Long life expectancy and low fertility rates are the main factors that have conditioned the increase on aged population and aged-related diseases, particularly dementias and specifically Alzheimer’s disease (AD). AD has been largely associated soluble oligomeric forms of β-amyloid peptide (Aβ) and its multiple neurotoxic effects. One of them relies on its ability to form non-selective pores on cell membranes inducing calcium overload and, in our hands, ATP leakage. In this review, we will summarize the Aβ-induced intracellular alterations that culminate with mitochondrial dysfunction, and how they determine the ultimate neurodegenerative conditions such as synaptic failure, and neuronal death. Identification of molecular o phenotypic changes on mitochondrial could represent a new start point to further address innovative experimental approaches to develop an appropriate therapy. The full understanding of the role and behavior of key fusion/fission protein will be relevant to define the no return point on the life/death cycle of neurons and synaptic network function.
AD, Aβ, mitochondrial dysfunction, mitochondrial dynamics
Aβ: amyloid-beta peptide; AD: Alzheimer’s disease; AICD: APP intracellular carboxy-terminal domain; APP: amyloid precursor protein; BACE1: β-site APP cleaving enzyme 1; CMT: Charcot–Marie–Tooth disease type II; Drp1: dynamin-related protein 1; ER: endoplasmic reticulum; GED: GTPase effector domain; HR: hydrophobic repeats; IMM: inner mitochondrial membrane; Mief1: mitochondrial elongation factor; mitoPLD: mitochondrial phospholipase D; Mfn: mitofusin; MMP: mitochondrial membrane potential; OPA1: optic atrophy 1; OMM: outer mitochondrial membrane; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; ROS: reactive oxygen species; SOAβ: soluble oligomeric forms of Aβ
Resulting from a tight interaction between genetic, epigenetic and environmental factors, aging entails a physiological phenomenon that reduces the efficiency of functioning organs, tissues and cells. Epigenetic changes play a central role on aging processes, as they may be one of the central mechanisms by which aging predisposes to many age-related diseases [1].
There are almost 900 million people aged 60 years and over living worldwide [2]. Declining fertility rates in most countries and life expectancy rise contribute to rapid increases in this number, and can be directly associated with higher prevalence of chronic or age-related diseases. One of the ageing-related group of disorders are dementias, syndromes caused by alterations and dysfunctions of important brain areas related with key cognitive functions (i.e., hippocampus and substantia nigra). These pathologies are characterized by a progressive, global deterioration in intellect including memory, learning, orientation, language, comprehension and judgment [3]. Dementia represents one of most important public health priorities given the physical, psychological, social and economic impact on caregivers, families and society.
Today, over 46 million people live with dementia, which accounts for more than total Spain’s population. This number is estimated to increase to 131.5 million by 2050, entailing huge economic impact evaluated in US $818 billion, and soon to become a trillion dollar cost by 2018 [4]. In terms of demographic implications, approximately 58% of all people with dementia live in countries currently classified by the World Bank as low or middle income countries, a proportion appraised to 63% by year 2030 and 68% by 2050 [2].
Alzheimer's disease (AD) is the most common cause of dementia and may contribute to 60–70% of cases. Today, nearly 44 million people have Alzheimer’s or a related dementia, a figure expected to double upon year 2040. As a neurodegenerative disease, AD comprises progressive memory loss and cognitive deterioration affecting memory, thinking, orientation, comprehension, calculation, learning capacity, language, and judgement. The impairment in cognitive function is commonly accompanied, and occasionally preceded, by loss of emotion control, social behavior, or motivation [3]. As yet, unfortunately, there is no reliable early peripheral biochemical marker for the AD and a definitive diagnosis can only be made on histologic examination of the brain at autopsy, where the two pathognomic hallmarks are found [5]. One of these distinctive lesions present within the diseased brain are the extracellular senile or amyloid plaques, shaped after progressive deposition of amyloid beta (Aβ) peptides and which, upon mature states, may be accompanied by degenerating neuronal processes and reactive glia [6]. The other main pathological feature relates to an intraneuronal filament-shaped structure called neurofibrillary tangle, composed by an hyperphosphorylated form of the microtubule-binding protein Tau [7].
Aβ peptide is produced by endoproteolysis of the amyloid precursor protein (APP), which is achieved by the sequential cleavage of APP by enzyme complexes termed β- and γ –secretases. APP can undergo proteolytic processing by one of two pathways that take place in different intra-cellular compartments. Most is processed through the non-amyloidogenic pathway, which occurs near or at the plasma membrane [8]; the first enzymatic cleavage is mediated by α-secretase which occurs within the Aβ domain, thereby preventing the generation of Aβ peptide and releasing instead a larger ectodomain (sAPPα) and a smaller carboxy-terminal fragment (C83). C83 can undergo an additional cleavage mediated by γ-secretase to generate extracellular peptide P3 [9] (Fig. 1.2). The alternative amyloidogenic pathway leading to Aβ generation comprises a β-secretase (β-site APP cleaving enzyme 1; BACE1) mediated cleavage, releasing an ectodomain (sAPPβ) and a 99-aminoacid-C-terminal fragment, which subsequently becomes a γ-secretase scission substrate for a 38 to 43 aminoacid product: the amyloid-beta peptide, along with its cytosolic counterpart AICD (APP Intra-Cellular Domain) [10] (Fig. 1.1). The amyloidogenic cascade takes place at endosomal/lysosomal structures [11], at Golgi-derived vesicles [12], and/or in the ER/intermediate compartment [13]. The γ-secretase has been identified as a complex of enzymes composed of presenilin 1 or 2, (PS1 and PS2), nicastrin, anterior pharynx defective and presenilin enhancer 2 [14]. Mutations in three genes — APP, PS1 and PS2 — are known to cause autosomal dominant AD or familial AD (FAD), which generally manifests with an early-onset pathogenesis [15].
Most of the Aβ peptide produced is 40 residues in length (Aβ1-40), whereas a small proportion (approximately 10%) is the 42 residue variant (Aβ1-42), the latter being more hydrophobic and prone to fibril formation [16]. Although the classical view according to Hardy’s amyloid cascade hypothesis is that Aβ is deposited extracellularly [17], emerging evidence from transgenic mice and human patients indicates that this peptide can also accumulate intraneuronally, which may contribute to disease progression [18]. In fact, and because there is a relatively poor correlation between the numbers of plaques and the degree of cognitive impairment, it has been proposed that different soluble oligomeric forms of Aβ (SOAβ) could be responsible for all the neurotoxic damage, synaptic failure and neurodegeneration [19].
Although the exact mechanism of SOAβ toxicity is not entirely defined, there is growing evidence suggesting that SOAβ excess is able to disrupt the membrane causing a pore-like structure formation (Fig. 1.3) which leads to ionic dyshomeostasis [20,21]. Furthermore, a proposed model that explains SOAβ-induced synaptotoxicity relies on its membrane perforation property, which can be associated to intracellular calcium overload and subsequent synaptic depletion [22]. More interestingly, membrane disruption not only relates to ionic influx but also appears to enable the leakage of some key neuronal metabolites; previous studies showed that SOAβ increased the extracellular level of ATP from cell cultures or slices [23,24]. Both leakage and depletion of ATP as well as cellular calcium dysregulation and cytosolic overload, involves prominent cellular alterations, including oxidative stress and free radical damage [25]. More importantly, energetic imbalance caused by ATP leakage [24], carries many detrimental effects on ATP-dependent neuronal processes, like exocytosis, neuronal transport and thus leading mitochondria to an impending dysfunctional state [26] (Fig. 1.4)
Mitochondria, the energy-generating organelles in eukaryotic cells, play essential roles in these fundamental cellular processes, such as the supply of metabolic intermediates supporting biosynthetic and bioenergetic needs of the cell [27], apoptosis [28], calcium homeostasis [29,30], cell signaling [31], among others. All of these functions were found to be altered in neurodegenerative diseases, particularly in early stages of AD. Wide evidence has reported the presence of increased intracellular concentrations of Aβ in different in vitro and in vivo studies [32,33], suggesting that Aβ could be the responsible for the induction of mitochondrial dysfunction typically observed in AD. Although Aβ shows no precise location within mitochondrial compartments, experimental evidence supports the idea that both local production and direct import either from ER or from cytosol, could be the source of Aβ, the latter via mitochondrial TOM/TIM (translocase of the outer membrane/ translocase of the inner membrane) protein-import machinery [34]. However, the mechanistic details of how Aβ gains access to the different mitochondrial sub-compartments, as well as how it exerts its mitotoxic effects remain currently unestablished. Therefore, mitochondria have been proposed as early sensors of cellular damage and as indicators of the “point of no return during apoptosis”.
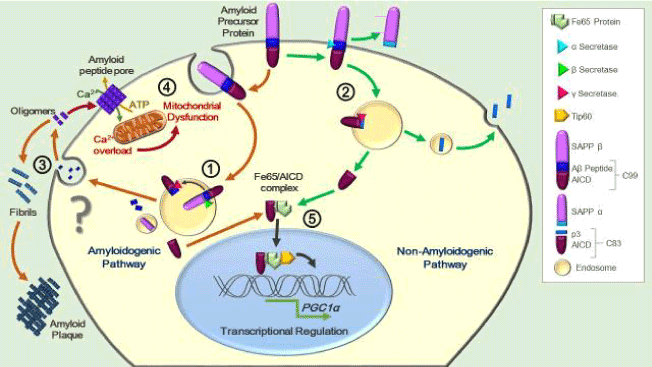
Figure 1. APP processing and its effect on mitochondrial biogenesis. In the amyloidogenic pathway (orange arrows), internalized APP on early endosomes gets sequentially cleaved by β-secretase, releasing the soluble extracellular domain of APP (sAPPβ), and then by the γ-secretase complex, which generates Aβ peptide and the APP intracellular carboxy-terminal domain (AICD) (1.1).By the other hand, during the non-amyloidogenic pathway (green arrows), a first cleavage by α-secretase prevents formation of Aβ, instead producing sAPPα and p3 peptide followed by a second cleavage by γ-secretase, producing AICD (1.2) Aβ peptide rapidly aggregates to form oligomers, protofibils and fibrils en route to the deposition of amyloid plaques (2.3). In its soluble oligomeric form (SOAβ), Aβ is able to disrupt the membrane by its insertion and formation of a non-specific pore-like structure that provokes calcium overload and ATP leakage, both impinging on mitochondrial function (2.4). AICD (generated in both pathways mentioned above), has been proposed to act as a transcriptional regulator through a mechanism involving interaction with the adaptor protein Fe65 and the action of Tip60, which could affect the PGC-1α gene expression –amongst others factors- having possible repercussions over mitochondrial function (2.5).
In this context, some molecular events on mitochondrial functioning are interesting to take into consideration, since experimental evidence shows that mitochondrial toxicity mechanisms encompass multiple deregulations across the organelle upon direct interactions between Aβ and all kinds of mitochondrial proteins; calcium dyshomeostasis [35], mitochondrial dynamics and morphology alteration [36], oxidative phosphorylation reduction [37], increase in ROS production [38], DNA/RNA mutation induction [39], apoptosis induction [40], etc. All of these dysfunctional features could provide as early biomarkers of AD’s onset.
Mitochondrial homeostasis is maintained through quality control measures that include balanced biogenesis of new mitochondria, autophagic degradation of dysfunctional mitochondria/mitophagy, mitochondria-specific protein refolding/turnover pathways, and mitochondrial fission/fusion events [41]. Mitochondrial biogenesis is a highly regulated cellular process controlled by the transcriptional coactivator PGC-1α, whose expression and activity regulation rely upon receptor tyrosine kinases, G protein–coupled receptors, and nitric oxide synthase through cGMP, AMPK activation, and SIRT1-mediated deacetylation [42].
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), as a transcriptional coactivator functions through physical interaction with transcription factors directly bound to DNA promoter regions. For example, PGC-1α N-terminus interacts with different hormone nuclear receptors, including PPARs, HNF4α, GR and ERRα. In addition, other protein components bind to transcription factors such as, MEF2C or FoxO1YY1 [43] and more importantly to nuclear respiratory factor (NRF) 1 and 2, leading to increased expression of mitochondrial transcription factor A and other nuclear-encoded mitochondrial proteins, for example ATP synthase subunits. Collectively, these PGC-1α effects culminate in the regulation of mitochondrial biogenesis and energy metabolism [44].
The mechanisms through which PGC-1α activates gene expression are poorly understood. Initial studies identified an extremely powerful autonomous transcriptional activity at the N-terminal region, for it shows to dock two other coactivators with acetyl transferase activity, SRC-1 and CBP/p300. Moreover, PGC-1α has been found in association with the GCN5 and TIP60 acetyl transferases complexes and many of their associated protein components [45]. GCN5 directly acetylates PGC-1α and negatively regulates its transcriptional activity through nuclear sub-localization. How spatially and temporally all these complexes are assembled to PGC-1α to control expression of genes is unknown. A current assembly model suggests that PGC-1α binds to specific transcription factors at promoters, then, additional recruitment of p300 and TRAP complexes would open the chromatin through histone acetylation thereby allowing initiation of RNA polII mediated transcription [43]. To terminate gene expression, GCN5 would acetylate PGC-1α resulting in relocalization to repressive subnuclear foci where it appears to co-localize with transcriptional repressor RIP140. Conversely, SIRT1 activation will maintain PGC-1α in a deacetylated active form bound to the chromatin and increasing rates of transcription [43].
Regarding AD pathogenesis, it has been widely associated with decreased expression and activity of proteins involved in mitochondrial bioenergetics, as it has been observed AD patients, an AD mouse model, and APP-mutated M17 cells [46]; additionally, mitochondrial dysfunction has confirmed in the triple transgenic AD mouse model where it was shown to precede plaque formation [47]. Mitochondrial dysfunction in AD may involve the action of APP and Aβ, and as previously mentioned, both were reported to target the mitochondria and impair mitochondrial function [25,48]. Moreover, APP-derived amyloid intracellular C-terminal domain AICD also appears to modulate mitochondrial functionality, for it has been proposed to act as a transcriptional regulator via a mechanism involving interaction with the adaptor protein Fe65 and the action of Tip60 [49,50] (Fig.1.5). In a recent study, AICD proved to upregulate PGC-1α promoter activity and mRNA levels on mutated MEF cells, the latter also reported in vivo in mouse brains. More importantly, AICD transcriptional effect was mediated by Fe65, and together they appear to have an impact on mitochondrial-associated features, such as ATP levels, oxygen consumption and mitochondrial membrane potential [51]. Taken together, these studies suggest that PGC-1α could stand as an important indicator of mitochondrial function, and changes on its expression or activity levels may represent an important event on induced toxicity.
Mitochondria form a dynamic cellular network that is tailored to the energetic and metabolic requirements of the cell. In many eukaryotic cell types, mitochondria continuously move along cytoskeletal tracks and frequently fuse and divide. These concerted activities control mitochondrial morphology, intracellular distribution and determine their cell type-specific appearance, all of them highly relevant in high demand sites, such as exocytotic synaptic sites. The antagonistic and balanced activities of the fusion and fission (collectively termed mitochondrial dynamics,) machineries [Fig.2.(B)] shape the mitochondrial compartment, bearing upon mitochondrial DNA inheritance and mitochondrial function maintenance [52]. A shift towards fusion favours the generation of interconnected mitochondria, whereas a shift towards fission produces numerous mitochondrial fragments. The large mitochondrial networks that are generated by fusion are beneficial in metabolically active cells, in which they contribute towards the dissipation of energy. By contrast, in quiescent cells mitochondria are frequently present as numerous morphologically and functionally distinct small spheres or short rods [53]. Mediators of mitochondrial dynamics have been highly conserved throughout evolution, which is in keeping with the critical regulatory roles of these proteins in both simple and complex organisms. The central groups of molecules involved in fission as well as in fusion are evolutionarily well-preserved large GTPases of the dynamin superfamily with a tripartite structure, a GTPase domain, a middle domain, and an assembly or GTPase effector domain (GED) at the C-terminus [54].
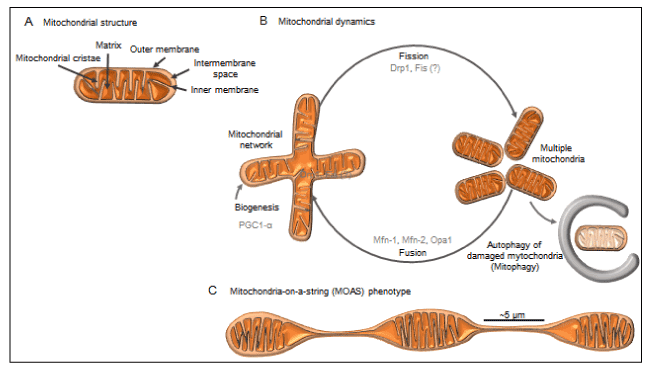
Figure 2. Mitochondrial structure and dynamics (2A). Mitochondrial structure, depicting its basic components: Outer and Inner mitochondrial membranes separated by an Intermembrane space, mitochondrial cristae, resulting from inner membrane invaginations and an interior matrix, containing the enzymes that are responsible for the citric acid cycle reactions. (2B). Mitochondrial dynamics are actively regulated processes designed to prevent cell damage and death through its multiple protective responses. PGC-1α–mediated mitochondrial biogenesis increases mitochondrial numbers and replaces damaged mitochondria. In addition, mitofusin-mediated fusion dilutes accumulated damage, including mitochondrial DNA mutations and oxidized proteins. When mitochondrial damage exceeds a critical level, Drp1-mediated asymmetric fission divides a mitochondrion into a larger, polarized healthy portion (which can reintegrate with the network) and a smaller depolarized, abnormal portion. This isolates the diseased mitochondria, allowing them to be packaged into autophagic vacuoles that are transported to lysosomes for elimination by mitophagy. Excessive fission causes cell death, whereas excessive mitophagy causes autophagic stress; both promote cell damage and death. (2C).Mitochondrial fission arrest phenotype that results in elongated interconnected organelles, “mitochondria-on-a-string” (MOAS), described by Zhang et al. [84] where multiple teardrop shaped mitochondria (~0.5 μm in diameter) were connected by thin double membranes extending up to 5 μm long. This unusual mitochondrial structure could be easily mistaken for multiple individual organelles if the membrane connections are missed.
Regarding fission machinery, Dynamin-related protein 1 (Drp1) is the only known large GTPase actively mediating mitochondrial fission, showing high expression levels in tissues with particular energy demands, such as brain and muscle in humans [55]. On the cellular level, almost the entire Drp1 pool is cytosolic with only about 3% of the total protein content residing at the mitochondrial surface. In order to induce organelle fission, cytosolic Drp1 requires activation and subsequent translocation to the mitochondrial outer membrane. Here, Drp1 dimers form ring-like multimeric structures on prospective outer mitochondrial membrane (OMM) fission sites through a self-assembling process [56], followed by GTP hydrolysis-driven conformational changes that—via ring constriction—lead to membrane severing and subsequent organelle division [57]. A hypothetic fission complex assembly model proposed by Otera et al. [58] outlined the OMM-bound mitochondrial fission factor (Mff) as an adaptor protein for Drp1 and mitochondrial elongation factor 1 (Mief1) as an enabler for its translocation. By blocking GTPase activity, Mief1 mediates Drp1GTP oligomerization. Drp1GTP multimers then promote the oligomerization of Mff with unknown factors into fission complexes, leading to the stimulation of Drp1 GTPase activity to induce mitochondrial fission [58]. According to a recently proposed model, expression of mammalian Fis1—another integral OMM protein—is able to partially reverse an elongated mitochondrial phenotype by sequestering Mief1 via unblocking Drp1 GTP hydrolysis, culminating in organelle fission [59]. Fis1 for a long time was supposed to mediate Drp1 anchoring on the OMM, as was shown for yeast, where the molecular components interacting in fission and fusion processes were primarily elucidated. However, Fis1 is evenly distributed along a mitochondrion, whereas Drp1 binding is patchy [54].
Drp1 translocation to the OMM and GTPase activation are provoked by a variety of stressors from outside and inside the cell inducing mitochondrial fragmentation as an early process in apoptosis or mitophagic clearance of dysfunctional parts. Also during mitosis, studies have shown enhanced fragmentation activated by Cdk1/cyclinB and aurora A-mediated phosphorylation at S616 (in human Drp1) [60]. On the other hand, fission can be inhibited by protein kinase A-based phosphorylation of a serine within the GED domain (S637 for human, S656 for rat Drp1) blocking GTPase activation and probably also Drp1 recruitment to the OMM [61], while dephosphorylation of S637 by the Ca2+-dependent phosphatase calcineurin promotes mitochondrial fission [62]. In addition, mitochondrial E3 ubiquitin ligases MARCH5 and Parkin participate in the regulation of mitochondrial division, the former being a positive regulator [63] whereas the latter promotes Drp1 proteasome-dependent degradation [64]. Finally, nytrosyslation and sumoylation stabilize the mitochondrial pool of Drp1 and enhance fission, further substantiating the importance of posttranslational modification in regulating morphology of the organelle [65].
Exceeding fission occurs for example, in the preparation of mitosis, or as a reaction to external stresses (e.g., exposure to elevated ROS levels [66], or uncouplers of oxidative phosphorylation [67] often as an initiation of apoptotic cell death, including neuronal degenerations or premature senescence. If fission results in mitochondrial fragments with different physiological performance, i.e., one with high and the other with reduced MMP), those with low MMP are no longer able to fuse [68] and are prone to autophagic degradation [Fig.2 (B)]. By this, fission is an important step in getting rid of dysfunctional mitochondria or mitochondrial components and could help to identify the point of no return on neuronal death.
On the other hand, Mitofusins (Mfn1 and Mfn2) are the motors driving OMM fusion. With an N-terminal GTPase domain and C-terminal coiled-coil regions toward the cytosol, their two transmembrane regions anchor to the OMM and thus mediate the tethering of two adjacent mitochondria through their hydrophobic repeats (HR2) [28]. IMM fusion is mediated by OPA1 (optic atrophy protein 1), an integral protein that faces the intermembrane space and controls IMM structure and its remodeling through cristae shaping [69] [Fig.2 (A)]. OPA1 is also required to maintain MMP in concert with Mfn1, as IMM fusion requires an intact MMP [67,70]. Ablation of Mfn2 in mice is embryonically lethal and MFN2 mutations in humans cause Charcot–Marie–Tooth type IIa (CMTIIa), a peripheral sensorimotor neuropathy characterized by degeneration of long peripheral axons, whereas OPA1 mutations cause autosomal-dominant optic atrophy [71]. Silencing of Mfn1 or Mfn2 results in mitochondrial fragmentation and increased sensitivity to apoptotic stimuli. Furthermore, overexpression of Mfn1 or 2, in addition to increasing mitochondrial connectivity, results in delayed Bax activation, cytochrome c release, and apoptotic death, suggesting a role for mitofusins in cell death [28].
Fusion and fission are guided by lipids generated by mitochondrial phospholipase D, notably phosphatidic acid [72]. The small, negatively charged lipid head group of phosphatidic acid causes negative curvature of lipid bilayers and recruits adaptor proteins, promoting fusion. However, phosphatidic acid can be hydrolyzed by lipin1, creating diacylglycerol, which promotes fission [73]. Furthermore, organelle interactions are crucial for mitochondrial lipid biosynthesis, which occurs at “mitochondria-associated membranes” (MAMs)—patches of ER membranes attached to mitochondria, containing several phospholipid and glycosphingolipid biosynthetic enzymes. Mitofusin2 is also located in the endoplasmic reticulum, where it alters morphologic features and promotes endoplasmic reticulum–mitochondrial tethering [68] , thereby enhancing mitochondrial calcium uptake and handling [74].
Membrane transport and fusion are intimately related processes that must be coordinately regulated to achieve proper organellar trafficking, and it is likely that molecular adaptor complexes at the outer mitochondrial membrane will serve dual roles in transport and fusion. In fact, a study performed by Misko et al. [75] revealed that Mfn2 is directly involved in and required for axonal mitochondrial transport, distinct from its role in mitochondrial fusion. Mfn2 disruption altered mitochondrial movement selectively, leaving transport of other organelles intact. Importantly, both Mfn1 and Mfn2 interact with mammalian Miro and Milton proteins, members of the molecular complex that link mitochondria to kinesin motors, but only Mfn2 knockdown provokes an alteration in attachment to microtubule based transport systems [75]. Stated briefly, mitofusins have been reported to perform numerous functions, including the regulation of mitochondrial fusion ER-mitochondrial tethering [74], apoptotic cell death and outer membrane permeability [28], oxidative phosphorylation and gradient coupling and microtubule-based mitochondrial transport.
In terms of the impact of fission and fusion in AD, as mentioned above, neurons are particularly vulnerable to mitochondrial dysfunction because of their high metabolic rate dependence and complex morphology. Mitochondria are of pivotal importance for synaptic development and plasticity, and changes in its distribution and/or function can result in synaptic dysfunction, loss of neurotransmitter release and active vesicular recycling [76]. During the past few years evidence has been emerging indicating that dysregulated mitochondrial structure and morphology may represent an important component in the complex pathogenesis of AD. Neurons that are either overexpressing APP or exposed to toxic Aβ peptides display abnormal levels of mitochondria shaping proteins in association with perinuclear aggregates [77,78]. These findings are paralleled by studies of AD patient brains reporting abnormal expression levels of mitochondrial morphogenic proteins, one specifically revealing abnormal interactions between Drp1 and Aβ, which could initiate mitochondrial fragmentation in neurons affected by AD [79]. Moreover, increased expression levels of the mitochondrial fission genes Drp1 and Fis1 and decreased expression levels of the mitochondrial fusion genes Mfn1, Mfn2, Opa1 and Tomm40 were reported in postmortem brain specimens from patients with AD at early, definite and severe stages of progression. Reduced levels of DRP1 and increased mitochondrial length were found in AD fibroblasts in one study [80]. Additionally, specific post-translational Drp1 modifications have recently been implicated with AD. For example, increased levels of Drp1 phosphorylation at position S616 are found in AD brains. Aβ-mediated DRP1 activation by S-nitrosylation was also observed [81], promoting mitochondrial fission.
Transport, distribution, and dynamics of mitochondria are all altered in AD patients and mouse models [77,82]. The progressive Aβ accumulation in mitochondria of APP mutant mice is associated with diminished activity of respiratory chain complexes III and IV, and a reduction in oxygen consumption [83], both considered preludes for oxidative stress.
Accordingly, to determine the role of FAD mutations in mitochondrial dynamics, a study that employed three transgenic mouse models revealed a consistent reduction of mitochondrial activity, where inhibition of axonal trafficking was among the earliest abnormalities detected before the onset of memory phenotype or formation of amyloid deposits. In transport-impaired neurons no correlation was found with Aβ levels, but the neurons were more susceptible to excitotoxicity. Interestingly, levels of fission and fusion proteins were not altered in any of these strains; however, mitochondrial length in APP and PSEN1 mutant mice showed a modest although inconsistent increase [84].
As pointed previously, Aβ has been shown to cause increased fission and decreased fusion, resulting in mitochondrial fragmentation, decreased mitochondrial density and, importantly, altered synaptic localization [78,80]. While changes in mitochondrial morphology affect the entire organelle, methods utilized to date to monitor mitochondrial dynamics primarily include fluorescence microscopy or/and conventional electron microscopy (EM), which reveals morphological changes in a thin section of the organelle. To account for a three-dimensional architecture of the brain tissue and organelles, a study developed 3-dimensional electron microscopy (3D EM) reconstruction of standard transmission electron microscopy (TEM) images and examined mitochondria in the CA1 hippocampal region from 5 transgenic mouse models carrying human FAD mutations and in postmortem brain tissue from AD patients. Surprisingly, instead of observing multiple small mitochondria produced by excessive fission previously reported for AD, an unknown mitochondrial phenotype was identified;
elongated interconnected organelles where multiple teardrop shaped mitochondria (~0.5 µm in diameter) were connected by thin double membranes extending up to 5 µm long [84] [Fig.2 (C)]. This new phenotype, later called M.O.A.S (as in “mitochondria on a string”) may emerge at the final stages of fission process (a fission arrest phenotype) and was not associated with altered levels of expression of fission/fusion proteins nor altered translocation of activated Drp1-S616 to mitochondria, but with reduced GTPase activity. Since MOAS formation was also observed in the brain tissue of WT mice in response to hypoxia or during chronological aging, fission arrest may represent fundamental compensatory adaptation to bioenergetic stress providing protection against mitophagy, which could preserve residual mitochondrial function. This study confers a major role of mitochondrial dynamics in regulating neuronal survival, as it reveals greater complexity of mitochondrial fission/fusion that includes sustained transition state dynamics, emphasizing the importance of utilizing advanced 3D tools to study complex tissues and organelles.
To develop new therapies for AD, we need a clearer understanding of how Aβ and mitochondria change during AD development and progression and how they affect each other. It is now generally accepted that the dysregulation of mitochondrial dynamics is either causative or at least a significant factor in the pathogenesis of a spectrum of human diseases, including neurodegenerative diseases. How mitochondrial dynamics is regulated under physiological and pathological conditions is not understood. Major goals of future research are to enlighten what governs the physiological regulation of mitochondrial dynamics, what determines the origin of a degenerative process in sporadic AD and the role mitochondria have in this process. To further address therapeutic targets related to fission and fusion imbalance, Drp1 role needs to be clearly defined regarding its participation in Aβ-induced mitochondria fragmentation.
Identification of a new M.O.A.S. phenotype by Trushina and co-workers [85], accounts for an undeniable contribution to the understanding of mitochondrial function and represents what could be a new predictor for the “no return point” regarding its fundamental role in maintaining synaptic functional integrity and neuronal survival. Taken together, this could represent a new start point ensuing innovative strategies and experimental approaches to develop an appropriate therapy.
- Ben-Avraham D, Muzumdar RH, Atzmon G (2012) Epigenetic genome-wide association methylation in aging and longevity. Epigenomics 4: 503-509. [crossref]
- Prince MJ, Wimo A, Guerchet MM, Ali GC, Wu YT et al. (2015) World Alzheimer Report 2015 - The Global Impact of Dementia An analysis of prevalence, incidence, cost and trends: Alzheimer's Disease International. [crossref]
- World Health Organization [WHO] (2016) Dementia Fact Sheet No. 362. [crossref]
- Alzheimer's disease International. (2009) World Alzheimer Report 2009. [crossref]
- Castellani RJ, Rolston RK, Smith MA (2010) Alzheimer disease. Dis Mon 56: 484-546. [crossref]
- De Strooper B, Annaert W (2000) Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci 113: 1857-1870. [crossref]
- Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, et al. (2011) Alzheimer's disease. The Lancet 377: 1019-1031. [crossref]
- Sisodia SS (1992) Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc Natl Acad Sci U S A 89: 6075-6079. [crossref]
- Haass C, Hung AY, Schlossmacher MG, Teplow DB, Selkoe DJ (1993) beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J Biol Chem 268: 3021-3024. [crossref]
- Thinakaran G, Koo EH (2008) Amyloid precursor protein trafficking, processing, and function. J Biol Chem 283: 29615-29619. [crossref]
- Koo EH, Squazzo SL (1994) Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 269: 17386-17389. [crossref]
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, et al. (1995) The Swedish mutation causes early-onset Alzheimer's disease by [beta]-secretase cleavage within the secretory pathway. Nat Med 1: 1291-1296. [crossref]
- Chyung AS, Greenberg BD, Cook DG, Doms RW, Lee VM (1997) Novel beta-secretase cleavage of beta-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment of NT2N cells. J Cell Biol 138: 671-680. [crossref]
- Wolfe MS (2008) Inhibition and modulation of gamma-secretase for Alzheimer's disease. Neurotherapeutics 5: 391-398. [crossref]
- Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362: 329-344. [crossref]
- Jarrett JT, Berger EP, Lansbury PT (1993) The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32: 4693-4697. [crossref]
2021 Copyright OAT. All rights reserv
- Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256: 184-185. [crossref]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, et al. (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46: 860-866. [crossref]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, et al. (1998) Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95: 6448-6453. [crossref]
- Arispe N, Rojas E, Pollard HB (1993) Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci U S A 90: 567-571. [crossref]
- Sepulveda FJ, Parodi J, Peoples RW, Opazo C, Aguayo LG (2010) Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS One 5: e11820. [crossref]
- Parodi J, Sepúlveda FJ, Roa J, Opazo C, Inestrosa NC, et al. (2010) Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol Chem 285: 2506-2514. [crossref]
- Kim SY, Moon JH, Lee HG, Kim SU, Lee YB (2007) ATP released from beta-amyloid-stimulated microglia induces reactive oxygen species production in an autocrine fashion. Exp Mol Med 39: 820-827. [crossref]
- Saez-Orellana F, Godoy PA, Bastidas CY, Silva-Grecchi T, Guzman L, et al. (2016) ATP leakage induces P2XR activation and contributes to acute synaptic excitotoxicity induced by soluble oligomers of beta-amyloid peptide in hippocampal neurons. Neuropharmacology 100: 116-123. [crossref]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, et al. (2006) Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15: 1437-1449. [crossref]
- Keating DJ (2008) Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J Neurochem 104: 298-305. [crossref]
- Weinberg SE, Sena LA, Chandel NS (2015) Mitochondria in the regulation of innate and adaptive immunity. Immunity 42: 406-417. [crossref]
- Suen DF, Norris KL, Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22: 1577-1590. [crossref]
- Celsi F, Pizzo P, Brini M, Leo S, Fotino C, et al. (2009) Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta 1787: 335-344. [crossref]
- Montero M, Alonso MT, Carnicero E, Cuchillo-Ibáñez I, Albillos A, et al. (2000) Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat Cell Biol 2: 57-61. [crossref]
- Tait SW, Green DR (2012) Mitochondria and cell signalling. J Cell Sci 125: 807-815. [crossref]
- Tillement L, Lecanu L, Papadopoulos V (2011) Alzheimer's disease: effects of β-amyloid on mitochondria. Mitochondrion 11: 13-21. [crossref]
- Muirhead KE, Borger E, Aitken L, Conway SJ, Gunn-Moore FJ (2010) The consequences of mitochondrial amyloid beta-peptide in Alzheimer's disease. Biochem J 426: 255-270. [crossref]
- Benek O, Aitken L, Hroch L, Kuca K, Gunn-Moore F, et al. (2015) A Direct interaction between mitochondrial proteins and amyloid-beta peptide and its significance for the progression and treatment of Alzheimer`s disease. Curr Med Chem. [crossref]
- Csordás G, Thomas AP, Hajnóczky G (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18: 96-108. [crossref]
- Wang X, Su B, Fujioka H, Zhu X (2008) Dynamin-Like Protein 1 Reduction Underlies Mitochondrial Morphology and Distribution Abnormalities in Fibroblasts from Sporadic Alzheimer’s Disease Patients. The American Journal of Pathology 173: 470-482. [crossref]
- Tillement L, Lecanu L, Yao W, Greeson J, Papadopoulos V (2006) The spirostenol (22R, 25R)-20alpha-spirost-5-en-3beta-yl hexanoate blocks mitochondrial uptake of Abeta in neuronal cells and prevents Abeta-induced impairment of mitochondrial function. Steroids 71: 725-735. [crossref]
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA (2005) Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem 93: 953-962. [crossref]
- Volicer L, Crino PB (1990) Involvement of free radicals in dementia of the Alzheimer type: a hypothesis. Neurobiol Aging 11: 567-571. [crossref]
- Li YP, Bushnell AF, Lee CM, Perlmutter LS, Wong SK (1996) Beta-amyloid induces apoptosis in human-derived neurotypic SH-SY5Y cells. Brain Res 738: 196-204. [crossref]
- Baker MJ, Tatsuta T, Langer T (2011) Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol 3. [crossref]
- Whitaker RM, Corum D, Beeson CC, Schnellmann RG (2016) Mitochondrial Biogenesis as a Pharmacological Target: A New Approach to Acute and Chronic Diseases. Annu Rev Pharmacol Toxicol, 56: 229-249. [crossref]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P (2008) Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett 582: 46-53. [crossref]
- Lin J, Handschin C, Spiegelman BM (2005) Metabolic control through the PGC-1 family of transcription coactivators. Cell Metabolism 1: 361-370. [crossref]
- Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, et al. (2006) GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab 3: 429-438. [crossref]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH (2011) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet 20: 4515-4529. [crossref]
- Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, et al. (2009) Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 106: 14670-14675. [crossref]
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci 26: 9057-9068. [crossref]
- McLoughlin DM, Miller CC (2008) The FE65 proteins and Alzheimer's disease. J Neurosci Res 86: 744-754. [crossref]
- Chang KA, Suh YH (2010) Possible roles of amyloid intracellular domain of amyloid precursor protein. BMB Rep 43: 656-663. [crossref]
- Robinson A, Grosgen S, Mett J, Zimmer VC, Haupenthal VJ, et al. (2014) Upregulation of PGC-1alpha expression by Alzheimer's disease-associated pathway: presenilin 1/amyloid precursor protein (APP)/intracellular domain of APP. Aging Cell 13: 263-272. [crossref]
- Ono T, Isobe K, Nakada K, Hayashi JI (2001) Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet 28: 272-275. [crossref]
- Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11: 872-884. [crossref]
- Bereiter-Hahn J (2014) Mitochondrial dynamics in aging and disease. Prog Mol Biol Transl Sci 127: 93-131. [crossref]
- Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol 143: 351-358. [crossref]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, et al. (2005) Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol 170: 1021-1027. [crossref]
- Lackner LL, Horner JS, Nunnari J (2009) Mechanistic analysis of a dynamin effector. Science 325: 874-877. [crossref]
- Otera H, Ishihara N, Mihara K (2013) New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta 1833: 1256-1268. [crossref]
- Zhao J, Liu T, Jin S, Wang X, Qu M, et al. (2011) Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J 30: 2762-2778. [crossref]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282: 11521-11529. [crossref]
- Chang CR, Blackstone C (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282: 21583-21587. [crossref]
- Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8: 939-944. [crossref]
- Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, et al. (2010) Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci 123: 619-626. [crossref]
- Wang H, Song P, Du L, Tian W, Yue W, et al. (2011) Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 286: 11649-11658. [crossref]
- Harder Z, Zunino R, McBride H (2004) Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol 14: 340-345. [crossref]
- Jendrach M, Mai S, Pohl S, Voth M, Bereiter-Hahn J (2008) Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion, 8: 293-304. [crossref]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, et al. (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177-189. [crossref]
- Rojo M, Legros F, Chateau D, Lombes A (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115: 1663-1674. [crossref]
- Olichon A, Guillou E, Delettre C, Landes T, Arnauné-Pelloquin L, et al. (2006) Mitochondrial dynamics and disease, OPA1. Biochim Biophys Acta 1763: 500-509. [crossref]
- Legros F, Lombès A, Frachon P, Rojo M (2002) Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 13: 4343-4354. [crossref]
- Oettinghaus B, Licci M, Scorrano L, Frank S (2012) Less than perfect divorces: dysregulated mitochondrial fission and neurodegeneration. Acta Neuropathol 123: 189-203. [crossref]
- Yang CY, Frohman MA (2012) Mitochondria: signaling with phosphatidic acid. Int J Biochem Cell Biol 44: 1346-1350. [crossref]
- Archer SL (2013) Mitochondrial Dynamics- Mitochondrial Fission and Fusion in Human Diseases. New England Journal of Medicine 369: 2236-2251. [crossref]
- de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605-610. [crossref]
- Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH (2010) Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30: 4232-4240. [crossref]
- Li Z, Okamoto KI, Hayashi Y, Sheng M (2004) The Importance of Dendritic Mitochondria in the Morphogenesis and Plasticity of Spines and Synapses. Cell 119: 873-887. [crossref]
- Wang X, Perry G, Smith MA, Zhu X (2010) Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener Dis 7: 56-59. [crossref]
- Wang X, Su B, Lee HG, Li X, Perry G, et al. (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci 29: 9090-9103. [crossref]
- Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 20: 2495-2509. [crossref]
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, et al. (2008) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 105: 19318-19323. [crossref]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, et al. (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324: 102-105. [crossref]
- Du H, Guo L, Yan S, Sosunov AA, McKhann GM, et al. (2010) Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A 107: 18670-18675. [crossref]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, et al. (2005) Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J 19: 2040-2041. [crossref]
- Zhang L, Trushin S, Christensen TA, Bachmeier BV, Gateno B, et al. (2016) Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s disease. Scientific Reports 6: 18725. [crossref]
- Trushina E, Nemutlu E, Zhang S, Christensen T, Camp J, et al. (2012) Defects in Mitochondrial Dynamics and Metabolomic Signatures of Evolving Energetic Stress in Mouse Models of Familial Alzheimer's Disease. PLoS ONE 7: e32737. [crossref]