able to overcome drug resistance when tested against MM cell lines resistant to conventional chemotherapy. Moreover, the combination, but not rapamycin alone, was able to overcome the growth advantage conferred on MM cells by interleukin-6 (IL-6), insulin-like growth factor-1 (IGF-1), or adherence to bone marrow stromal cells (BMSCs). Combining rapamycin and CC-5013 induced apoptosis of MM cells. Differential signaling cascades, including the mitogen-activated protein kinase (MAPK) and the phosphatidylinositol 3'-kinase / Akt kinase (PI3K /Akt) pathways [64-67], were targeted by these drugs individually and in combination, suggesting the molecular mechanism by which they interfere with MM growth and survival. These studies, therefore, provide the framework for clinical evaluation of mTOR inhibitors combined with IMiDs to improve patient outcome in MM.
IMiDs down-regulate CCAAT/enhancer-binding protein-β (C/EBPβ) resulting in abrogation of cell proliferation [68]. Overexpression of C/EBPβ rescued MM cells from IMiD-induced inhibition of proliferation, indicating that C/EBPβ is critical in mediating antiproliferative effects. IMiD-induced decrease of C/EBPβ protein led to impaired transcription of interferon regulatory factor 4 (IRF4).
Down-regulation of IRF4 by lenalidomide was confirmed by longitudinal studies of bone marrow samples from 23 patients obtained before and during lenalidomide treatment using CD138⁺/IRF4⁺ double labeling. In contrast to down-regulation of C/EBPβ protein, IMiD compounds did not alter C/EBPβ mRNA levels or protein stability, suggesting translational regulation of C/EBPβ. We could demonstrate that C/EBPβ protein expression is under eIF4E-translational control in MM. Furthermore, inhibition of the eIF4E-C/EBPβ axis by IMiD compounds was not observed in IMiD-resistant MM cells.
However, targeting translation at a different level by inhibiting eukaryotic translation initiation factor 4E-binding protein 1 phosphorylation overcame resistance, suggesting that this pathway is critical and might be a target to overcome drug resistance. MM cell lines and primary MM cells strongly expressed C/EBPβ, whereas normal B cells and plasma cells had little or no detectable levels of C/EBPβ [69]. Silencing of C/EBPβ led to down-regulation of transcription factors such as IRF4, XBP1, and BLIMP1 accompanied by a strong inhibition of proliferation. Further, silencing of C/EBPβ led to a complete down-regulation of antiapoptotic B-cell lymphoma 2 (BCL2) expression. In chromatin immunoprecipitation assays, C/EBPβ directly bound to the promoter region of IRF4, BLIMP1, and BCL2. C/EBPβ is involved in the regulatory network of transcription factors that are critical for plasma cell differentiation and survival. Targeting C/EBPβ may provide a novel therapeutic strategy in the treatment of multiple myeloma.
Exposure plasma cells to lenalidomide activated the Wnt/β-catenin pathway and its downstream targets such as cyclin D1 and MYC [70]. The accumulation of β-catenin during treatment with lenalidomide might be cause of drug resistance [70]. N-cadherin-based interaction between MM cells and osteoblasts block MM cell growth. Therefore, the high levels of N-cadherin expression in osteoblasts confers strong proliferation block on MM cells. Since β-catenin associates with N-cadherin at the cell membrane, N-cadherin adhesion is disrupted, β-catenin is released and will translocate to the nucleus leading to the transcription of target genes, and ultimately, cell proliferation [71]. Resistance of myeloma to lenalidomide is an emerging clinical problem, and though it has been associated in part with activation of Wnt/β-catenin signaling, the mediators of this phenotype remained undefined. Lenalidomide-resistant models were found to overexpress the hyaluronan (HA)-binding protein CD44, a downstream Wnt/β-catenin transcriptional target [72]. Consistent with a role of CD44 in cell adhesion-mediated drug resistance (CAM-DR), lenalidomide-resistant myeloma cells were more adhesive to bone marrow stroma and HA-coated plates. Blockade of CD44 with monoclonal antibodies, free HA or CD44 knockdown reduced adhesion and sensitized to lenalidomide. Wnt/β-catenin inhibition by FH535, a compound that suppresses both Wnt/β-catenin and peroxisome proliferator-activated receptor (PPAR) signaling, enhanced the activity of lenalidomide, as did interleukin-6 neutralization with siltuximab.
Cereblon as the direct target protein of IMiDs
Ito, et al. [73-76] developed a new affinity bead technology for isolating ligand-binding proteins. Polymer-coated beads were constructed that allow single-step purification of ligand target molecules. These beads include styrene-glycidyl-methacrylate (SG) beads and ferrite-glycidyl-methacrylate (FG) beads. FG beads were used for the purification of thalidomide-binding proteins from various cell extracts. Thalidomide-modified beads were incubated with cell extracts and then washed with buffer. Bound proteins were eluted with free thalidomide and analyzed by gel electrophoresis. Only two specific protein bands were detected (55 kDa and 127 kDa). These proteins were identified as CRBN and DDB1. As we described in Abstract and Introduction, CRBN, like DDB1, is a component of the cullin 4 ring E3 ubiquitin ligase complex (CRL4). Auto-ubiquitination of CRBN was inhibited by thalidomide in vitro, suggesting that thalidomide is an inhibitor of E3 ubiquitin ligase [77].
Cereblon as a primary target for thalidomide teratogenicity
The zebrafish was adopted as a model animal for in vivo study of thalidomide teratogenicity [73-76]. Zebrafish have a protein zCrbn, which is 70% homologous to human CRBN and possesses thalidomide binding activity. Thalidomide exposure induces fin and ear (otic vesicle) defects in zebrafish. A reduction in in the size of otic vesicle was observed in zebrafish following thalidomide exposure. Zebrafish fins and tetrapod limbs are homologous with respect to early patterning and gene expression, although skeletal structures in adult fish and tetrapods are quite different. Shortening of the pectoral fins along the proximo-distal axis and the inhibition of fibroblast growth factor 8 (Fgf8) gene expression were found in thalidomide-treated zebrafish embryos. Finally, chicks were employed for the investigation of the conserved role of CRBN. Down-regulation of Fgf8 and Fgf10 cause multiple birth defects and limb deformities.
The role of Ikaros family proteins in IMiDs and CRBN mechanism
Using distinct but complementary proteomic techniques and systems, three groups have recently simultaneously reported that IMiDs induced the CRBN-dependent proteasomal degradation of IKZF1 (Ikaros) and IKZF3 (Aiolos) [50,78,79]. Schema is shown in Figure 2. IKZF1, a zinc finger transcription factor initially discovered as a regulator of the T cell receptor, is required for hematopoiesis, particularly lymphocyte development and plasma cell maturation. Loss of function mutations of IKZF1 and IKZF3 are associated with acute lymphoblastic leukemia, consistent with a tumor suppressor function. On the other hand, IKZF1 and IKZF3 are required for the viability of many MM cell lines. IKZF1 and IKZF3 are also involved in the complex process of chromatin remodeling, and the nature of their interactions is poorly understood.
IKZF1 bound and activated the IRF4 gene promoter and loss of IKZF1 led to decreased IRF4 and MYC expression. However, lenalidomide could also inhibit MM cell lines with high basal levels of IRF4 unchanged by drug treatment, suggesting that other IKZF1/3 targets can play a role in the therapeutic response to IMiDs. IKZF1/3 are known repressors of IL-2 gene promoter. The degradation of IKZF1/3 in response to IMiDs explains enhanced T cell IL-2 production. Hence, many of the effects of IMiDs can be explained by a unified mechanism: IMiDs re-target the cullin 4 ring E3 ubiquitin ligase activity toward IKZF1/3 in a change-of-function effect (Figure 2).
An alternative CRL4CRBN substrate in the lenalidomide-responsive myelodysplastic syndrome with del(5q)
Krӧnke, et al [18] identified a novel target casein kinase1A1 (CSNK1A1) by quantitative proteomics in the myeloid cell line KG-1. CSNK1A1 is encoded in the del(5q) commonly deleted region and the gene is haploinsufficient. Lenalidomide treatment leads to increased ubiquitination of CSNK1A1 and decreased protein abundance (Figure 3). CSNK1A1 was shown as a therapeutic target in a murine model of AML [80,81] and in MDS with del(5q) [82,83]. CSNK1A1 negatively regulates β-catenin which drives stem cell self-renewal and CSNK1A1 haploinsufficiency causes the initial clonal expansion in patients with the del(5q) MDS and contributes to the pathogenesis of del(5q) MDS. Further inhibition of CSNK1A1 in del(5q) MDS is associated with del(5q) cells apoptosis and p53 activation. The inhibition of CSNK1A1 reduced RPS6 phosphorylation, induced p53 expression, and triggered myeloid differentiation program. TP53-null leukemia did not respond to CSNK1A1 inhibition, strongly supporting the importance of the p53 expression for the yield of CSNK1A1 inhibition. CSNK1A1 mutations have been recently found in 5-18% of MDS patients with del(5q) [82,84-87]. These mutations are associated similarly to the effect of TP53 mutations with rise to a poor prognosis in del(5q) MDS [87].
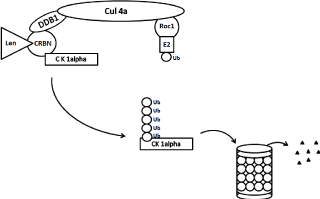
Figure 3. Schematic diagram of ubiquitination and degradation of casein kinase 1α by CRL4cereblon E3 ubiquitin ligase and proteasomes. Binding of cereblon (CRBN) by lenalidomide induces ubiquitination (marking CK1α) and degradation of casein kinase 1α. CRBN functions as a substrate recognition component (substrate receptor) of this E3 ubiquitin ligase enzyme complex. CRL4cereblon complex consists of cullin 4A, RING finger protein regulator of cullins (Roc1), and DNA damage binding protein 1 (DDB1).
While CSNK1A1 is CRL4CRBN target in del(5q) MDS, CRL4CRBN targets in lower risk non-del(5q) remain to be determined. The mechanism of action of lenalidomide is still unclear in non-del(5q) MDS cells.
Importance of valosin-containing protein/p97 for the degradation of all known CRL4CRBN targets
Valosin-containing protein (VCP)/p97 is ATPase which delivers ubiquitinated proteins for degradation in proteasomes. VCP is required for degradation of all known CRL4CRBN targets (IKZF1, IKZF3, casein kinase 1α, and the translation termination factor GSPT1) [88]. VCP promotes also degradation of glutamine synthetase (GS). GS is important for de novo synthesis of glutamine and functions tn detoxification of glutamate and ammonia. GS is associated with diseases including cancer, Alzheimer´s disease and Huntington disease [89].
Immunomodulatory drugs disrupt the cereblon-CD147-MCT1 axis to exert anti-cancer activity and teratogenicity in a ubiquitin-independent way
CRBN promotes the activation of the CD147 (basigin; BSG) - MCT1 (solute carrier family 16 members 1; SLC16A1) transmembrane complex. This complex activatesvarious biological functions, including angiogenesis, proliferation, invasion and lactate export. Binding of IMiDs to CRBN leads to destabilization of the CD147-MCT1 complex and to the inhibition of tumor growth [90].
Measurement of cereblon mRNA and cereblon protein levels as biomarkers for IMiDs response
The measurement of CRBN mRNA by quantitative RT-PCR typically uses commercial assays, such as predesigned TaqMan assays, where primers and probe are optimized with respect to known gene splicing information. For CRBN mRNA, the current “best coverage assay” detects the exon 8-exon 9 junction and exon 10 as a measure of gene expression [91]. This assay (Hs00372271_m1; Applied Biosystems, Life Technologies Corp.) measures all CRBN mRNA variants that are translated to functional protein with exception of the variants with removed exon 10 (part of IMIDs binding region). The presence of multiple CRBN mRNA splice variants complicates the transcript measurement by Affymetrix array.
We have found that MDS patients with isolated del(5q) (the so-called 5q minus syndrome) have higher levels of full-length CRBN mRNA than other patients with lower risk MDS, linking higher levels of a known lenalidomide target CRBN and an MDS subgroup known to be especially sensitive to lenalidomide [92].
Currently available commercial antibodies are neither sensitive nor specific for reliable detection of CRBN protein levels. Gandhi et al. [91] characterized a monoclonal antibody CRBN65 and compared its properties with the commonly used, currently available commercial antibodies against CRBN. This antibody is the most sensitive and specific and can detect as little as 200 pg of CRBN protein via Western blot [91].
Appropriate antibodies and validated assays for cereblon protein detection and CRBN gene expression that account for the known gene splicing information are needed for CRBN measurements in the clinic.
While it has been shown that CRBN and IRF4 levels correlate with lenalidomide responsiveness in MM patients, previous in vitro investigations using cytogenetically discrete human myeloma cell lines have not been able to replicate this phenomenon [93,94]. The implications of this apparent discordance are two-fold. First, these results show that it is important to investigate CRBN and IRF4 gene expression in both CD138- and CD138+ myeloma cells. Second, because there are multiple isoforms of CRBN, it may be important to study expression levels of each one in the context of both in vivo and in vitro settings [91].
Conclusion and perspectives
The studies showing IMiDs induced the CRBN-dependent proteasomal degradation of IKZF1, IKZF3, and caein kinase 1α [18,50,78,79] have greatly advanced our understanding of the mechanism of action of IMiDs in MM and MDS with del(5q). The small-molecule drug lenalidomide modulates the activity of the CRBN-CRL4 E3 ubiquitin ligase complex to increase ubiquitination of two transcription factors, IKZF1 and IKZF3, or casein kinase 1α. It does so by specific binding to one component of the system, cereblon. Crystal structures of these complexes were studied in detail [83,94,95]. All these studies may have wider implications for the targeting of E3 ubiquitin ligases in drug discovery for other diseases. There are very few approved and experimental drugs that modulate the ubiquitin system like lenalidomide in this case. Other small molecules could be developed to alter the specificity of ubiquitination complexes with the aim of inducing the specific degradation of previously “undruggable” oncoproteins such as Ras and Myc in many different types of cancer [96-99]. Nevertheless, there are still gaps in our understanding of the mechanism of action of IMiDs in MM and other hematological malignancies. For example, the proteasomal inhibitor bortezomib (VelcadeTM, PS-341) is used for the treatment of MM. Bortezomib is used efficiently also in combination with lenalidomide. There is an apparent paradox, because inhibition of proteasomal destruction of IKZF1 and IKZF3 by bortezomib contradicts the preposed mechanism. Lu et al. [80] hypothesize that since the proteasomal inhibition by bortezomib is incomplete with therapeutic dosing, this might allow sufficient destruction of IKZF1 and IKZF3 while retaining other therapeutic effect of bortezomib. Alternatively, they hypothesize that IKZF1 and IKZF3 once polyubiquitylated, may be inactive or act as dominant-negatives.
Future dissection of CRBN direct down-stream substrates and CRBN indirect down-stream factors will help to identify mechanisms of IMiD action and find new biomarkers for prediction of IMiD response and IMiD resistence as well as developing a new therapy to treat the patients with MM.
It is also possible to speculate whether or not anti-myeloma activity of IMiDs is associated with the interaction between CRBN and AMPK (AMP-activated protein kinase) or KCNT1 (Potassium Sodium-Activated Channel Subfamily T Member 1). Both AMPK and KCNT1 are potential substrates of the CRBN-CRL4 E3 ubiquitin ligase complex, but they may not be associated with downstream signaling that leads to anti-myeloma activity of IMiDs.
Acknowledgements
This work was supported by the project for conceptual development of research organisation No 00023736 (Institute of Hematology and Blood Transfusion) from the Ministry of Health of the Czech Republic.
References
- Smithells RW, Newman CG (1992) Recognition of thalidomide defects. J Med Genet 29: 716-723. [Crossref]
- Vargesson N (2009) Thalidomide-induced limb defects: resolving a 50-year-old puzzle. Bioessays 31: 1327-1336. [Crossref]
- Franks ME, Macpherson GR, Figg WD (2004) Thalidomide. Lancet 363: 1802-1811. [Crossref]
- Delforge M, Bladé J, Dimopoulos MA, Facon T, Kropff, M, et al. (2010) Treatment-related peripheral neuropathy in multiple myeloma: the challenge continues. Lancet Oncol 11: 1086-1095.
- Richardson PG, Delforge M, Beksac M, Wen P, Jongen JL, et al. (2012) Management of treatment-emergent peripheral neuropathy in multiple myeloma. Leukemia 26: 595-608.
- Andhavarapu S, Roy V (2013) Immunomodulatory drugs in multiple myeloma. Expert Rev Hematol 6: 69-82.
- Saini N, Mahindra A (2013) Novel immunomodulatory compounds in multiple myeloma. Expert Opin Investig Drugs 22: 207-215. [Crossref]
- Davies F, Baz R (2010) Lenalidomide mode of action: linking bench and clinical findings. Blood Rev 24 Suppl 1: S13-19. [Crossref]
- Quach H, Kalff A, Spencer A (2012) Lenalidomide in multiple myeloma: Current status and future potential. Amer J Hematol 87: 1089-1095.
- Chan AC, Neeson P, Leeansyah E, Tainton K, Quach H, et al. (2013) Natural killer T cell defects in multiple myeloma and the impact of lenalidomide therapy. Clin Exper Immunol 175: 49-58.
- Strati P, Keating MJ, Wierda WG, Badoux XC, Calin S, et al. (2013) Lenalidomide induces long-lasting responses in elderly patients with chronic lymphocytic leukemia. Blood 122: 734-737.
- Semeraro M, Vacchelli E, Eggermont A, Galon J, Zitvogel L, et al. (2013) Trial Watch: Lenalidomide-based immunochemotherapy. Oncoimmunology 2: e26494. [Crossref]
- Chang X, Zhu Y, Shi C, Stewart AK (2014) Mechanism of immunomodulatory drugs action in the treatment of multiple myeloma. Acta Biochim Biophys Sin 46: 240-253.
- Keevan J, Figg WD (2014) New mechanism of lenalidomide activity. Cancer Biol Ther 15: 968-969. [Crossref]
- Desai M, Newberry K, Ou Z, Wang M, Zhang L (2014) Lenalidomide in relapsed or refractory mantle cell lymphoma: overview and perspective. Ther Adv Hematol 5: 91-101. [Crossref]
- Gribben JG, Fowler N, Morschhauser F (2015) Mechanisms of Action of Lenalidomide in B-Cell Non-Hodgkin Lymphoma. J Clin Oncol 33: 2803-2811. [Crossref]
- Ghosh N, Grunwald MR, Fasan O, Bhutani M (2015) Expanding role of lenalidomide in hematologic malignancies. Cancer Management Res 7: 105-119.
- Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN (2015) Lenalidomide induces ubiquitination and degradation of CK1a in del(5q) MDS. Nature 523: 183-188.
- Guirguis AA, Ebert BL (2015) Lenalidomide: deciphering mechanisms of action in myeloma, myelodysplastic syndrome and beyond. Curr Opin Cell Biol 37: 61-67.
- Liang L, Zhao M, Zhu YC, Hu X, Yang LP, et al. (2016) Efficacy of lenalidomide in relapsed/refractory chronic lymphocytic leukemia patient: a systematic review and meta-analysis. Ann Hematol 95: 1473-1482.
- Arora M, Gowda S, Tuscano J (2016) A comprehensive review of lenalidomide in B-cell non-Hodgkin lymphoma. Ther Adv Hematol 7: 209-221. [Crossref]
- Lindner S, Krönke J (2016) The molecular mechanism of thalidomide analogs in hematologic malignancies. J Mol Med (Berl) 94: 1327-1334. [Crossref]
- Manasanch EE (2016) Recommend maintenance therapy with lenalidomide in multiple myeloma. Semin Oncol 43: 712-713. [Crossref]
- Holstein SA, McCarthy PL (2017) Immunomodulatory Drugs in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Drugs 77: 505-520. [Crossref]
- Itchaki G, Brown JR (2017) Lenalidomide in the treatment of chronic lymphocytic leukemia. Expert Opin Investig Drugs 26: 633-650. [Crossref]
- Stahl M, Zeidan AM (2017) Lenalidomide use in myelodysplastic syndromes: Insights into the biologic mechanisms and clinical applications. Cancer 123: 1703-1713. [Crossref]
- Lacy MQ, McCurdy AR (2013) Pomalidomide. Blood 122: 2305-2309. [Crossref]
- Richardson PG, Mark TM, Lacy MQ (2013) Pomalidomide: new immunomodulatory agent with potent antiproliferative effects. Crit Rev Oncol Hematol 88 Suppl 1: S36-44. [Crossref]
- Chanan-Khan AA, Swaika A, Paulus A, Kumar SK, Mikhael JR, et al. (2013) Pomalidomide: the new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J 3: e143. [Crossref]
- McCurdy AR, Lacy MQ (2013) Pomalidomide and its clinical potential for relapsed or refractory multiple myeloma: an update for the hematologist. Ther Adv Hematol 4: 211-216. [Crossref]
- Mark TM, Coleman M, Niesvizky R (2014) Preclinical and clinical results with pomalidomide in the treatment of relapsed/refractory multiple myeloma. Leuk Res 38: 517-524.
- Fouquet G, Bories C, Guidez S, Renaud L, Herbaux C, et al. (2014) Pomalidomide for multiple myeloma. Expert Rev Hematol 7: 719-731. [Crossref]
- Jones JR, Pawlyn C, Davies FE, Morgan GJ (2016) The safety of pomalidomide for the treatment of multiple myeloma. Expert Opin Drug Saf 15: 535-547. [Crossref]
- Touzeau C, Moreau P (2016) Pomalidomide in the management of relapsed multiple myeloma. Future Oncol 12: 1975-1983. [Crossref]
- Hagner PR, Man HW, Fontanillo C, Wang M, Couto S, et al. (2015) CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood 126: 779-789.
- Nowakowski GS1 (2015) Evolution: IMiDs to PPMs, revolution in DLBCL? Blood 126: 698-700. [Crossref]
- Camicia R, Winkler 2021 Copyright OAT. All rights reservonalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: a comprehensive review. Mol Cancer 14: 207. [Crossref]
- Cubillos-Zapata C, Córdoba R, Avendaño-Ortiz J, Arribas-Jiménez C, Hernández-Jiménez E, et al. (2016) CC-122 immunomodulatory effects in refractory patients with difuse large B-cell lymphoma. Oncoimmunology 5: e1231290.
- Cubillos-Zapata C, Córdoba R, Avendaño-Ortiz J, Lopez-Collazo E (2017) Thalidomide analog CC-122 induces a refractory state in monocytes from patients with difuse large B-cell lymphoma. Leuk Lymphoma 58: 1999-2001.
- Matyskiela ME1, Zhang W1, Man HW1, Muller G1, Khambatta G1, et al. (2017) A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J Med Chem. [Crossref]
- Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, et al. (2016) A novel cereblon modulator recruits GSPT1 to the CRL4 (CRBN) ubiquitin ligase. Nature 535: 252-257.
- Hansen JD, Condroski K, Correa M, Muller G, Man HW, et al. (2017) Protein Degradation via CRL4CRBN Ubiquitin Ligase: Discovery and Structure-Activity Relationships of Novel Glutarimide Analogs That Promote Degradation of Aiolos and/or GSPT1. J Med Chem. [Crossref]
- Quach H, Ritchie D, Stewart AK, Neeson P, Harrison S, et al. (2010) Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 24: 22-32. [Crossref]
- Maiga S, Gomez-Bougie P, Bonnand S, Grates C, Moreau P, et al. (2013) Paradoxical effect of lenalidomide on cytokine/growth factor profiles in multiple myeloma. Br J Cancer 108: 1801-1806.
- Geitz H, Handt S, Zwingenberger K (1996) Thalidomide selectively modulates the, density of cell surface molecules involved in the adhesion cascade. Immunopharmacology 31: 213-221.
- Muller GW, Chen R, Huang SY, Corral LG, Wong LM, et al. (1999) Amino-substituted thalidomide analogs: potent inhibitors of TNF-alpha production. Bioorg Med Chem Lett 9: 1625-1630.
- Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC (2001) The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene 20: 4519-4527.
- Handa K, Suzuki R, Matsui H, Shimizu Y, Kumagai K (1983) Natural killer (NK) cells as a responder to interleukin 2 (IL 2). II. IL 2-induced interferon gamma production. J Immunol 130: 988-992.
- Haslett PA, Colrral LG, Albert M, Kaplan G (1998) Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J Exp Med 187: 1885-1892.
- Gandhi AK, Kang J, Havens CG, Conklin T, Yuhong N, et al. (2014) Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4CRBN. Br J Haematol 164: 811-821.
- Chang DH, Liu D, Klimek V, Hassoun H, Mazumder A, et al. (2006) Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide.: therapeutic implications. Blood 108: 618-621.
- Galustian C, Meyer B, Labarte MC, Dredge K, Klaschka D, et al. (2009) The anti-cancer agents lenalidomide and pomalidomide inhibits the proliferation and function of T regulatory cells. Cancer Immunol Immunother 58: 1033-1045.
- Dredge K, Marriott JB, Macdonald CD, Man HW, Chen R, et al. (2002) Novel thalidomide analogues display anti-angiogenic activity independently of immunomodulatory effects. Br J Cancer 87: 1166-1172.
- Dredge K, Horsfall R, Robinson SP, Zhang LH, Lu L, et al. (2005) Orally administrated lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitro. Microvascular Res 69: 56-63.
- Dankbar B, Paadro T, Leo R, Feldman B, Kropff M, et al. (2000) Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood 95: 2630-2636.
- Escoubet-Lozach L, Lin IL, Jensen-Pergakes K, Brady HA, Gandhi AK, et al. (2009) Pomalidomide and lenalidomide induce p21 WAF-1 expression in both lymphoma and multiple myeloma through a LSD1-mediated epigenetic mechanism. Cancer Res 69: 7347-7356.
- Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, et al. (2002) Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: Therapeutic implications. Blood 99: 4525-4530.
- Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, et al. (1997) Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kB control. Proc Natl Acad Sci USA 94: 10057-10062.
- Kreuz S, Siegmund D, Scheurich P, Wajant H (2001) NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of dech receptor signaling. Mol Cell Biol 21: 3964-3973.
- Li ZW, Chen H, Campbell RA, Bonavida B, Berenson JR (2008) NF-kappaB in the pathogenesis and treatment of multiple myeloma. Curr Opin Hematol 15: 391-399. [Crossref]
- Lopez-Girona A, Heintel D, Zhang LH, Mendy D, Gaidarova S, et al. (2011) Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4 providing a potential mechanistic link for predicting response. Br J Haematol 154: 325-336.
- Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, et al. (2008) IRF4 addiction in multiple myeloma. Nature 454: 226-231. [Crossref]
- Raje N, Kumar S, Hideshima T, Ishitsuka K, Chauhan D, et al. (2004) Combination of the mTOR inhibitor rapamycin and CC-5013 has synergistic activity in multiple myeloma. Blood 104: 4188-4193.
- Steinbrunn T, Stühmer T, Sayehli C, Chatterjee M, Einsele H, et al. (2012) Combined targeting of MEK/MAPK and PI3K/Akt signalling in multiple myeloma. Br J Haematol 159: 430-440. [Crossref]
- Yang J, He J, Wang J, Cao Y, Ling J, et al. (2012) Constitutive activation of p38 MAPK in tumor cells contributes to osteolytic bone lesions in multiple myeloma. Leukemia 26: 2114-2123.
- Tu Y, Gardner A, Lichtenstein A (2000) The phosphatidylinositol 3-kinase/AKT kinase pathway in multiple myeloma plasma cells: roles in cytokine-dependent survival and proliferative responses. Cancer Res 60: 6763-6770.
- Younes H, Leleu X, Hatjiharissi E, Moreau AS, Hideshima T, et al. (2007) Targeting the phosphatidylinositol 3-kinase pathway in multiple myeloma. Clin Cancer Res 13: 3771-3775. [Crossref]
- Li S, Pal R, Monaghan SA, Schafer P, Ouyang H, et al. (2011) IMiD immunomodulatory compounds block C/EBPß translation through eIF4E down-regulation resulting in inhibition of MM. Blood 117: 5157-5165.
- Pal R, Janz, M, Galson DL, Gries M, Li S, et al. (2009) C/EBPß regulates transcription factors critical for proliferation and survival of multiple myeloma cells. Blood 114: 3890-3898.
- Bjorklund CC, Ma W, Wang ZQ, Davis RE, Kuhn DJ, et al. (2011) Evidence of a role for activation of Wnt/ß-catenin signaling in the resistance of plasma cells to lenalidomide. J Biol Chem 13: 11009-11020.
- Sadler NM, Harris BR, Metzger BA, Kirshner J (2013) N-cadherin impedes proliferation of the multiple myeloma cancer stem cells. Am J Blood Res 3: 271-285.
- Bjorklund CC, Baladandazuthapani V, Lin HY, Jones RJ, Kuiatse I, et al. (2014) Evidence of a role for CD44 and cell adhesion in mediating resistance to lenalidomide in multiple myeloma: therapeutic implications. Leukemia 28: 373-383.
- Ito T, Ando H, Suzuki T, Ogura T, Hotta K, et al. (2010) Identification of a primary target of thalidomide teratogenicity. Science 327: 1345-1350.
- Ito T, Ando H, Handa H (2011) Teratogenic effects of thalidomide: molecular mechanisms. Cell Mol Life Sci 68: 1569-1579.
- Ito T, Handa H (2012) Deciphering the mystery of thalidomide teratogenicity. Congenit Anom (Kyoto) 52: 1-7. [Crossref]
- Ito T, Handa H (2016) Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs. Int J Hematol 104: 293-299.
- Liu Y, Huang X, He X, Zhou Y, Jiang X, et al. (2015) A novel effect of thalidomide and its analogs: suppression of cereblon ubiquitination enhances ubiquitin ligase function. FASEB J 29: 4829-4839.
- Kr?nke J, Udeshi ND, Narla A, Grauman P, Hurst SN, et al. (2014) Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343: 301-305.
- Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, et al. (2014) The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343: 305-309.
- Stegmaier K (2014) Targeting Csnk1a1 in leukemia. J Exp Med 211: 594. [Crossref]
- Järås M, Miller PG, Chu LP, Puram RV, Fink EC, et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. (2014) J Exp Med 211: 605-612.
- Schneider RK, Ademà V, Heckl D, Järås M, Mallo M, et al. (2014) Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 26: 509-520.
- Petzold G, Fischer ES, Thomä NH (2016) Structural basis of lenalidomide-induced CK1a degradation by the CRL4(CRBN) ubiquitin ligase. Nature 532:127-130.
- Heuser M, Meggendorfer M, Cruz MM, Fabisch J, Klesse S, et al. (2015) Frequency and prognostic impact of casein kinase 1A1 mutations in MDS patients with deletion of chromosome 5q. Leukemia 29: 1942-1945.
- Bello E, Pellagatti A, Shaw J, Mecucci C, Kušec R, et al. (2015) CSNK1A1 mutations and gene expression analysis in myelodysplastic syndromes with del(5q). Br J Haematol 171: 210-214.
- Boultwood J, Pellagatti A (2015) Clinical associations of CSNK1A1 mutation in myelodysplastic syndrome. Lancet Haematol 2: e182-183.
- Smith AE, Kulasekararaj AG, Jiang J, Mian S, Mohamedali A, et al. (2015) CSNK1A1 mutations and isolated del(5q) abnormality in myelodysplastic syndrome: a retrospective mutational analysis. Lancet Haematol 2: e212-221.
- Van Nguyen T, Li J, Lu CCJ, Mamrosh JJ, Lu G, et al. (2017) p97/VCP promotes degradation of CRBN substrate glutamine synthetase and neosubstrates. Proc Natl Acad Sci USA 114: 3565-3571.
- Kim HK, Ko TH, Nyamaa B, Lee SR (2016) Cereblon in health and disease. Pflugers Arch 468: 1299-1309. [Crossref]
- Eichner R, Heider M, Fern8ndey/S8iy V, van Bebber F, Gary AK, et al. (2016) Immunomodulatory drugs disrupt the cereblon-CD147-MCT1 axis to exert antitumor activity and teratogenicity. Nat Med 22: 735-743.
- Gandhi AK, Mendy D, Waldman M, Chen G, Rychak E, et al. (2014) Measuring cereblon as a biomarker of response or resistance to lenalidomide and pomalidomide requires use of standardized reagents and understanding of gene complexity. Br J Haematol 164: 233-244. [Crossref]
- Jonasova A, Bokorova R, Polak J, Vostry M (2015) High level of full-length cereblon mRNA in lower risk myelodysplastic syndrome with isolated 5q deletion is implicated in the efficacy of lenalidomide. Eur J Haematol 95: 27-34. [Crossref]
- Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, et al. (2012) Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 26: 2326-2335.
- Greenberg AJ, Walters DK, Kumar SK, Vincent Rajkumar S, Jelinek DF, et al. (2013) Responsiveness of cytogenetically discrete human myeloma cell lines to lenalidomide: lack of correlation with cereblon and interferon regulatory factor 4 expression levels. Eur J Haematol 91: 504-513.
- Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, et al. (2014) Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512: 49-53. [Crossref]
- Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, et al. (2014) Structure of the human cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Str Mol Biol 21: 803-809.
- Bulatov E, Ciulli A (2015) Targeting cullin-ring E3 ubiquirin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochem J 467: 365-386.
- Huang X, Dixit VM (2016) Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res 26: 484-498.
- Collins I, Wang H, Caldwell JJ, Chopra R (2017) Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochem J 474: 1127-1147. [Crossref]