Neuronal voltage gated calcium channels (VGCCs) are well known for its importance in synaptic transmission and signaling pathway in the specific circuits. On the other hand, the role of different VGCCs in the cognitive functions has not been studied. Although it has been reported that fear extinction requires Cav2.1-regulated N-methyl-D-aspartate (NMDA) receptor signaling, Cav2.2-regulated synaptic function in extinction remains unknown. This study examined whether Cav2.2-mediated signaling plays role in consolidation of extinction. Mice received intracerebroventricular injection of Cav2.2 blocker (ω-conotoxin GVIA, 5 pg/side) showed impaired extinction behavior and decreased expression of CREB-dependent gene Arc in medial prefrontal cortex (mPFC). Intra-mPFC injections of ω-conotoxin GVIA (5 pg/midline) blocked extinction. These results indicate that Cav2.2-mediated signaling is critical in the mPFC-dependent fear extinction.
Cav2.2, fear extinction, neuronal circuits, ω-conotoxin GVIA
Extinction procedures do not erase the original fear memory, but yield a new safety memory that inhibits fear under certain conditions [1]. Animals learn to associate a previously neutral or conditioned stimulus (CS) with an aversive or unconditioned stimulus (US) during fear conditioning. Subsequent re-exposure to the CS alone triggers two competing processes. Brief re-exposure to the CS initiates a reconsolidation process that serves to stabilize or maintain the original CS-US memory [2]. In contrast, more prolonged re-exposure to the CS leads to the formation of an inhibitory extinction (CS-no US) memory [3]. It is interesting to understand the neural circuit mechanism underlying fear extinction.
Ca2+ signaling through voltage gated calcium channels (VGCCs) mediates Ca2+ entry into cells in response to membrane depolarization and thus transduces electrical signals into chemical signals [4]. Neuronal VGCCs including Cav2.1 (P/Q-type), Cav2.2 (N-type), and Cav2.3 (R-type) channels mediate a number of neuronal functions including neuronal excitation, neurite outgrowth, synaptogenesis, neurotransmitter release, neuronal survival, differentiation, plasticity, and regulation of gene expression [4-6]. On the other hand, the role of different VGCCs in the neural circuits underlying fear extinction has not been studied.
Cav2.1 channels mediate the presynaptic machinery for glutamate release [7]. Cav2.1 and NMDA receptors and express broadly in the central nervous system [8-11]. A previous study reported that fear extinction requires a distributed neural circuit in the brain, especially the medial prefrontal cortex (mPFC) [12,13]. Blocking protein synthesis in the mPFC prevents the formation of extinction memory, and activation of cAMP-responsive element-binding protein (CREB)-mediated transcription is induced in the mPFC in the consolidation phase of extinction [14]. Our previous study has showed that Cav2.1-regulated NMDA receptor signaling in the mPFC regulates the CREB cascade involved in extinction memory [15]. The administration of a Cav2.1 blocker induced dysfunctional fear extinction [15]. However, the physiological role of Cav2.2-regulated extinction has not yet been determined. Cav2.2 also express broadly in the central nervous system [8]. Thus, administration of a Cav2.2 blocker should result in dysfunctional fear extinction, because precise regulation of neurotransmitter release through Cav2.2 also plays an important role in neuronal circuits [4-6].
There has been a growing interest in the neural mechanisms of fear extinction [16,17], as extinction may be a potential target for the treatment of neuropsychiatric diseases. In the present study, to examine the relationship between Cav2.2-mediated signaling in the mPFC and extinction memory formation, we investigated the extinction of conditioned fear using the wild-type mice received intracerebroventricular or intra-mPFC injection of Cav2.2 blocker (ω-conotoxin GVIA). We used immunohistochemical analysis to characterize the relationship between Cav2.2-mediated signaling and the expression of the CREB-dependent gene activity-regulated cytoskeleton-associated protein (Arc) after prolonged re-exposure. The studies presented here demonstrate the importance of mPFC-dependent Cav2.2-mediated signaling in the consolidation of fear extinction.
Mice
All animal procedures were approved by the Animal Experiments Committee of Shanghai Jiao Tong University and RIKEN. The C57BL/6J mice was provided by Charles River Japan (Kanagawa, Japan) The mice were given free access to water and food pellets (CRF-1; Oriental Yeast Co. Ltd., Tokyo, Japan) and were housed under a 12/12-h light/dark cycle (lights on from 08:00 to 20:00) at 23 ± 1°C and 55 ± 5% humidity. Testing was performed during the light phase of the cycle. All experiments were conducted blind to the treatment condition of the mouse.
Contextual fear conditioning
Mice were trained and tested in conditioning chambers (17.5 × 17.5 × 15 cm) that had a stainless steel rod floor through which footshocks could be delivered (Med Associates, Inc., St. Albans, VT, USA). Training consisted of placing a mouse in the chamber and delivering a series of unsignaled three footshocks at 1-min intervals (first footshock at 148 s after placement in the chamber). Mice were returned to their home cages 30 s after the final footshock. Twenty four hours after training, mice were re-exposed to the conditioning context for 30 min. One day following the extinction session, the mice were tested (5 min in context). Freezing behavior was automatically measured (Winroof version 5.5 software; Mitani Corporation, Tokyo, Japan).
Infusion
For the infusion studies, Cav2.2 blocker, ω-conotoxin GVIA (10, 50, or 100 pg/μL, Peptide Institute, Osaka, Japan) were dissolved in saline (vehicle). Under anesthesia and using standard stereotaxic procedures, stainless-steel guide cannulae (22-gauge) were implanted into the lateral ventricle (posterior to bregma, -0.34 mm; lateral to midline, ±0.9 mm; ventral from the dura, −2.3 mm) or mPFC (anterior to bregma, +1.9 mm; lateral to midline, ±0 mm; ventral from the dura, −2.3 mm). Mice were allowed to recover for at least 1 week following surgery. The mice were briefly anesthetized with isoflurane to facilitate insertion of the injection cannula (26-gauge). Infusions into the lateral ventricle (0.1 μL/side) or mPFC (0.1 μL/midline) including prelimbic cortex (PLC) and infralimbic cortex (ILC) are accomplished at a rate of 0.05 μL/min immediately after the re-exposure session. The injection cannula was left in place for 2 min following the infusion. The drug doses were determined according to previous report [15,18]. Mice that were not treated with drugs received an equivalent volume of vehicle.
Immunocytochemistry
Mice that received intracerebroventricular injection immediately after the re-exposure session were anesthetized with sodium pentobarbitone 90 min after the re-exposure session and were perfused with phosphate-buffered saline (PBS)/0.1 mm sodium fluoride (NaF) containing 4% paraformaldehyde. The brains were then removed, fixed overnight, and transferred into 30% sucrose. Coronal sections (30 μm) were cut on a cryostat. Consecutive sections were incubated overnight with an anti-Arc rabbit polyclonal primary antibody (1:1,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) in the blocking solution. The sections were incubated with biotinylated goat anti-rabbit IgG (SAB-PO kit; Nichirei Biosciences, Tokyo, Japan), followed by incubation at room temperature in streptavidin-biotin-peroxidase complex (SAB-PO kit). Quantification of Arc-positive cells in sections (bregma between 2.00 and 1.88 mm) of PLC and ILC was determined with a computerized image analysis system (Winroof version 5.5 software). Immunoreactive neurons were counted bilaterally with a fixed sample window across (200 × 200 μm) at least three sections by an experimenter blind to the treatment condition.
Data analysis
Data are presented as means ± standard error of the mean (SEM). Statistical analyses for the behavioral and immunocytochemical studies were conducted using Excel Statistics 2006 (SSRI, Tokyo, Japan). Data were analyzed using repeated measures analysis of variance (ANOVA) followed by Tukey’s post-hoc tests.
Intracerebroventricular injection effects of ω-conotoxin GVIA on freezing percentage
We examined intracerebroventricular injection effects of ω-conotoxin GVIA on the consolidation of extinction. Our experimental design is shown in Figure 1A. We used four groups of male mice (n=10 each) given systemic injections of 0 (vehicle), 1, 5, or 10 pg/side ω-conotoxin GVIA. There were no significant differences among groups in the extinction training session [F(15, 216)=0.8, P > 0.05] (Figure 1B). However, the groups significantly differed in the percentage of freezing during the test [F(3, 36)=155.7, P < 0.01] (Figure 1C). The mice given 5 or 10 pg/side ω-conotoxin GVIA showed greater freezing behavior than the mice given vehicle. These results shows that blockade of Cav2.2-mediated signaling impairs consolidation of extinction. Correct placement of the guide cannula was verified by ink injection after experiments (data not shown).
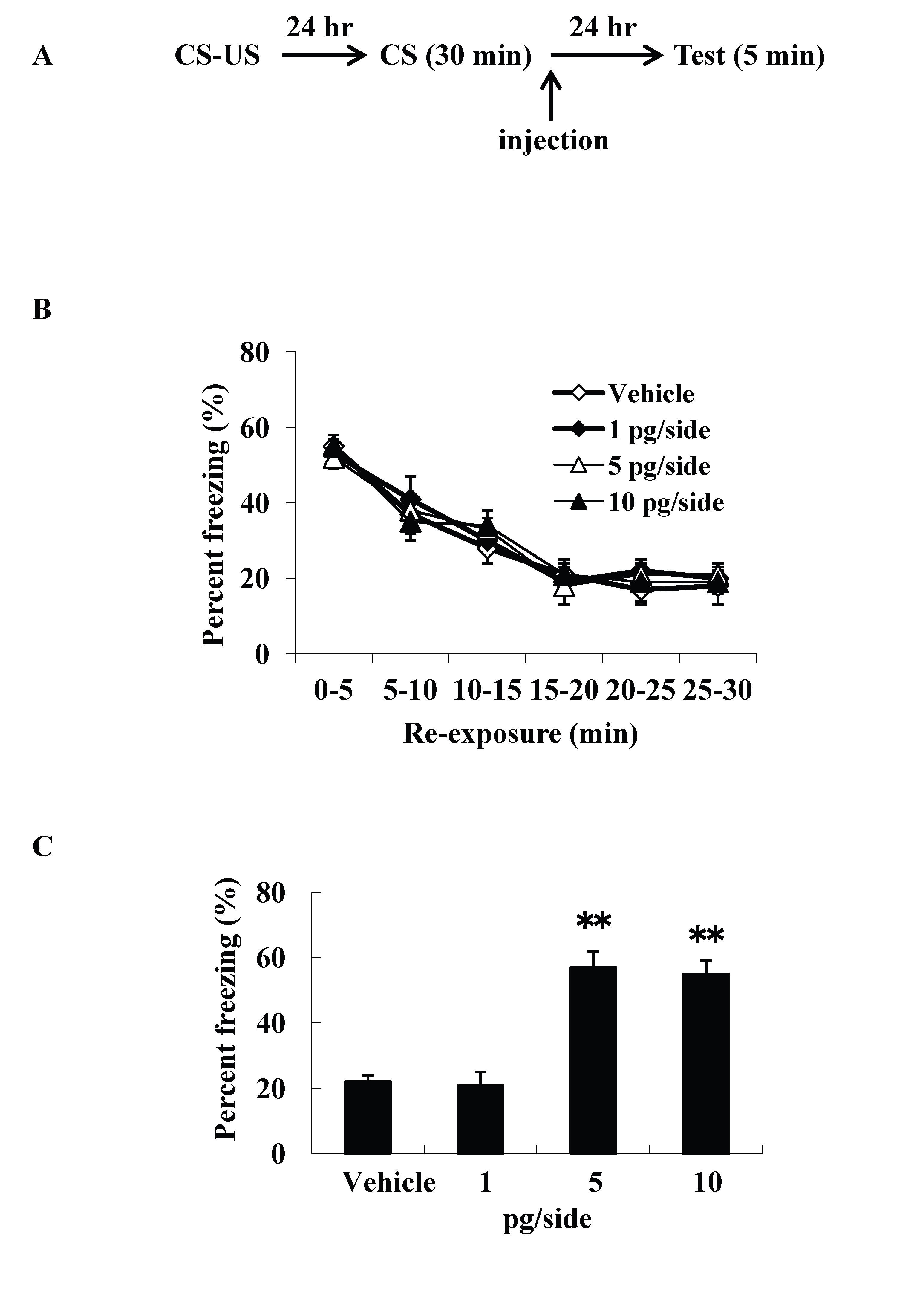
Figure 1. Intracerebroventricular injection effects of ω-conotoxin GVIA on freezing percentage. The experimental design shown (A) was used to collect the data presented below. The effect of ω-conotoxin GVIA on the freezing percentage is shown during the extinction training session (B) and on the test (C). ** P < 0.01 compared with the appropriate control (Tukey’s test).
Intracerebroventricular injection effects ofω-conotoxin GVIA on Arc expression in the mPFC
The intracerebroventricular injection study is difficult to evaluate the site of action of the cascade in the brain. Because previous study reported that fear extinction requires CREB expression in the mPFC [14], we examined Arc expression patterns using mice (n=4 each) given the injections of vehicle or 5 pg/side ω-conotoxin GVIA immediately after re-exposure session. The experimental design is shown in Figure 2A. All groups showed decreasing levels of freezing over the 30-min re-exposure session [F(5, 84)=0.7, P> 0.05]. After the 90-min re-exposure session, significantly higher Arc expression was detected in mPFC regions, including the prelimbic cortex (PLC) and the infralimbic cortex (ILC), in the vehicle-injected mice compared with the ω-conotoxin GVIA-injected mice [PLC: F(1, 14)=84.6, P < 0.01, ILC: F(1,14)=20.2, P < 0.01] (Figure 2B and 2C). These results indicate that Cav2.2-mediated signaling in the mPFC is necessary for consolidation of extinction.
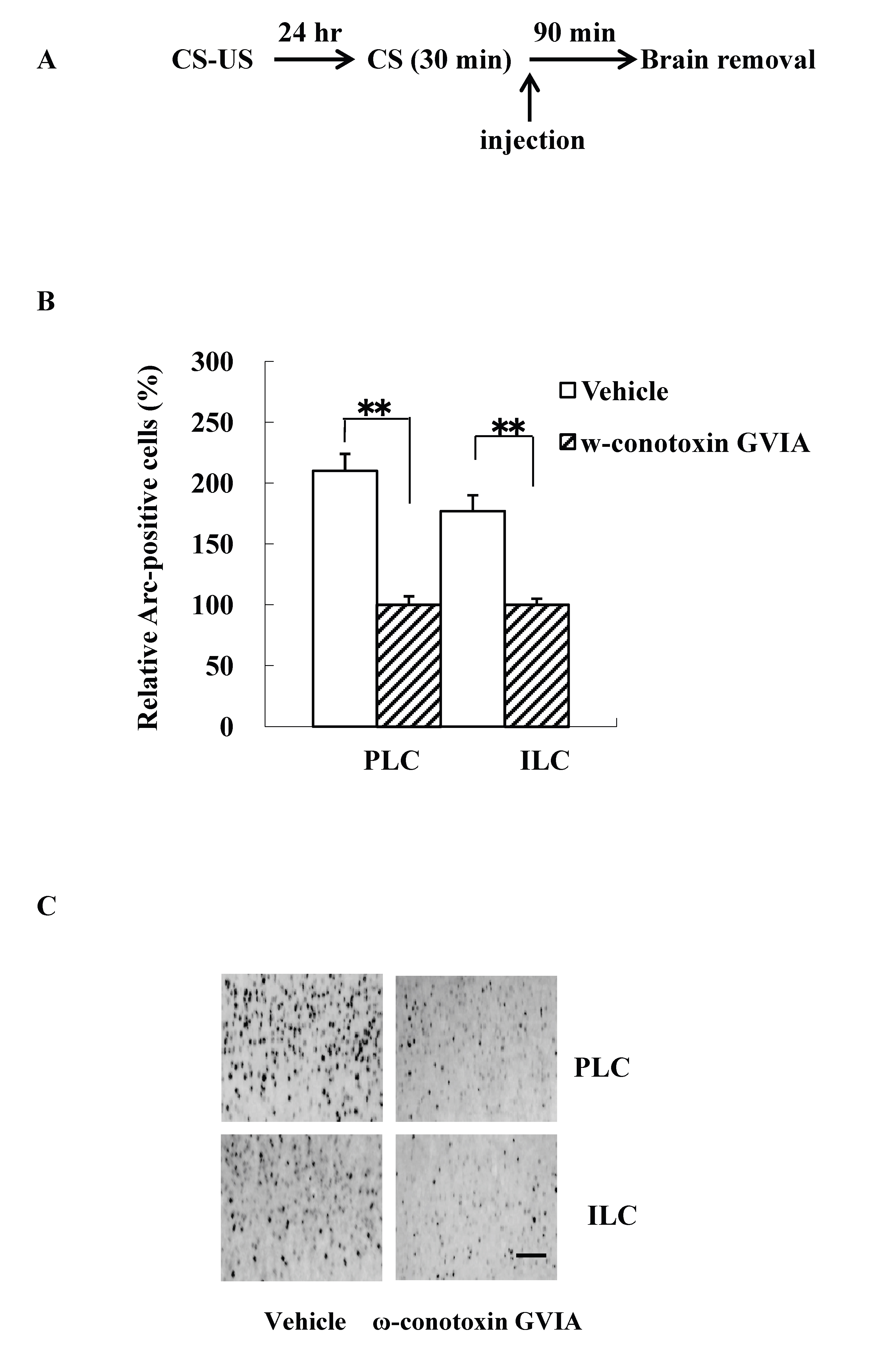
Figure 2. Intracerebroventricular injection effects of ω-conotoxin GVIA on Arc expression. The experimental design shown (A) was used to collect the data presented below. The amount of Arc expression in the mPFC, including the prelimbic cortex (PLC) and infralimbic cortex (ILC), was calculated relative to the amount of Arc present in the vehicle injected mice (B); representative immunocytochemistry in the PLC and ILC is shown (C). Quantification was based on the average of five independent experiments. ** P < 0.01 compared with the appropriate control (Tukey’s test). Scale bar, 50 μm.
Intra-mPFC injection effects of ω-conotoxin GVIA on freezing percentage
We examined intra-mPFC injection effects of ω-conotoxin GVIA on the consolidation of extinction. Our experimental design is shown in Fig. 3A. We used four groups of male mice (n=10 each) given systemic injections of 0 (vehicle), 1, 5, or 10 pg/midline ω-conotoxin GVIA. There were no significant differences among groups in the extinction training session [F(15, 216)=0.8, P > 0.05] (Fig. 3B). However, the groups significantly differed in the percentage of freezing during the test [F(3, 36)=146.9, P < 0.01] (Figure 3C). Figure 3D shows the infusion cannula placement in the mPFC, including the PLC and ILC. The mice given 5 or 10 pg/midline ω-conotoxin GVIA showed greater freezing behavior than the mice given vehicle. These results shows that blockade of Cav2.2-mediated signaling in the mPFC impairs consolidation of extinction.
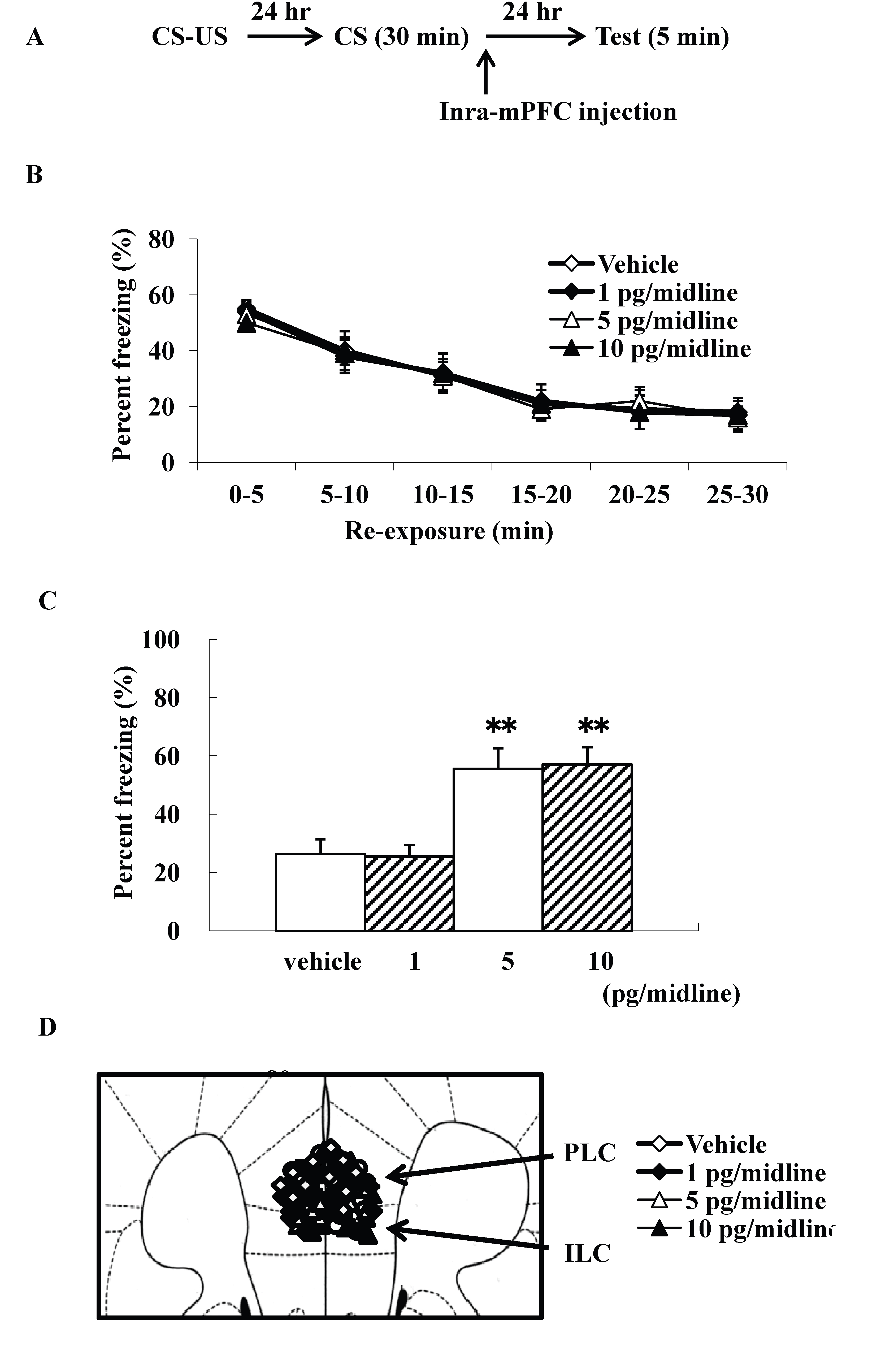
Figure 3. Intra-mPFC injection effects of ω-conotoxin GVIA on freezing percentage. The experimental design shown (A) was used to collect the data presented below. Effect of ω-conotoxin GVIA on the freezing percentage is shown during the extinction training session (B) and on the test (C). This coronal drawing shows the location of the ω-conotoxin GVIA injection aimed at the mPFC, including the prelimbic cortex (PLC) and infralimbic cortex (ILC) (D). ** P < 0.01 compared with the appropriate control (Tukey’s test).
A major goal in extinction research is to understand the molecular and circuit mechanisms that underlie each of the phases of the extinction experience. Previous studies using administered NMDA receptor antagonists have demonstrated that NMDA receptors are not necessary during the extinction training session (extinction acquisition), but are necessary for long-term retention of extinction (extinction consolidation) [19,20]. It has been reported that Cav2.1 blockers inhibit glutamatergic synaptic transmissions [17,21] and consolidation of extinction (i.e., after extinction training) [15]. It has been reported that ω-conotoxin GVIA inhibits presynaptic Cav2.2 function [15] and reduces glutamate release [21]. The role of different VGCCs in the cognitive functions has not been studied. Therefore, we examined whether the Cav2.2-mediated signaling is important for consolidation of extinction
We showed that intracerebroventricular injection of ω-conotoxin GVIA blocked extinction memory, suggesting that the signal cascade activated by Cav2.2 plays a role in the consolidation of extinction. Because Cav2.2 channels are present at a variety of synapses [8], it is impossible that intracerebroventricular injection study identifies the specific brain regions involved in the formation of extinction memory. In this regard, studies of expression and microinfusion in the specific neural region would be useful for elucidating the relevant neuronal mechanisms.
Activation of CREB-mediated transcription is induced in the mPFC during the consolidation of the extinction phase [14]. Previous reports have indicated that the mPFC is engaged after extinction training occurs [22-25]. Our results showed that intracerebroventricular injection of ω-conotoxin GVIA immediately after the re-exposure session blocks Arc expression in the mPFC. The results indicate that the mPFC is an important region in extinction memory formation, particularly in consolidation of the extinction phase, and that information within the mPFC is likely processed by Cav2.2-regulated transmission. Indeed, the present study showed that blocking Cav2.2-regulated signaling in the mPFC induces dysfunctional fear extinction. These results indicate that Cav2.2-mediated signaling pathway in the mPFC plays an important role after extinction training in the acquisition of a vital aspect of the extinction experience, by putting together relationships between different input source(s) and the mPFC. This, in turn, serves to shift behavioral strategies to rapidly inhibit the previously learned fear response during extinction retrieval. Within the mPFC, the PLC and ILC have been implicated in the expression of fear [26], and extinction memory formation in the consolidation phase is associated with activated neuronal function in both the PLC and ILC [14]. The PLC projects mainly to excitatory neurons involved in fear expression [27-29], whereas the ILC projects mainly to inhibitory neurons involved in suppressing fear after extinction [30-32]. The microinfusion of NMDA receptor antagonists or GABAA receptor agonists impairs extinction memory [24,33], whereas GABAA receptor antagonists facilitate extinction memory [34]. These results indicate that both glutamatergic and GABAergic synaptic transmission within the mPFC contribute to extinction learning. It has been reported that Cav2.2 mediates glutamatergic [35] and GABAergic [36] synaptic transmissions. In the present study, neuronal activity in both the PLC and ILC of ω-conotoxin GVIA injected mice was reduced. Although direct evidence of the neural circuit mechanisms of extinction learning and recall within the mPFC and between the mPFC and other regions have yet to be examined, these information would aid in understanding the nature and cause of extinction impairments that contribute to psychopathology.
In this study, we showed that Cav2.2-mediated signaling is critical to mPFC-dependent consolidation of extinction behavior of conditioned fear. However, we didn’t examine whether there is a relationship between Cav2.2-mediated signal transduction and glutamatergic system, between Cav2.2-mediated signal transduction and GABAergic system, and between Cav2.1-mediated signal transduction and Cav2.2-mediated signal transduction in the extinction memory. We plan to identify functional signaling pathways in specific neuronal circuits underlying fear extinction.
The authors declare no competing interests.
WL and ET designed and supervised the research, and wrote the manuscript. YZ and KN performed the behavioral and biochemical experiments. All authors read and approved the final version of the manuscript.
2021 Copyright OAT. All rights reserv
- Milad MR, Rosenbaum BL, Simon NM (2014) Neuroscience of fear extinction: Implications for assessment and treatment of fear-based and anxiety related disorders. Behav Res Ther 62: 17-23. [Crossref]
- Frankland PW, Ding HK, Takahashi E, Suzuki A, Kida S, et al. (2006) Stability of recent and remote contextual fear memory. Learn Mem 13: 451-457. [Crossref]
- Myers KM, Davis M (2002) Behavioral and neural analysis of extinction. Neuron 36: 567-584. [Crossref]
- Catterall WA, Few AP (2008) Calcium channel regulation and presynaptic plasticity. Neuron 59: 882-901. [Crossref]
- Evans RM, Zamponi GW (2006) Presynaptic Ca2+ channels--integration centers for neuronal signaling pathways. Trends Neurosci 29: 617-624. [Crossref]
- Jarvis SE, Zamponi GW (2007) Trafficking and regulation of neuronal voltage-gated calcium channels. Curr Opin Cell Biol 19: 474-482. [Crossref]
- Lee CY, Chen CC, Liou HH (2009) Levetiracetam inhibits glutamate transmission through presynaptic P/Q-type calcium channels on the granule cells of the dentate gyrus. Br J Pharmacol 158: 1753-1762. [Crossref]
- Tanaka O, Sakagami H, Kondo H (1995) Localization of mRNAs of voltage-dependent Ca(2+)-channels: four subtypes of alpha 1- and beta-subunits in developing and mature rat brain. Brain Res Mol Brain Res 30: 1-16. [Crossref]
- Ishibashi H, Rhee JS, Akaike N (1997) Effect of nilvadipine on high-voltage activated Ca2+ channels in rat CNS neurons. Neuroreport 8: 853-857. [Crossref]
- Gourley SL, Kedves AT, Olausson P, Taylor JR (2009) A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology 34: 707-716. [Crossref]
- Paoletti P (2011) Molecular basis of NMDA receptor functional diversity. Eur J Neurosci 33: 1351-1365. [Crossref]
- Maren S, Quirk GJ (2004) Neuronal signalling of fear memory. Nat Rev Neurosci 5: 844-852. [Crossref]
- Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ (2007) Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron 53: 871-880. [Crossref]
- Mamiya N, Fukushima H, Suzuki A, Matsuyama Z, Homma S, et al. (2009) Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J Neurosci 29: 402-413. [Crossref]
- Niimi K, Han Y, Zhou Y, Yoshimoto T, Dai F, et al. (2014) Blockade of Cav2.1-mediated NMDA receptor signaling disrupts conditioned fear extinction. Behav Brain Res 259: 45-49. [Crossref]
- Quirk GJ, Mueller D (2008) Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 33: 56-72. [Crossref]
- Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, et al. (2010) Neuronal circuits of fear extinction. Eur J Neurosci 31: 599-612. [Crossref]
- Ogura H, Furuya Y, Teramoto T, Niidome T, Nishizawa Y, et al. (1998) Peptide N- and P/Q-type Ca2+ blockers inhibit stimulant-induced hyperactivity in mice. Peptides 19: 1017-1022. [Crossref]
- Santini E, Muller RU, Quirk GJ (2001) Consolidation of extinction learning involves transfer from NMDA-independent to NMDA-dependent memory. J Neurosci 21: 9009-9017. [Crossref]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, et al. (2004) Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci 24: 4787-4795.[Crossref]
- Kimura M, Katayama K, Nishizawa Y (1999) Role of glutamate receptors and voltage-dependent calcium channels in glutamate toxicity in energy-compromised cortical neurons. Jpn J Pharmacol 80: 351-358. [Crossref]
- Quirk GJ, Garcia R, González-Lima F (2006) Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry 60: 337-343. [Crossref]
- Rauch SL, Shin LM, Phelps EA (2006) Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research--past, present, and future. Biol Psychiatry 60: 376-382. [Crossref]
- Sotres-Bayon F, Cain CK, LeDoux JE (2006) Brain mechanisms of fear extinction: historical perspectives on the contribution of prefrontal cortex. Biol Psychiatry 60: 329-336. [Crossref]
- Quirk GJ, Mueller D (2008) Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 33: 56-72. [Crossref]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ (2011) Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36: 529-538. [Crossref]
- McDonald AJ, Mascagni F, Guo L (1996) Projections of the medial and lateral prefrontal cortices to the amygdala: A Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience 71: 55-75.
- Likhtik E, Pelletier JG, Paz R, Paré D (2005) Prefrontal control of the amygdala. J Neurosci 25: 7429-7437. [Crossref]
- Burgos-Robles A, Vidal-Gonzalez I, Quirk GJ (2009) Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J Neurosci 29: 8474-8482. [Crossref]
- Rosenkranz JA, Grace AA (2002) Dopamine-mediated modulation of odour-evoked amygdala potentials during pavlovian conditioning. Nature 417: 282-287. [Crossref]
- Paré D, Quirk GJ, Ledoux JE (2004) New vistas on amygdala networks in conditioned fear. J Neurophysiol 92: 1-9. [Crossref]
- Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Paré D (2008) Amygdala intercalated neurons are required for expression of fear extinction. Nature 454: 642-645. [Crossref]
- Laurent V, Westbrook RF (2008) Distinct contributions of the basolateral amygdala and the medial prefrontal cortex to learning and relearning extinction of context conditioned fear. Learn Mem 15: 657-666. [Crossref]
- Thompson BM, Baratta MV, Biedenkapp JC, Rudy JW, Watkins LR, et al. (2010) Activation of the infralimbic cortex in a fear context enhances extinction learning. Learn Mem 17: 591-599. [Crossref]
- Li DP, Chen SR2 (2014) Nitric oxide stimulates glutamatergic synaptic inputs to baroreceptor neurons through potentiation of Cav2.2-mediated Ca(2+) currents. Neurosci Lett 567: 57-62. [Crossref]
- Murakami N, Ishibashi H, Katsurabayashi S, Akaike N (2002) Calcium channel subtypes on single GABAergic presynaptic terminal projecting to rat hippocampal neurons. Brain Res 951: 121-129. [Crossref]