Abstract
Alzheimer’s disease is characterized by progressive memory loss, death of hippocampal, cortical pyramidal and basal forebrain cholinergic neurons, and formation of amyloid-beta (Aβ) plaques and neurofibrillary tangles. Neurotoxic fragments of amyloid-beta protein precursor (AβPP), such as Aβ1-42, are generated by amyloidogenic processing via β- and γ- secretases. However, recent findings suggest that full length AβPP is also toxic to neurons, although the mechanism by which the non-cleaved protein induces cell death is presently unclear. Here, we utilize a transient transfection strategy to show that overexpression of wild type (WT) AβPP in mouse hippocampal HT22 cells induces caspase-dependent apoptosis. Cell death induced by AβPP is independent of the mitochondrial permeability transition but requires the activation of Bax. Incubation with β- or γ-secretase inhibitors has no effect on AβPP content or apoptosis and the mechanism of AβPP-induced cell death in HT22 cells is distinct from that of Aβ1-42 overexpression. Importantly, a mutant of AβPP that does not localize to mitochondria fails to induce apoptosis in HT22 cells. Finally, ρ0 SH-SY5Y neuroblastoma cells lacking functional mitochondria are resistant to AβPP-induced apoptosis. These findings demonstrate that the localization of full length AβPP to functional mitochondria is a prerequisite for this molecule to induce Bax-dependent apoptosis of hippocampal neuronal cells.
Key words
Amyloid-beta protein precursor (AβPP), amyloid-beta (Aβ), apoptosis, Bax, mitochondria
Abbreviations
AD: Alzheimer’s disease, ABAD: Amyloid beta binding alcohol dehydrogenase, Aβ: amyloid-beta, ANT: adenine nucleotide translocator, AβPP: Amyloid-beta protein precursor, AICD: Amyloid-beta protein precursor intracellular domain, AβPP 3M: Amyloid-beta protein precursor triple mutant, BSA: bovine serum albumin, CHO: Chinese hamster ovarian cells, CsA: cyclosporine, Cox-IV: cytochrome c oxidase subunit 4 isoform 1, cyto: cytosolic, DTT: dithiothreitol, ETC: electron transport chain, GAPDH: glyceraldehyde 3-phosphate dehydrogenase, GSH: glutathione, HBSS: Hank’s balanced buffer solution, mito: mitochondrial, MOS: mitochondrial oxidative stress, mPTP: mitochondrial permeability transition pore, OPA1: optic atrophy, ROS: reactive oxygen species, ρ0: rho negative-lack mitochondrial DNA, ρ+: rho positive- contains mitochondrial DNA, TIM: translocase of the inner mitochondrial membrane, TOM: translocase of the outer mitochondrial membrane, VDAC: voltage dependent anion channel, WT: wild-type
Introduction
Alzheimer’s disease (AD) is the most common form of dementia involving both progressive and debilitating memory loss induced by the death of hippocampal, cortical pyramidal and basal forebrain cholinergic neurons. With an aging population, a risk rate of 40% for people over the age of 80, and therapies with low efficacy, AD is a major health care concern [1,2]. Furthermore, many purported therapeutic targets have been identified to no avail because the underlying pathologic mechanism in AD is not well understood.
Multiple studies have pointed to a prominent role for mitochondrial dysfunction and mitochondrial oxidative stress (MOS) in the pathogenesis of AD. For example, in platelets collected from AD patients, there is reduced activity of cytochrome c oxidase. Similar results have been observed in the frontal, temporal and parietal cortices from autopsied AD patients [3]. Decreases in the distribution of healthy mitochondria were found in hippocampal pyramidal neurons from AD patients compared to areas of the brain that are not affected in this disease [4]. Indices of increased MOS have also been found in brains of AD patients, such as increased oxidative damage to mitochondrial DNA and oxidative modification of the mitochondrial protein, VDAC1 [5,6]. Additional studies utilizing cybrid cells from AD patients showed an increase in reactive oxygen species (ROS) production as well as decreases in ATP production and cytochrome oxidase activities, compared to cybrid cells from healthy individuals [7]. Furthermore, in vivo studies have shown that transgenic AD mice (3xTg-AD) display mitochondrial dysfunction and MOS prior to the detection of overt AD pathology [8]. Finally, aberrant production of ROS by aging dysfunctional mitochondria triggers a vicious cycle by inducing amyloidogenic processing of AβPP, a process which can be suppressed by the mitochondrial transcriptional regulator TFAM [9,10]. Together, these studies suggest that mitochondrial dysfunction and MOS are significant and early events in the pathogenesis of AD.
The amyloid cascade hypothesis has dominated the field of AD research and is founded on substantial evidence that toxic Aβ species contribute to the pathology of AD, particularly at the level of the mitochondria. For example, Aβ has been shown to localize to mitochondria and to interact with amyloid-beta binding alcohol dehydrogenase (ABAD) within these organelles in brain tissue from AD patients and AD transgenic mice [11,12]. Aβ has also been shown to inhibit multiple complexes of the electron transport chain (ETC) and consequently induces MOS and organelle dysfunction [13,14].
In addition to Aβ, full length AβPP also localizes to mitochondria. AβPP inserts into the mitochondrial translocases, TOM 40 and TIM 23, and as a result has the potential to induce mitochondrial dysfunction [15]. In this context, we have previously shown that overexpression of full length WT AβPP induces intrinsic apoptosis in Chinese hamster ovary (CHO) cells, independently of Aβ production [16]. Our previous study is in agreement with others showing that full length WT AβPP can induce apoptosis in the human SH-SY5Y neuroblastoma and differentiated NBP2 neuroblastoma cell lines [17,18]. Furthermore, an in vivo study using WT AβPP overexpressing mice showed that these mice develop memory deficits independently of Aβ accumulation in the hippocampus [19]. Collectively, the above studies suggest that full length AβPP may contribute to AD pathogenesis; however, whether its localization to mitochondria is necessary to induce neuronal cell death is currently unknown.
In the present study, we examined the effects of WT AβPP overexpression in the mouse HT22 hippocampal cell line. Overexpression of full length WT AβPP induces Bax- and caspase-dependent apoptosis in HT22 cells. AβPP protein levels and AβPP-induced apoptosis were unaffected by inhibitors of either β- or γ- secretases and the mechanism of AβPP-induced cell death is distinct from that of Aβ1-42 overexpression, suggesting that the full length protein is intrinsically toxic to HT22 cells. Finally, we show that AβPP localization to functional mitochondria is required for this protein to induce neuronal apoptosis.
Materials and methods
Cell culture
Immortalized mouse hippocampal cells (HT22) were plated on 35-mm diameter plastic dishes in DMEM low glucose (with L-glutamine) containing 10% fetal bovine serum and penicillin/streptomycin (100 Units/mL/100 μg/mL). SH-SY5Y (both ρ+ and ρ0) cells were plated on 35-mm diameter plastic dishes in DMEM high glucose (with L-glutamine) containing 10% fetal bovine serum, penicillin/streptomycin (100 Units/mL/100 μg/mL), 50 μg/mL uridine and 100 μg/mL sodium pyruvate. Cells were cultured overnight at 37°C in 10% CO2. The following day cells were transfected and treated, at which point cultures were 80-90% confluent.
Reagents
Hoechst dye, kanamycin sulfate, and staurosporine were purchased from Sigma Aldrich (St. Louis, MO). Dulbecco’s modified eagle’s medium (low glucose), lipofectamine 2000, Opti-MEM medium and Image-iT live mitochondrial transition pore assay kit was purchased from Invitrogen (Carlsbad, CA). Cyclosporin A, Q-VD-OPh non-o-methylated, active Bax (6A7) monoclonal antibody, and glutathione monoethylester were purchased from Calbiochem (SanDiego, CA). Active caspase-3 antibody was purchased from Promega (Madison, WI). β-secretase inhibitor IV and Bax inhibiting peptide were purchased from Millipore (Billerica, MA). The γ-secretase inhibitor XXIII was purchased from EMD (Darmstadt, Germany). The β-secretase and γ-secretase inhibitors were initially prepared as concentrated stock solutions in DMSO. The final DMSO concentration used on the cells was 0.1%. The AβPP antibody was purchased from Biolegend (San Diego, CA). The OPA1 antibody was purchased from BD (Franklin Lakes, NJ). The mitochondrial/cytosolic fractionation kit was purchased from BioVision (Mountain View, CA). Site directed mutagenesis kit was purchased from Agilent Technologies (Santa Clara, CA). Primers for mutagenesis were purchased from Integrated DNA Technologies (Coralville, IA). The DsRed2-WT AβPP (N-terminus tagged) plasmid was a generous gift from Dr. Xiongwei Zhu from Case Western Reserve University (Cleveland, OH) and it encoded the human AβPP695 isoform.
Plasmid preparation
DsRed2 and DsRed2-WT AβPP were transformed using 50 ng of plasmid in JM109 Eschericia coli (E. coli) and were subsequently grown on LB agar plates containing 35 μg/mL kanamycin sulfate at 37°C overnight. Starter cultures were grown in LB broth with 35 μg/mL kanamycin sulfate for 6-8 h at 37°C, and diluted 1:250 into overnight cultures. Plasmid purification was performed using the Qiagen Maxi Prep Kit (Valencia, CA) per the manufacturer’s instructions. DNA concentrations were determined using an average of three trials on a Thermo Scientific NanoDrop 2000.
Transfection
DsRed2 and DsRed2-WT AβPP were used at a concentration of 5μg/mL. Plasmids were transfected using a standard Lipofectamine 2000 protocol. Cell cultures were incubated with the plasmid-Lipofectamine 2000 mixture in Opti-MEM for 6 h at 37°C and 10% CO2. Transfection media was removed from cell cultures after 6 h and cells were placed in 1mL of culture medium and the indicated treatments were administered. Cells were then incubated overnight at 37°C and 10% CO2.
Mitochondrial, cytosolic, and plasma membrane subcellular fractionation
Medium was aspirated and cells were washed 1× in ice-cold phosphate buffered saline solution (PBS, pH 7.4). A 200 μL aliquot of cytosolic buffer (provided in the kit, diluted 1:5 in ddH2O, with added protease inhibitor cocktail and 1 mM DTT, as per the manufacturer's recommendations) was added to the cells and allowed to incubate on ice for 20 min. Cells were scraped, harvested, and then homogenized with 40 passes of a dounce homogenizer. Samples were spun down at 720 rcf for 10 min at 4°C. The supernatant from each sample was transferred to a new tube labeled “mitochondrial fraction” and spun at 10,000 rcf for 30 min at 4°C. The supernatant was then transferred to a new tube labeled “cytosolic fraction” and the pellet in the mitochondrial fraction tube was resuspended in 100 μl of mitochondrial buffer (provided in the kit, with added protease inhibitor cocktail and 1 mM DTT, as per the manufacturer's recommendations).
Immunoblotting
Immunoblot analysis was performed as previously described [20]. The antibody used for western blotting for AβPP was BioLegend Clone C1/6.1 and was raised against the conserved carboxyl-terminal 20 residues of AβPP (residues 676-695 of AβPP695). This antibody was used at a final dilution of 1mg/ml for immunoblotting. Additional antibodies used for western blotting included Cox-IV, GAPDH, Na/K ATPase, OPA1, and β-tubulin. Each of these antibodies was used at the dilution recommended by the manufacturer.
Immunocytochemistry and fluorescence imaging
HT22 cells were washed once in 1X PBS and fixed with 1 mL 4% paraformaldehyde for 1 h at RT. Cells were permeabilized and blocked with a 5% BSA solution in 0.2% Triton X-100-1X PBS (PBS-T) for 1h at RT. Primary antibody was diluted in a 2% BSA solution PBS-T and incubated with cells overnight at 4°C. The following day cells were washed 5x with 1X PBS-T over 30 min. Cells were incubated for 1h at RT in Hoechst (1:500) and secondary antibody at a dilution of 1:250 in 2% BSA solution in PBS-T. Cells were washed 5x in 1X PBS-T over 30 min, and placed in anti-quench solution (1X PBS containing p-phenylenediamine). Fluorescent images were captured using a 40X objective on a Zeiss Axiovert-200 microscope.
Mitochondrial permeability transition pore (mPTP) assay
At 24 h post-transfection, HT22 cells were washed once with Hank’s balanced buffered salt solution (HBSS; pH 7.4) and cells were placed in 1 mL of Hank’s buffer. Calcein AM (1mM), Hoechst (1mM), and cobalt (II) chloride hexahydrate (1 M) were added at 1 μl each per well of cells. Cells were incubated at 37ºC and 10% CO2 for 15 min. Next, cells were washed once with HBSS and imaged live by fluorescence microscopy.
Site-directed mutagenesis
AβPP 3M was generated by mutagenesis of the DsRed-tagged AβPP in four site-directed mutagenesis reactions, each creating a point mutation. Sense primer sequences were as follows; ATGAATGTCCAGAATGGGAACTGGGATTCAGATCC (AAG to AAC or Lys to Asn) TGTGGCAGACTGAACATGAACATGAATGTCCAGAATG (CAC to AAC or His to Asn) GATTGCCATGTTCTGTGGCAAACTGAACATGAACATGAA (AGA to AAA or Arg to Lys) ATTGCCATGTTCTGTGGCAACCTGAACATGAACATGAATG (AAA to AAC or Lys to Asn). Primers were generated using Agilent Technologies web-based QuikChange Primer Design Program. Site direction reactions were set up as per the manufacturer’s recommendations in the QuickChange Lightning Site directed mutagenesis kit.
Statistical analysis and quantification
Recorded values of apoptosis, including caspase-3 activation and DNA fragmentation, and mPTP opening were quantified from 15 images per experiment, for each treatment. Data are represented as mean ± SEM for the number (n) of independent experiments performed. Only transfected cells (DsRed2 fluorescent) were counted in each field and values are shown as a percentage of total transfected cells. Experiments are representative of at least n=3 trials. Statistical analysis was performed using one-way analysis of variance (ANOVA) and a post hoc Tukey’s test. Differences were considered significant with a p value <0.05.
Results
Overexpression of AβPP induces apoptosis in HT22 cells
To assess if overexpression of AβPP induces apoptosis in a hippocampal cell line, we transfected HT22 cells with a DsRed2 WT AβPP construct or a DsRed2 empty vector. Typical transfection efficiency was between 20-30%. HT22 cells transfected with the control DsRed2 vector appeared healthy, with intact large nuclei and little-to-no active caspase-3 staining (Figure1A, left panels). In contrast, cells expressing WT AβPP (DsRed2 fluorescence) displayed an increase in active caspase-3 staining as well as an increase in fragmented and condensed nuclei (Figure 1A, right panels). HT22 cells which were not transfected with WT AβPP did not appear apoptotic. Quantification of these data showed that cells transfected with WT AβPP had a significant increase in condensed nuclei compared to cells transfected with DsRed2 alone (Figure 1B). Furthermore, WT AβPP induced a significant increase in cells which displayed both condensed nuclei and active caspase-3 staining compared to DsRed2 alone (Figure 1B).
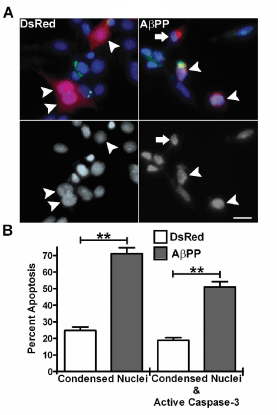
Figure 1. Overexpression of AβPP induces apoptosis of HT22 hippocampal cells. A) HT22 cells at 24 h post-transfection with either DsRed2 (left) or DsRed2- AβPP (right). Top panels show fluorescence of DsRed2 (red), active caspase-3 (green) and Hoechst stain (blue). Arrowheads indicate healthy transfected cells with large nuclei in the left panels. In the right panels arrowheads indicate transfected cells with apoptotic nuclei and active caspase-3 staining and the arrow indicates a cell displaying a condensed nucleus without active caspase-3 staining. Bottom panels are decolorized Hoechst fluorescence to emphasize neuclear morphology. Images are representative of all experiments n=15. Scale bar represents 10 microns. B) Quantitative assessment of HT22 cell apoptosis at 24 h post-transfection with either DsRed2 empty vector or DsRed2-AβPP. Cells were counted and scored as having apoptotic nuclei (condensed or fragmented morphology) or having both apoptotic nuclei and active caspase-3 staining. Only transfected cells were quantified. Data are expressed as the mean ± SEM, n=15 independent experiments each performed in duplicate. ** indicates p<0.01 compared to DsRed2 control using an unpaired student’s T-test.
Apoptosis induced in HT22 cells by the overexpression of AβPP is caspase-dependent
To further support that AβPP overexpression induces caspase-dependent apoptosis, we treated AβPP overexpressing cells with a pan-caspase inhibitor, Q-VD-OPh (Q-VD). Q-VD treated AβPP overexpressing cells showed significantly reduced active caspase-3 staining and larger, healthier nuclei compared to AβPP overexpressing cells left untreated. Quantification of these experiments indicated that Q-VD essentially completely protected HT22 cells from apoptosis induced by AβPP overexpression (Figure 2A). These data suggest that AβPP induces a caspase-dependent apoptotic cascade when overexpressed in HT22 cells.
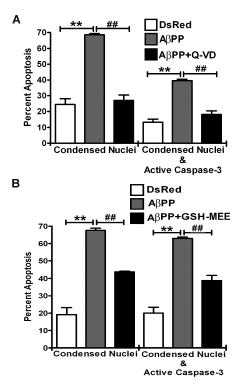
Figure 2. A pan-caspase inhibitor and glutathione (GSH) each protect HT22 cells from apoptosis induced by AβPP. A) Quantitative assessment of HT22 cell apoptosis at 24 h post- transfection with DsRed2-empty vector, DsRed2-AβPP, or DsRed2-AβPP treated with 20 μM Q- VD. Cells were counted and scored as either having apoptotic nuclei (condensed or fragmented morphology) or having both apoptotic nuclei and active caspase-3 staining. Only transfected cells were quantified. Data are expressed as the mean ± SEM, n=3. B) Quantitative assessment of HT22 cell apoptosis at 24 h post-transfection with DsRed2-empty vector, DsRed2-AβPP, or DsRed2-AβPP treated with 2 mM GSH monoethylester (GSH-MEE). Transfected cells were counted and scored as in (A). Data are expressed as the mean ± SEM, n=3. ** indicates p<0.01 compared to DsRed2, ## indicates p<0.01 compared to AβPP as determined using one-way ANOVA with a post hoc Tukey’s test.
GSH affords partial protection against apoptosis induced by AβPP
Next we examined if the mechanism of apoptosis induced by the overexpression of AβPP is due to MOS induction, by determining if a cell permeable form of glutathione (GSH) protected HT22 cells from apoptosis induced by AβPP overexpression. GSH is a tri-peptide antioxidant with a large cytosolic reservoir and a discrete mitochondrial pool that plays an essential role in protecting these organelles from MOS. If MOS is substantially induced by AβPP overexpression, then addition of exogenous GSH should protect from cell death. As shown in Figure 2B, GSH afforded only partial protection from apoptosis induced by the overexpression of AβPP. These data suggest that AβPP is likely inducing some amount of MOS; however, the apoptotic mechanism within this system is also dependent on other factors.
AβPP induces apoptosis in an mPTP-independent manner
Based on a partial involvement of MOS in the cell death induced by overexpression of AβPP, we next examined the role of the mitochondrial permeability transition pore (mPTP). Previous studies have shown that MOS can induce a calcium-dependent opening of the mPTP [21]. Here, we used a cobalt/calcein assay, in which if the mPTP is activated, CoCl2 is released from the matrix of the mitochondria into the cytosol allowing it to react with and quench the fluorescence (green) signal of calcein which is localized in the cytoplasm. In our previous study, we showed that AβPP overexpression activates the mPTP in CHO cells [16]. However, in HT22 cells no significant activation of the mPTP was observed with AβPP overexpression (Figure 3A, right panel). Ionomycin was used as a positive control for mPTP opening and as expected, cells treated with this calcium ionophore showed substantial activation of the mPTP (Figure 3A, bottom panel). Quantification of these data confirmed that compared to DsRed2 vector transfection, the overexpression of AβPP failed to activate the mPTP (Figure 3B). To provide further evidence that the mPTP is not involved in the cell death induced by the overexpression of AβPP in HT22 cells, the effects of cyclosporine A (CsA) on cell viability were examined. CsA has been shown to bind to cyclophilin D and prevent mPTP opening [22]. Indeed, CsA failed to protect cells overexpressing AβPP from apoptosis compared to AβPP transfection controls which were not treated with CsA (Figure 3C). These results demonstrate that the apoptotic cascade induced by the overexpression of AβPP is independent of mPTP activation.
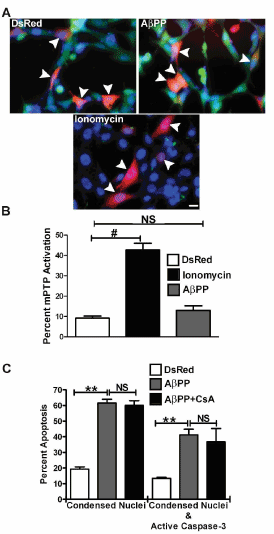
Figure 3. Overexpression of AβPP does not trigger mitochondrial permeability transition pore (mPTP) opening in HT22 cells. A) Live cell images of HT22 cells at 24 h post- transfection using a cobalt-calcein stain for mPTP activation. Top left panel shows DsRed2- empty vector transfected cells while the top right panel shows DsRed2-AβPP transfected cells. Bottom panel shows DsRed2 transfected cells treated with 2.5 μM ionomycin. Arrowheads indicate transfected cells. Images are representative of all experiments n=4. Scale bar represents
10 microns B) Quantitative analysis of mPTP opening expressed as the percentage of DsRed2 positive cells which were negative for calcein fluorescence. Data are expressed as the mean ± SEM, n=4. ## indicates p<0.01 compared to DsRed2, NS=not significant, as determined using one-way ANOVA with a post hoc Tukey’s test. C) Quantitative assessment of HT22 cell apoptosis at 24 h post-transfection with DsRed2-empty vector, DsRed2-AβPP, or DsRed2-AβPP treated with 10 μM cyclosporine A (CsA). Transfected cells were counted and scored as apoptotic as described in the legend to Figure 1B. Data are expressed as the mean ± SEM, n=3.
** indicates p<0.01 compared to DsRed 2 control, NS= not significant as determined using one- way ANOVA with a post hoc Tukey’s test.
The overexpression of AβPP in HT22 cells induces Bax-dependent apoptosis
Bcl-2 associated protein X (Bax) is a pro-apoptotic protein which induces pore formation of the outer mitochondrial membrane in response to specific apoptotic stimuli [23,24]. Specifically, increased ROS can stimulate the activation of Bax through either direct or indirect mechanisms. Therefore, we investigated if the overexpression of AβPP led to the activation of Bax using an active conformation epitope- specific antibody (monoclonal 6A7). We observed a significant increase in active Bax in AβPP overexpressing cells that was not observed in DsRed2 transfected cells (Figure 4A). Treatment of DsRed2 empty vector-transfected cells with staurosporine, a known inducer of Bax mediated apoptosis, showed a similar magnitude of Bax activation as AβPP transfected cells (Figure 4B). Finally, we examined the ability of a Bax-inhibitory peptide to protect HT22 cells from apoptosis induced by AβPP overexpression. In AβPP transfected cells, the Bax-inhibitory peptide afforded significant protection from apoptosis, compared to a negative control peptide (Figure 4C). Overall, these data indicate that the apoptotic mechanism induced by AβPP overexpression is dependent on the activation of Bax.
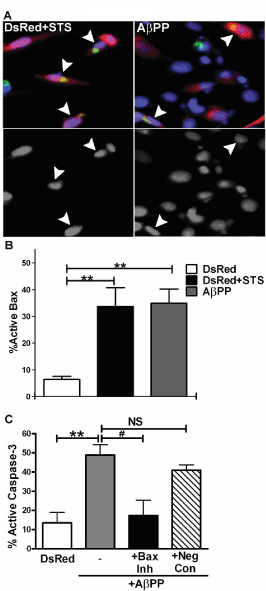
Figure 4. AβPP-induced apoptosis of HT22 cells is Bax-dependent. A) HT22 cells at 24 h post-transfection with DsRed2 treated with 100 nM staurosporine (left) and DsRed2-AβPP (right). Top panels show fluorescence for DsRed2 (red), active Bax (green) and Hoechst stain (blue). Arrowheads indicate transfected cells with active Bax staining (yellow). Bottom panels are decolorized Hoechst fluorescence to show nuclear morphology. n=3, scale bar represents 10 microns. B) Quantitative assessment of active Bax staining at 24 h post-transfection with DsRed2-AβPP or DsRed2 treated with 100 nM staurosporine. Transfected cells were counted as having active Bax staining. Data are shown as mean +/- SEM, n=3. ** indicates p<0.01 compared to DsRed2 control as determined using a one-way ANOVA with post hoc Tukey’s test. C) Quantitative assessment of apoptosis at 24 h post-transfection with DsRed-2 empty vector, DsRed2-AβPP, DsRed2-AβPP treated with 150 μM Bax inhibitor or DsRed-2 AβPP
treated with 150 μM negative control Bax inhibitor. Transfected cells were counted and scored as apoptotic as described in the legend to Figure 1B. Data are shown as mean ± SEM, n=3. ** indicates p<0.01 compared to DsRed2 control, * indicates p<0.05 compared to DsRed2-AβPP, NS=not significant as determined using a one-way ANOVA with post hoc Tukey’s test.
The apoptotic pathway induced by AβPP occurs in the absence of its proteolytic processing
Amyloid-beta fragments, the AβPP intracellular domain (AICD), or full length AβPP, are each hypothesized to cause AD pathology; however, there are disagreements as to which is primarily responsible for neuronal cell death. To determine which of these was responsible for the HT22 cell apoptosis induced by the overexpression of AβPP, gamma and beta secretase inhibitors were used. Surprisingly, we observed no protection from apoptosis with inhibitors of either gamma or beta secretase in AβPP overexpressing cells (Figure 5A). This suggests that Aβ and AICD fragments do not play a significant role in mediating the observed cell death in this system. To support this conclusion, we examined full length AβPP protein levels in cells treated with or without the secretase inhibitors and observed little-to-no change in the amount of full length DsRed2-AβPP protein levels (Figure 5B). In addition, we treated AβPP-transfected HT22 cells with the gamma and beta secretase inhibitors and then performed western blots for C- terminal fragments, as well as an ELISA for human Aβ1-42 (Invitrogen). In these experiments we were unable to detect either C-terminal fragments (C83/C99) or Aβ1-42 (data not shown), suggesting that either the HT22 cells do not generate detectable quantities of these peptides following transfection with WT AβPP or due to the relatively low transfection efficiency (~20%), we are simply unable to detect these peptides from cell lysates.
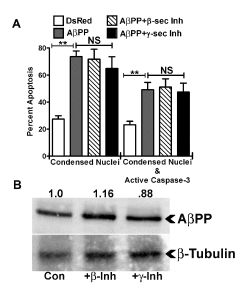
Figure 5. β- and γ- secretase inhibitors have no effect on HT22 cell apoptosis induced by AβPP. A) Quantitative assessment of HT22 cell apoptosis at 24 h post-transfection with DsRed2-empty vector, DsRed2-AβPP, DsRed2-AβPP treated with 250 nM β-secretase inhibitor, or DsRed2-AβPP treated with 500 nM γ-secretase inhibitor. Transfected cells were counted and scored as apoptotic as described in the legend to Figure 1B. Data are expressed as the mean ± SEM, n=3. ** indicates p<0.01 compared to DsRed2-control, NS=not significant, as determined using one-way ANOVA with a post hoc Tukey’s test. B) Western blot analysis of HT22 whole cell lysates after 24 h DsRed2 AβPP transfection Con or treated with β-secretase inhibitor (+β- Inh) or γ-secretase inhibitor (+γ-Inh). Top blot shows AβPP. Bottom blot shows β-tubulin loading control, n=3. Densitometry of AβPP band density normalized to β-tubulin band density is shown with the AβPP control being set to 1.0.
Finally, we compared the mechanism of AβPP-induced apoptosis in HT22 cells to that of cell death induced by Aβ1-42 overexpression. In the current study, we observed that HT22 cells overexpressing WT AβPP were completely protected by the pan-caspase inhibitor Q-VD and significantly, but only partially, protected by GSH-MEE (Figure 2). In contrast, HT22 cells transfected with Aβ1-42 (cloned into the pIRES2 DsRed-Express 2 bicistronic vector) underwent marked cell death that was not significantly attenuated by Q-VD but which was completely protected against by GSH-MEE (Table 1). These stark differences in the apparent mechanism of cell death induced by overexpression of full length WT AβPP versus Aβ1-42 peptide suggest that the toxic effects observed in this cell system with the full length molecule do indeed occur independently of its proteolytic processing to toxic fragments like Aβ1-42. However, we cannot completely rule out the possible toxic contribution(s) of some other fragment(s) that we cannot detect by western blotting or ELISA.
Table 1. Effects of Q-VD and GSH-MEE on HT22 cell death induced by Aβ42
Treatment |
% Apoptosis (mean±SEM; n=3 experiments, each in duplicate) |
pIRES2 DsRed |
22±5% |
pIRES2 DsRed/Aβ42 |
48±6%* |
Aβ42+Q-VD |
37±7% |
Aβ42+GSH-MEE |
18±4%# |
*Significantly different from DsRed control (p<0.05; one way ANOVA with post hoc Tukey’s test)
#Significantly different from Ab42 control (p<0.05; one way ANOVA with post hoc Tukey’s test)
AβPP localizes to mitochondrial-enriched fractions of HT22 cells
An increasing body of literature has shown that in both cell culture and brain tissue from AD patients, a significant amount of AβPP localizes to the mitochondria [15,25]. Consistent with these previous findings, we observed punctate localization of DsRed2 fluorescence specifically in AβPP overexpressing cells (Figure 6A).
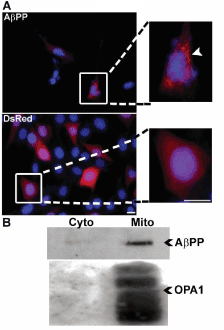
Figure 6. Overexpressed DsRed2-AβPP shows a punctate distribution indicative of its localization to mitochondria. A) HT22 cells at 24 h post-transfection with either DsRed2- empty vector or DsRed2-AβPP. Panels show fluorescence of DsRed2 (red) and Hoechst stain (blue). Right panels show 3X magnification of images from the boxed area in right panels. Arrowhead indicates the punctate nature of DsRed2-AβPP localization. Scale bar represents 10 microns. B) Western blot analysis of mitochondrial/ cytosolic fractionation of HT22 cells at 24 h post-transfection with DsRed2-AβPP. Top blot shows AβPP. Cytosolic fraction (Cyto) is shown on the left and mitochondrial fraction (Mito) is shown on the right. Bottom blot of OPA1 is to shown to indicate enrichment of mitochondrial fractionation.
Upon immunoblotting cellular fractions from AβPP transfected cells, it was found that AβPP localized substantially to the mitochondrial-enriched fraction as opposed to the cytosolic fraction (Figure 6B). OPA1, a mitochondrial protein indicates pure subcellular fractions.
Localization of AβPP to mitochondria is required for its induction of apoptosis
In order to determine if AβPP was inducing apoptosis in a mitochondrial-dependent manner, the three positive residues indicated in Figure 7A, were mutated via site-directed mutagenesis into uncharged amino acids. These particular mutations have previously been shown to prevent the mitochondrial localization of AβPP [15]. Indeed, compared to AβPP, the 3 residue mutant of AβPP (AβPP 3M) failed to localize to mitochondrial- enriched fractions in HT22 cells (Figure 7B). Neither AβPP nor AβPP 3M were observed in cytosolic fractions, however both were found in plasma membrane fractions. In addition, it has been previously shown that both AβPP and the 3M mutant AβPP are secreted into the extracellular space from cells [15]. Cox-IV, GAPDH, and Na/K ATPase are shown to indicate pure subcellular fractions (Figure 7B). Finally, HT22 cells overexpressing AβPP displayed increased indices of apoptosis, as shown in previous figures; however, the overexpression of AβPP 3M caused no significant induction of apoptosis compared to DsRed2 control overexpression (Figure 7C). These data show that the apoptotic mechanism employed by the overexpression of AβPP is strictly dependent on its localization to mitochondria.
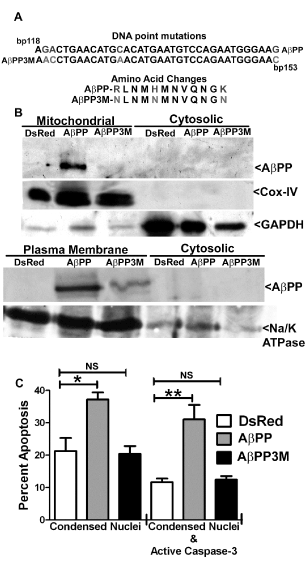
Figure 7. An AβPP mutant that cannot localize to mitochondria (AβPP 3M) fails to induce apoptosis in HT22 cells. A) Schematic displaying the site directed mutagenesis completed to generate AβPP 3M. Base pair and amino acid changes are indicated using different shades of type.. AβPP and AβPP 3M sequences are shown. B) Western blot analysis of mitochondrial/cytosolic and plasma membrane/cytosolic fractionations of HT22 cells at 24 h post-transfection with DsRed2-empty vector, DsRed2-AβPP, or DsRed2- AβPP 3M. Cox-IV, GAPDH and Na/K ATPase blots indicate the relative purity of the mitochondrial, cytoplasmic and plasma membrane fractions, respectively. C) Quantitative assessment of HT22 cell apoptosis at 24 h post-transfection with DsRed2-empty vector, DsRed2-AβPP, or DsRed2-AβPP 3M. Transfected cells were counted and scored as apoptotic as described in the legend to Figure 1B. Data are expressed as the mean ± SEM, n=3. * indicates p<0.05 compared to DsRed2, **indicates p<0.01 compared to DsRed2, NS=not significant, as determined using one-way ANOVA with a post hoc Tukey’s test.
Functional mitochondria are required for AβPP to induce apoptosis in the SH-SY5Y neuroblastoma cell line
2021 Copyright OAT. All rights reserv
Several cell lines devoid of mitochondrial DNA have been developed and are termed rho zero (ρ0) cell lines. While most mitochondrial genes are coded for within the nucleus, several components of the ETC remain genetically encoded within the mitochondrial matrix. Therefore, ρ0 cell lines lack functional electron transport while mitochondrial membranes are still present. Here, we used an SH-SY5Y ρ0 cell line in comparison with an SH-SY5Y ρ+ cell line to determine if the mechanism of AβPP-induced apoptosis at mitochondria is dependent on a functional ETC. First, we observed that the localization of AβPP was mitochondrial in both ρ+ and ρ0 cell lines (Figure 8A-C). This is indicated by both punctuate DsRed2 fluorescence (Figure 8A) and immunoblots of subcellular fractions. Cox-IV and GAPDH immunoblots demonstrate pure subcellular fractions (Figures 8B,C). Furthermore, the overexpression of AβPP induced a significant increase in apoptosis compared to DsRed2 transfection in the ρ+ cell line, while no discernible difference in apoptosis between DsRed2 and AβPP overexpression was observed in the ρ0 cell line (Figure 8D). These results indicate that the apoptotic mechanism induced by AβPP overexpression requires a functional ETC at the level of the mitochondria.
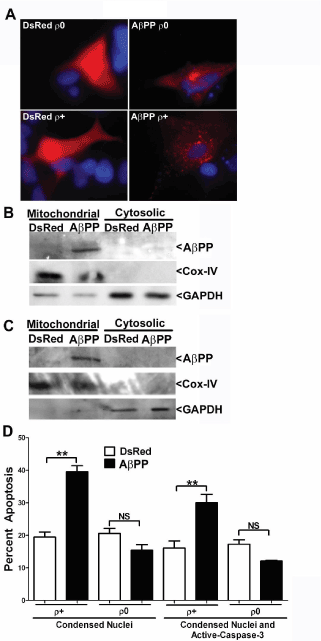
Figure 8. Overexpression of AβPP does not induce apoptosis of mitochondrial DNA- deficient ρ0 SH-SY5Y neuroblastoma cells. A) SH-SY5Y ρ0 or ρ+ cells at 24 h post- transfection with either DsRed2-empty vector or DsRed2-AβPP. Panels show fluorescence of DsRed2 (red) and Hoechst stain (blue). Scale bar represents 10 microns. B) Western blot analysis of mitochondrial/ cytosolic fractionation of SH-SY5Y ρ+ cells at 24 h post-transfection with DsRed2-empty vector or DsRed2-AβPP. Cox-IV and GAPDH blots indicate pure mitochondrial and cytosolic fractions, respectively. C) Western blot analysis of mitochondrial/ cytosolic fractionation of SH-SY5Y ρ0 cells at 24 h post-transfection with DsRed2-empty vector or DsRed2-AβPP. Cox-IV and GAPDH blots indicate pure mitochondrial and cytosolic fractions, respectively. D). Quantitative assessment of SH-SY5Y ρ0 and ρ+ cell apoptosis at 24 h post- transfection with DsRed2-empty vector or DsRed2-AβPP. Transfected cells were counted and scored as apoptotic as described in the legend to Figure 1B. Data are expressed as the mean ± SEM, n=3. ** indicates p<0.01 compared to DsRed2, NS=not significant, as determined using one-way ANOVA with a post hoc Tukey’s test.
Discussion
The amyloid cascade hypothesis states that the proteolytic processing of AβPP into Aβ fragments is the underlying cause in the pathogenesis of AD [26]. While it has been well documented that Aβ fragments induce cell death both in vitro and in vivo, there is emerging evidence that AβPP and its intracellular domain (AICD) may also contribute to AD pathogenesis [16-19,27,28]. In this study, the overexpression of AβPP in a mouse hippocampal cell line induced a caspase-dependent and Bax-mediated apoptotic pathway which appeared to be largely independent of AβPP processing. Therefore, our data are consistent with the emerging hypothesis that AβPP may also contribute to the pathogenic mechanism in AD.
While mutations in AβPP and/or presenilins, which enhance amyloidogenic processing and production of Aβ peptides, underlie the onset and progression of familial AD, sporadic AD fails to recapitulate these genetic mutations yet shares many attributes of the disease phenotype [29-32]. This suggests that other factors besides increased Aβ production contribute to the pathogenesis of sporadic AD. In this context, patients with Down Syndrome typically develop early onset AD that is most often attributed to expressing an extra copy of the AβPP gene which is localized to chromosome 21 [33-36]. The overexpression of AβPP has been shown to induce neuronal apoptosis in a variety of in vitro and in vivo models [16-19,37]. For example, Simón, et al. (2009) [19] observed that the overexpression of WT AβPP in mice led to memory deficits and hippocampal neurodegeneration which were independent of Aβ accumulation. On the other hand, mutant AβPP expressing mice displayed high levels of Aβ in hippocampus, but they failed to show overt evidence of neuronal degeneration in the hippocampus [19]. Our current study in undifferentiated HT22 cells is in agreement with both in vitro and in vivo observations demonstrating that AβPP overexpression can lead to neuronal cell death due to apoptosis. Moreover, in additional experiments we observed that overexpression of WT AβPP similarly induced greater than 50% apoptosis in HT22 cells which underwent a differentiation protocol prior to transfection (data not shown), indicating that the effects documented in the present study are not restricted to undifferentiated cell lines [38]. Thus, although AβPP undoubtedly has important physiological functions, in cases where it is overexpressed (e.g., WT AβPP transgenic mice or Down Syndrome), aberrantly high levels of AβPP may contribute to neurotoxicity.
The activation of intrinsic apoptosis culminates in the permeabilization of the outer mitochondrial membrane allowing for the release of pro-apoptotic factors such as cytochrome c. Two mechanisms could allow for the permeabilization of the outer mitochondrial membrane, the first being the activation of the mPTP. Controversy remains about the exact components of the mPTP, but it is largely accepted that VDAC, ANT, and cyclophilin D are required for its activation [39]. Here, we show that the overexpression of AβPP in HT22 cells does not induce apoptosis in an mPTP-dependent manner. Therefore, we examined the second mechanism typically responsible for outer mitochondrial membrane permeabilization, activation of the pro-apoptotic protein Bax. Previous studies have shown that Bax has the ability to form pores within lipid membranes and can trigger the release of cytochrome c from isolated mitochondria [40,41]. Our data indicate that Bax plays an essential role in the induction of apoptosis caused by the overexpression of AβPP.
Oxidative stress plays a major role in both aging and AD, and the strongest identified risk factor for AD is increased age [42-44]. Indices of oxidative stress-induced damage have been observed in both aging individuals and AD patients. Therefore, if AβPP plays a critical role in the neuronal cell death underlying AD, then oxidative stress should be a component of the cell death mechanism induced by its overexpression. To determine if oxidative stress was a contributing factor in our model, HT22 cells were treated with a cell-permeable ester of GSH. We observed significant protection with the addition of GSH from apoptosis induced by AβPP overexpression; however, only partial attenuation of cell death occurred. Therefore, while oxidative stress does appear to contribute to the apoptotic mechanism of AβPP overexpression within this cell system, additional factors must also be involved.
Mitochondrial abnormalities in AD are supported by numerous lines of evidence. Deficiencies in ETC complexes, mitochondrial DNA perturbations, and MOS have all been observed in AD patients as well as in vitro and in vivo models of this disease [3-8,45]. Previous studies have shown that AβPP localizes to mitochondria and therefore, we examined if this localization was required for apoptosis induced by AβPP overexpression [15]. Here, we used a triple mutant of AβPP (mutations within the acidic domain) that lacked the ability to localize to mitochondria, but which retained its localization to the plasma membrane. Interestingly, overexpression of this mutant failed to induce apoptosis within the HT22 cell line. These data indicate that the mitochondrial localization of AβPP is required for its induction of apoptosis within this system, but they do not directly implicate mitochondrial respiratory function in the mechanism of AβPP- induced apoptosis.
Therefore, we next examined the contribution of the ETC to the apoptotic mechanism of AβPP overexpression. To test if the ETC was a required component of the apoptotic pathway, we employed a cell system which is devoid of mitochondrial DNA known as ρ0 cells. Without mitochondrial DNA, the proper function of the ETC cannot occur because certain genetic components of Complexes I, III, IV and V are encoded within the mitochondrial genome [46]. Therefore, the ρ0 cells depend exclusively on glycolysis for ATP production. Our data show that overexpression of AβPP in the SH-SY5Y ρ0 cell line did not induce apoptosis, while its overexpression in an SH-SY5Y ρ+ cell line led to a significant induction of apoptosis. It could be argued that because ρ0 cell lines depend on glycolysis for ATP production, lower sustained ATP levels might prevent apoptosome formation which is required for intrinsic apoptosis. However, other insults such as staurosporine and H2O2 have the ability to induce apoptosis within ρ0 cells [47,48]. Therefore, ρ0 cells retain the ability to undergo apoptosis, so we can conclude that mitochondrial localization and a functional ETC are required for AβPP to induce apoptosis within this system.
Several models exist by which AβPP could lead to apoptosis in a mitochondrial dependent manner. First, AβPP localized to mitochondria has been shown to have a high affinity for the mitochondrial translocases (TOM and TIM) where AβPP could induce the disruption of mitochondrial protein import and failure of ETC function [25]. Prolonged failure of mitochondrial protein import could lead to mitochondrial dysfunction, MOS, and apoptosis. Second, AβPP could also exert pro-apoptotic effects at the level of the mitochondria through its observed interaction with the pro-survival protein Bcl-2 [49]. Beyond its anti- apoptotic functions, Bcl-2 has also been shown to be a GSH-binding protein and to contribute to the regulation of mitochondrial GSH transport [20,50]. If the interaction between Bcl-2 and AβPP leads to an inhibitory effect on the antioxidant-like functions of Bcl-2, then mitochondrial GSH depletion might be induced which would result in organelle damage and decreased function of the ETC. The inhibitory effects of AβPP on Bcl-2 would culminate in mitochondrial dysfunction, MOS, and apoptosis. Therefore, the potential toxic effects of AβPP at the level of the mitochondria could be executed via two distinct but not mutually exclusive, mechanisms. While our data do not conflict with either of these hypotheses, the observation that a functional ETC is necessary for AβPP to induce mitochondrial apoptosis indicates that deficits in protein import which are toxic to mitochondria likely involve additional proteins beyond ETC components.
In summary, our findings demonstrate that AβPP overexpression induces intrinsic apoptosis in HT22 cells via a Bax-dependent mechanism. We observed substantial differences in the apparent mechanism of cell death induced by overexpression of full length WT AβPP versus Aβ1-42 peptide, suggesting that the toxic effects caused in this cell system by the full length molecule likely occurs independently of its proteolytic processing to toxic fragments like Aβ1-42. Furthermore, the apoptotic mechanism of AβPP is not only dependent on its localization to mitochondria, but it also requires a functional ETC. These data suggest that the full length AβPP molecule, acting specifically at the level of the mitochondria, should be considered a likely contributor to the hippocampal neuronal cell death that underlies the pathogenesis of AD.
Author contributions
DL conceived and coordinated the study and wrote the paper. KM, EM, HW, RM, and EI designed, performed and analyzed the experiments. HW, RS, and DP revised the article critically for important intellectual content. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
These studies were funded by a grant from the Knoebel Institute for Healthy Aging at the University of Denver. This work was also supported by the Mitochondrial Genomics and Metabolism Core of the University of Kansas Alzheimer’s Disease Center (P30AG035982).
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- Deak F, Freeman WM, Ungvan Z, Csiszar A, Sonntag WE (2016) Recent developments in understanding brain aging: implications for Alzheimer's disease and vascular cognitive impairment. J Gerontol A Biol Sci Med Sci 71: 13-20. [crossref]
- Mohamed T, Shakeri A, Rao PP (2016) Amyloid cascade in Alzheimer's disease: Recent advances in medicinal chemistry. Eur J Med Chem 113: 258-272. [crossref]
- Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, et al. (1992) Brain cytochrome oxidase in Alzheimer's disease. J Neurochem 59: 776-779. [crossref]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, et al. (2001) Mitochondrial abnormalities in Alzheimer's disease. J of Neurosci 21: 3017-3023. [crossref]
- Mecocci P, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol 36: 747-751. [crossref]
- Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, et al. (2006) Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis 22: 76-87. [crossref]
- Swerdlow RH, Khan SM (2004) A "mitochondrial cascade hypothesis" for sporadic Alzheimer's disease. Med Hypotheses 63: 8-20. [crossref]
- Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, et al. (2009) Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. P Nat Acad Sci USA 106: 14670-14675. [crossref]
- Leuner K, Schütt T, Kurz C, Eckert SH, Schiller C, et al. (2012) Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 16: 1421-1433. [crossref]
- Oka S, Leon J, Sakumi K, Ide T, Kang D, et al. (2016) Human mitochondrial transcriptional factor A breaks the mitochondria-mediated vicious cycle in Alzheimer's disease. Sci Rep 6: 37889. [crossref]
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, et al. (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304: 448-452. [crossref]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, et al. (2006) Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15: 1437-1449. [crossref]
- Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA (2002) Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem 80: 91-100. [crossref]
- Fang D, Zhang Z, Li H, Yu Q, Douglas JT, et al. (2016) Increased electron paramagnetic resonance signal correlates with mitochondrial dysfunction and oxidative stress in an Alzheimer's disease mouse brain. J Alzheimer's Dis 51: 571-580. [crossref]
- Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani, NG (2003) Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. Journal Cell Biol 161: 41-54. [crossref]
- Bartley MG, Marquardt K, Kirchhof D, Wilkins HM, Patterson D, et al. (2012) Overexpression of amyloid-beta protein precursor induces mitochondrial oxidative stress and activates the intrinsic apoptotic cascade. J Alzheimer's Dis 28: 855-868. [crossref]
- Hanson AJ, Prasad JE, Nahreini P, Andreatta C, Kumar B, et al. (2003) Overexpression of amyloid precursor protein is associated with degeneration, decreased viability, and increased damage caused by neurotoxins (prostaglandins A1 and E2, hydrogen peroxide, and nitric oxide) in differentiated neuroblastoma cells. J Neurosci Res 74: 148-159. [crossref]
- Matsumoto K, Akao Y, Yi H, Shamoto-Nagai M, Maruyama W, et al. (2006) Overexpression of amyloid precursor protein induces susceptibility to oxidative stress in human neuroblastoma SH-SY5Y cells. J Neural Transm 113: 125-135. [crossref]
- Simón AM, Schiapparelli L, Salazar-Colocho P, Cuadrado-Tejedor M, Escribano L, et al. (2009) Overexpression of wild-type human APP in mice causes cognitive deficits and pathological features unrelated to Abeta levels. Neurobiol Dis 33: 369-378. [crossref]
- Zimmermann AK, Loucks FA, Schroeder EK, Bouchard RJ, Tyler KL, et al. (2007) Glutathione binding to the Bcl-2 homology-3 domain groove: a molecular basis for Bcl-2 antioxidant function at mitochondria. J Biol Chem 282: 29296-29304. [crossref]
- Kantrow SP, Tatro LG, Piantadosi CA (2000) Oxidative stress and adenine nucleotide control of mitochondrial permeability transition. Free Rad Biol Med 28: 251-260. [crossref]
- Halestrap AP, Davidson AM (1990) Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J 268: 153-160. [crossref]
- Sharpe JC, Arnoult D, Youle RJ (2004) Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta 1644: 107-113. [crossref]
- Renault TT, Manon S (2011) Bax: Addressed to kill. Biochimie 93: 1379-1391. [crossref]
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci 26: 9057-9068. [crossref]
- Hardy J (1997) Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci 20: 154-159. [crossref]
- Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, et al. (2009) Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A 106: 18367-18372. [crossref]
- Wang X, Wang Z, Chen Y, Huang X, Hu Y, et al. (2014) FoxO mediates APP-induced AICD-dependent cell death. Cell Death Dis 5: e1233. [crossref]
- Selkoe DJ (1998) The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol 8: 447-453. [crossref]
- Lin MT, Beal MF (2006) Alzheimer's APP mangles mitochondria. Nat Med 12: 1241-1243. [crossref]
- Mancuso M, Orsucci D, LoGerfo A, Calsolaro V, Siciliano G (2010) Clinical features and pathogenesis of Alzheimer's disease: involvement of mitochondria and mitochondrial DNA. Adv Exp Med Biol 685: 34-44. [crossref]
- Roher AE, Maarouf CL, Kokjohn TA (2016) Familial presenilin mutations and sporadic Alzheimer's disease pathology: is the assumption of biochemical equivalence justified? J Alzheimer's Dis 50: 645-658. [crossref]
- Devenny DA, Wegiel J, Schupf N, Jenkins E, Zigman W, et al. (2005) Dementia of the Alzheimer's type and accelerated aging in Down syndrome. Sci Aging Knowledge Environ 2005: dn1. [crossref]
- Zigman WB, Lott IT (2007) Alzheimer's disease in Down syndrome: neurobiology and risk. Ment Retard Dev D R 13: 237-246. [crossref]
- Patterson D (2009) Molecular genetic analysis of Down syndrome. Hum Genet 126: 195-214. [crossref]
- Wiseman FK, Al-Janabi T, Hardy J, Karmiloff-Smith A, Nizetic D, et al. (2015) A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci 16: 564-574. [crossref]
- Calvo-Rodriguez M, Garcia-Durillo M, Villalobos C, Núñez L (2016) Aging enables Ca2+ overload and apoptosis by amyloid- oligomers in rat hippocampal neurons: neuroprotection by non-steroidal anti-inflammatory drugs and R-flurbiprofen in aging neurons. J Alzheimer's Dis 54: 207-221. [crossref]
- He M, Liu J, Cheng S., Xing Y, Suo WZ (2013) Differentiation renders susceptibility to excitotoxicity in HT22 neurons. Neural Regen Res 8: 1297-1306. [crossref]
- Crompton M, Barksby E, Johnson N, Capano M (2002) Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 84: 143-152. [crossref]
- Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, et al. (1998) Bax directly induces release of cytochrome c from isolated mitochondria. P Nat Acad Sci USA 95: 4997-5002. [crossref]
- Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC (2000) Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J 345 Pt 2: 271-278. [crossref]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, et al. (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropath Exp Neur 60: 759-767. [crossref]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, et al. (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429: 883-891. [crossref]
- Wirz KT, Keitel S, Swaab DF, Verhaagen J, Bossers K (2014) Early molecular changes in Alzheimer's disease: can we catch the disease in its presymptomatic phase? J Alzheimer's Dis 38: 719-740. [crossref]
- Santos RX, Correia SC, Zhu X, Smith MA, Moreira PI, et al. (2013) Mitochondrial DNA oxidative damage and repair in aging and Alzheimer's disease. Antioxid Redox Signal 18: 2444-2457. [crossref]
- Baker BM, Haynes CM (2011) Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci 36: 254-261. [crossref]
- Cardoso SM, Santos S, Swerdlow RH, Oliveira CR (2001) Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J 15: 1439-1441. [crossref]
- Morais Cardoso S, Swerdlow RH, Oliveira, CR (2002) Induction of cytochrome c- mediated apoptosis by amyloid beta 25-35 requires functional mitochondria. Brain Res 931: 117-125. [crossref]
- Yang TT, Hsu CT, Kuo YM (2009) Amyloid precursor protein, heat-shock proteins, and Bcl-2 form a complex in mitochondria and modulate mitochondria function and apoptosis in N2a cells. Mech Ageing Dev 130: 592-601. [crossref]
- Wilkins HM, Marquardt K, Lash LH, Linseman DA (2012) Bcl-2 is a novel interacting partner for the 2-oxoglutarate carrier and a key regulator of mitochondrial glutathione. Free Radical Bio Med 52: 410-419. [crossref]