Abstract
Polycythemia vera (PV) is a chronic myeloproliferative disorder characterized by abnormal growth of erythroid precursors in the bone marrow. Almost all patients with PV, around 97%, have a mutation in Janus kinase 2 (JAK2). It is through the activation of JAK/Signal Transducers and Activators of Transcription (STAT) protein signaling pathway that the JAK2 mutation is thought to induce cellular proliferation, growth, hematopoiesis and immune response in PV patients. A summary of the current role of immunotherapy in the treatment of PV is provided. There are new JAK kinase inhibitors that are currently being evaluated and are at various stages of clinical trials and development, as well as the previously studied cytokines therapies. At the present time, ruxolitinib and interferon-α (IFN-α) are the only United States Food and Drug Administration (FDA) approved drugs for the management of advanced PV. JAK kinase inhibitors are better tolerated and less problematic than the interferons. Still, allogeneic stem cell transplantation is the only potentially curable method for end-stage PV. Additional genetic mutations have been implicated in PV pathogenesis. In this perspective, targeting different pathways might be required. Further investigations are needed to evaluate the promising role of immunotherapy in PV whether alone or in combination with other modalities.
Key words
Polycythemia vera, Kinase inhibitors, cytokines, stem cells
Abbreviations
AE: Adverse Events; Bcl-x: B-cell lymphoma-extra; CMML: Chronic Myelomonocytic Leukemia; CYP: Cytochrome P; ET: Essential Thrombocythemia; FDA: Food and Drug Administration; HSC: Hematopetic Stem Cells; IFN-α: Interferon alfa; JAK2: Janus Kinase 2; JAK/STAT: Janus Kinase /Signal Transducers and Activators of Transcription; MPDs: Myeloproliferative Disorders; MU: Million Units; NF-E: Nuclear Factor Erythroid-Derived 2; PEG: Polyethylene Glycol; PD/PK: Pharmacodynamics/Pharmacokinetics; PMF: Primary Myelofibrosis; PV: Polycythemia Vera; PRV-1: Polycythemia Rubra Vera 1; Tpo-R: Thrombopoietin Receptor; Wt: Wild Type
Introduction/Epidemiology
Myeloproliferative disorders (MPDs) are classified according to the most affected type of blood cells. There are four main types of MPDs, which include Polycythemia Vera (PV), Essential Thrombocythemia (ET), Primary Myelofibrosis (PMF), and Chronic Myelomonocytic Leukemia (CMML).
PV is a myeloproliferative disorder, which presents as an abnormal increase in the number of red blood cells [1]. It is the most common form of the MPDs. Its incidence in the US is estimated to be 1.9/100,000, with an increase associated with ageing. The mean age at diagnosis of PV is approximately 60 years old. However, in 2010 the age adjusted prevalence of PV was 57.15/100,000 [2]. PV is very rare in children.
Etiology/predisposing risk factors
The cause of chronic MPDs remains unknown. A mutation in a specific protein, Janus kinase 2 (JAK2) is found in a large number * of people with MPDs. It is detected in about 95% of those with PV, in approximately 50–70% with ET, and 40–50% with PMF [3]. There are several risk factors, which increases the risk of chronic MPDs. They include the following:
Age and sex
Although one third of the cases diagnosed are in those below the age of 50, PV is most commonly diagnosed at 60 years of age and above [4]. Similarly, the incidence of MPDs and specifically CMML increases with age [5,6]. The median age at diagnosis of CMML patients ranges between 65 and 75 years [7]. PV, as well as all MPDs in general, are more prevalent in men [5].
Cancer treatment
Prior treatment with chemotherapy appear to increase the risk of CMML [8]. However, the risk of CMML after cancer chemotherapy is not as high as that of other hematological malignancies, such as myelodysplastic syndromes and acute myeloid leukemia [9].
Exposure to carcinogenic agents
Petrochemicals such as benzene and toluene, and ionizing radiation increase the risk of PV and PMF [10,11]
Pathophysiology/molecular basis
A mutation in a specific gene, Janus kinase 2 (JAK2) is found in a large percentage of people with MPDs. Figure 1 describes a working model for genetic events and other mechanisms, possibly involved in the pathophysiology of MPDs [12].
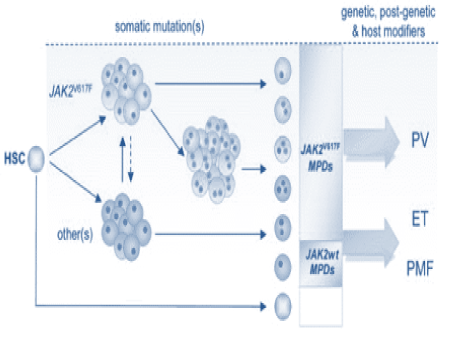
Figure 1: A model of genetic events and other mechanisms possibly involved in the pathophysiology of MPDs HSC: Hematopoetic Stem Cells; MPDs: Myeloproliferative Disorders; PV: Polycythemia Vera; ET: Essential Thrombocythemia; Wt: Wild Type; PMF: Primary Myelofibrosis.
Hematopoietic growth factor responses mediated by the JAK/STAT Pathway
The JAK/STAT pathway [13] plays a vital role in the initiation of signal transduction through hematopoietic growth factor receptors. JAK/STAT is also a target for identifying the molecular abnormalities in PV. Furthermore, in erythropoietin-independent differentiation of erythroid progenitors in PV, where constitutive activation of STAT3 has been reported, STAT3 was found to be repressed by inhibitors of JAK2 [14,15]. Recently, several groups have identified a consistent, single somatic activating mutation in the JAK2 gene in the majority of patients with PV [16].
JAK2 mutation in the PV
Several studies showed that the majority of patients (65%-97%) with PV have the JAK2-V617F mutation [17,18]. A report by Kralovics et al. [19] showed that patients with JAK2-V617F had a significantly longer duration of disease, more treatment with cyto-reducing agents and higher rates of complications (i.e., myelofibrosis, thrombosis and bleeding) than those with wild-type JAK2. Other biologic and epigenetic markers present in PV patients are deregulated expression of Bcl-x (B-cell lymphoma-extra, an inhibitor of apoptosis) [20], overexpression of the PRV-1 (polycythemia rubra vera 1) and transcription factor NF-E2 (nuclear factor erythroid-derived 2) genes [21,22], and impaired expression of Tpo-R (thrombopoietin receptor) [23]. No mutations have yet been detected in these genes and recent evidence indicates that altered expression of these markers is due to activation of the JAK/STAT pathway through the JAK2–V617F mutation [24]. Therefore, abnormal expression of these molecules appears to be a secondary consequence of the primary JAK2 mutation [25].
Molecular Re-classification of the MPDs
From the well-known phenotypic overlap and transitions that occur during the natural history of the classic MPDs, it is also possible that patients with clinically diagnosed ET or myelofibrosis, who have the JAK2–V617F mutation, actually represent variants of PV [25,26]. This possibility is supported by the finding that up to 30% of patients with PV, but very few patients with ET, appear to be homozygous for JAK2–V617F [25]. Therefore, mutation-positive ET patients may, in fact, be individuals with “inapparent PV”. These patients may have variable degrees of plasma volume expansion which might mask an absolute increase in red cell mass, unless these are directly measured [27], the distinction is not always perceptible. They may also represent early or late (spent) phases of PV.
Immunotherapy for PV
Kinase inhibitors
Ruxolitinib: An orally bioavailable JAK inhibitor with potential antineoplastic and immunomodulating activities approved by FDA in the management of PV. Ruxolitinib specifically binds to and inhibits protein tyrosine kinases, JAK1 and JAK2, which may lead to a reduction in inflammation and an inhibition of cellular proliferation. The JAK/STAT pathway plays a key role in the signaling of many cytokines and growth factors and is involved in cellular proliferation, growth, hematopoiesis and the immune response; JAK kinases may be up-regulated in inflammatory diseases, MPDs and various malignancies.
Ruxolitinib is FDA approved for the treatment of patients with PV, who have had an inadequate response to or are intolerant to therapy with hydroxyurea. Ruxolitinib has also been indicated for the treatment of intermediate or high-risk myelofibrosis, including primary myelofibrosis, post-PV myelofibrosis and post-ET myelofibrosis and PV in patients who have had an inadequate response to or are intolerant to hydroxyurea.
Orally administered ruxolitinib is rapidly absorbed with Cmax achieved within 1-2 hours post-administration. The mean volume of distribution at steady-state is 75 L with an inter-subject variability of 23%. Ruxolitinib is metabolized by cytochrome P3A4 (CYP3A4) with a mean elimination half-life of approximately 3 hours. Adverse events may include thrombocytopenia, anemia and neutropenia, risk of infections, and non-melanoma skin cancer. However, the most common hematologic adverse reactions are thrombocytopenia and anemia with the most common non-hematologic adverse reactions reported being bruising, dizziness, and headache [28].
Momelotinib: Momelotinib is an orally bioavailable small-molecule inhibitor of JAK1/2 with potential antineoplastic activity, which is being investigated in a phase II clinical trial (Table 1). Momelotinib competes with JAK1/2 for ATP binding, which may result in the inhibition of JAK1/2 activation, and therefore, inhibition of the JAK/STAT signaling pathway, thus promoting induction of apoptosis and a reduction of tumor cell proliferation in JAK1/2-expressing tumor cells.
Drug |
Clinical trial identifier no. |
Phase |
Study Design |
Target |
Momelotinib |
NCT01998828 |
Phase II |
Randomized, Safety/Efficacy study, open label |
JAK1/2 |
Table 1. Non-FDA Approved kinase inhibitors [29]
Cytokine therapy
Interferon-alfa (IFN-α): INF-α is an analogue of consensus interferon containing an additional methionyl-amino-acid residue. Consensus interferon (also known as interferon alfacon-1; rCon-IFN, and CIFN) is a genetically engineered synthetic interferon created from the most common amino acid sequences found in the naturally occurring alfa interferons. Alfa interferons bind to specific cell-surface receptors, resulting in the transcription and translation of genes whose protein products have antiviral, antiproliferative, anticancer, and immune-modulating effects.
IFN-α suppresses the proliferation of hematopoietic progenitors and has a direct inhibiting effect on bone marrow fibroblast progenitor cells. Additionally, IFN-α antagonizes the action of platelet-derived growth factor, transforming growth factor-β and other cytokines that may be involved in the development of myelofibrosis. IFN-α should be administered at the dose of 3 million units (MU) daily, until a hematocrit response is reached (hematocrit < 0.45 [45%]); then, maintenance therapy has to be adjusted to the lowest weekly doses that control the hematocrit at that response level. During the first month of therapy a complete hemogram must be recorded weekly; for the second month the test is required, every 2 weeks, then monthly, and, in steady-state in responding patients, every 3-4 months. IFN-α is contraindicated in patients with thyroid and/or mental disorders. The most common adverse effects associated with IFN-α are weakness, myalgia, weight and hair loss, severe depression, and gastrointestinal and cardiovascular symptoms [30,31].
PEGASYS: PEGASYS is a covalent conjugate of recombinant IFN-α, subtype 2a and polyethylene glycol (PEG),used as an antiviral and antineoplastic agent and presently being tested in a phase II clinical trial (Table 2). The biological activity of this agent is derived from its IFN-α-2a protein moiety. IFN-α binds to specific cell-surface receptors leading to the transcription and translation of genes whose protein products mediate antiviral, antiproliferative, anticancer and immune-modulating effects. The PEG moiety lowers the clearance of IFN-α-2a, thereby extending the duration of its therapeutic effects, but may also reduce interferon-mediated stimulation of an immune response.
Table 2. Non-FDA Approved IFN-α [32,33]
Drug |
Clinical trial identifier no. |
Phase |
Study Design |
Target |
PEGASYS |
NCT01259856 |
Phase III |
Randomized, Safety/Efficacy study, open label |
Cell surface receptors of the malignant clone in PV |
PEGASYS |
NCT01259817 |
Phase II |
Non Randomized, single arm, open label, Safety/Efficacy Study |
Cell surface receptors of the malignant clone in PV |
Stem cell transplantation: Currently, allogeneic stem cell transplantation is the only potentially curative treatment for advanced PV. Analysis of 250 consecutive patients was performed with an initial diagnosis of PV (n=120) who underwent transplantation due to progression of myelofibrosis (n=193) or acute myeloid leukemia (n=57) and who were reported to the European Group for Blood and Marrow Transplantation registry between 1994 and 2010. The median age was 56 years (range, 22–75), and the interval between diagnosis and transplantation was 10 years or more in 52 % of the cases. With a median follow-up from transplantation of 13 months, the 3-year overall survival rate and relapse incidence were 55% and 32%, respectively. In a univariate analysis, the main parameters that negatively affected post-transplantation outcomes were age (>55 years), a diagnosis of acute myeloid leukemia at transplant and obtaining a suitable transplant from an unrelated donor. The overall 3-year cumulative incidence of non-relapse mortality was 28%, but was significantly higher in older patients than in younger ones (>55 years, 35% versus 20%, P=0.032), in those transplanted from an unrelated donor rather than a related donor (34% versus 18%, P=0.034) and in patients with a diagnosis of acute myeloid leukemia compared to myelofibrosis (29% versus 27%, P=0.045). This large retrospective study confirmed that transplantation is potentially curative for patients with end-stage PV progressing to myelofibrosis or acute myeloid leukemia.
I looked up the topic and I could not find planned clinical trials involving the use of vaccination the management of PV or MPDs (except CML) [34].
Conclusions and future perspectives
Our success in treating hematological malignancies is increasing and advancing day by day with improved knowledge of how the immune system becomes dysfunctional in these blood disorders. The identification of the JAK2–V617F mutation in patients diagnosed and progressing to chronic MPDs has stimulated a great deal of effort in screening for MPDs and for developing specific JAK1/2 and other inhibitors for clinical therapy. It is certain that the next few years will bring further developments in this fast-evolving field. Researchers are still challenged to explore innate and adaptive immune systems and immunotherapy has become a promising development in the past few years in the treatment of various cancers. These recent advances have increased our understanding of the tumor microenvironment, and of various immunotherapeutic modalities or combination therapies such as chemotherapy with immunotherapy although the effects of such modalities in combination with immunotherapy in cancer patients are still in the exploratory phase. In summary, the complete perspective of immunotherapy treatment has not been realized and/or utilized and in that respect proper preclinical and clinical designs are the important pillars for understanding the future of immunotherapy in treating cancer patients and individuals with hematological disorders.
References
- Kremyanskaya M, Najfeld V, Mascarenhas J, et al. (2012) The polycythemias Hematology: Basic Principles and Practice. (6th edn.) Saunders Elsevier, USA 67: 998-1033.
- Mesa RA, Jyotsna M, Wang H, Wang Y, Iqbal U, et al. (2012) Epidemiology of Myeloproliferative Disorders in US – a real world analysis. Abstract 2834.
- Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OA (2011) New mutations and pathogenesis of myeloproliferative neoplasms. Blood 118: 1723-1735. [Crossref]
- Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, et al. (2013) Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 27: 1874-1881. [Crossref]
- Rollison DE, Howlader N, Smith MT, Strom SS, Merritt WD, et al. (2008) Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001-2004, using data from the NAACCR and SEER programs. Blood 112: 45-52. [Crossref]
- Lichtman MA, Tefferi A (2010) Primary myelofibrosis. (8th edn.) Williams Hematology 91: 2219-2246.
- Parikh SA, Tefferi A (2013) Chronic myelomonocytic leukemia: 2013 update on diagnosis, risk stratification, and management. Am J Hematol 88: 967-974. [Crossref]
- Ueki K, Sato S, Tamura J, Sawamura M, Murakami H, et al. (1991) Three cases of multiple myeloma developing into melphalan-related chronic myelomonocytic leukemia. J Med 22: 157-161. [Crossref]
2021 Copyright OAT. All rights reserv
- Pedersen-Bjergaard J, Andersen MK, Christiansen DH (2000) Therapy-related acute myeloid leukemia and myelodysplasia after high-dose chemotherapy and autologous stem cell transplantation. Blood 95: 3273-3279. [Crossref]
- Kaplan SD (1986) Update of a mortality study of workers in petroleum refineries. J Occup Med 28: 514-516. [Crossref]
- Najean Y1, Rain JD, Billotey C (1998) Epidemiological data in polycythaemia vera: a study of 842 cases. Hematol Cell Ther 40: 159-165. [Crossref]
- Vannucchi AM, Guglielmelli P (2008) Molecular pathophysiology of Philadelphia-negative myeloproliferative disorders: Beyond JAK2 and MPL mutations. Haematologica 93: 972-976.
- Rawlings JS, Rosler KM, Harrison DA (2004) The JAK/STAT signaling pathway. J Cell Sci 117: 1281-1283. [Crossref]
- Ugo V, Marzac C, Teyssandier I, Larbet F, Lécluse Y, et al. (2004) Multiple signaling pathways are involved in erythropoietin-independent differentiation of erythroid progenitors in polycythemia vera. Exp Hematol 32: 179-187. [Crossref]
- Röder S, Steimle C, Meinhardt G, Pahl HL (2001) STAT3 is constitutively active in some patients with Polycythemia rubra vera. Exp Hematol 29: 694-702. [Crossref]
- Kaushansky K (2005) On the molecular origins of the chronic myeloproliferative disorders: it all makes sense. Blood 105: 4187-4190. [Crossref]
- James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, et al. (2005) A unique clonal JAK2 mutation leading to constitutive signaling causes polycythaemia vera. Nature 434: 1144-1148. [Crossref]
- Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, et al. (2005) Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 106: 2162-2168. [Crossref]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, et al. (2005) A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352: 1779-1790. [Crossref]
- Silva M, Richard C, Benito A, Sanz C, Olalla I, et al. (1998) Expression of Bcl-x in erythroid precursors from patients with polycythemia vera. N Engl J Med 338: 564-571. [Crossref]
- Temerinac S, Klippel S, Strunck E, Röder S, Lübbert M, et al. (2000) Cloning of PRV-1, a novel member of the uPAR receptor superfamily, which is overexpressed in polycythemia rubra vera. Blood 95: 2569-2576. [Crossref]
- Goerttler PS, Kreutz C, Donauer J, Faller D, Maiwald T, et al. (2005) Gene expression profiling in polycythaemia vera: overexpression of transcription factor NF-E2. Br J Haematol 129: 138-150. [Crossref]
- Moliterno AR, Hankins WD, Spivak JL (1998) Impaired expression of the thrombopoietin receptor by platelets from patients with polycythemia vera. N Engl J Med 338: 572-580. [Crossref]
- Kralovics R, Teo SS, Buser AS, Brutsche M, Tiedt R, et al. (2005) Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2. Blood 106: 3374-3376. [Crossref]
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, et al. (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365: 1054-1061. [Crossref]
- Wolanskyj AP, Lasho TL, Schwager SM, McClure RF, Wadleigh M, et al. (2005) JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 131: 208-213. [Crossref]
- Lamy T, Devillers A, Bernard M, Moisan A, Grulois I, et al. (1997) Inapparent polycythemia vera: an unrecognized diagnosis. Am J Med 102: 14-20. [Crossref]
- Verstovsek S, Vannucchi AM, Griesshammer M, Masszi T, Durrant S, et al. (2016) Ruxolitinib versus best available therapy in patients with Polycythemia Vera: 80 Week follow up from the RESPONSE trial. Haematologica. [Crossref]
- Gilead Sciences; Gilead Sciences. Safety and Efficacy of Momelotinib in subjects with Polycythemia vera or Essential thrombocythemia. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US): ID# NCT01998828. 2016 May 9.
- Lengfelder E, Berger U, Hehlmann R (2000) Interferon alpha in the treatment of polycythemia vera. Ann Hematol 79: 103-109. [Crossref]
- Silver RT (2006) Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 107: 451-458. [Crossref]
- Ronald Hoffman; Ronald Hoffman, Mount Sinai School of Medicine. Randomized Trial of Pegylated Interferon Alfa-2a Versus Hydroxyurea in Polycythemia Vera (PV) and Essential Thrombocythemia (ET). In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). ID# NCT01259856. 2016 May 9.
- Ronald Hoffman; Ronald Hoffman, Mount Sinai School of Medicine. Pegylated Interferon Alfa-2a Salvage Therapy in High Risk Polycythemia Vera (PV) or Essential Thrombocythemia (ET).In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). ID# NCT01259817. 2016 May 9
- Lussana F, Rambaldi A, Finazzi MC, van Biezen A, Scholten M, et al. (2014) Allogeneic hematopoietic stem cell transplantation in patients with polycythemia vera or essential thrombocythemia transformed to myelofibrosis or acute myeloid leukemia: a report from the MPN Subcommittee of the Chronic Malignancies Working Party of the European Group for Blood and Marrow Transplantation. Haematologica 99: 916-921. [Crossref]