Neuronal voltage-gated calcium (Ca2+) channels (VGCCs) are well known for their important role in the activities of neurotransmitter systems and signaling pathways in specific circuits. Disturbances in the neuronal circuits can contribute to the manifestation of neuropsychiatric disorders. For example, sensorimotor gating deficits are observed in a majority of patients with schizophrenia. The integrity of sensorimotor gating mechanisms is often assessed with the prepulse inhibition (PPI) test but the various roles of different VGCCs during PPI remain unclear. Thus, the present study aimed to examine whether Cav2.2-mediated signaling is associated with the symptoms of schizophrenia using behavioral and biochemical tests. Mice that received intracerebroventricular (i.c.v.) injections of ω-Conotoxin GVIA (5 pg/side), a Cav2.2 blocker, exhibited deficits in PPI and decreased baseline levels of dopamine and serotonin within the striatum and frontal cortex. These findings indicate that abnormalities in Cav2.2-mediated neuronal circuits might contribute to the basic pathophysiological mechanisms underlying schizophrenia.
cav2.2, neuronal circuits, ω-conotoxin GVI, prepulse inhibition, schizophrenia
Calcium (Ca2+) signaling via voltage-gated Ca2+ channels (VGCCs) mediates the entry of Ca2+ into cells following membrane depolarization and initiates the transduction of electrical signals into chemical signals [1]. Neuronal VGCCs such as the Cav2.1 (P/Q-type), Cav2.2 (N-type), and Cav2.3 (R-type) channels mediate a number of neuronal functions, including neuronal excitation, neurite outgrowth, synaptogenesis, neurotransmitter release, neuronal survival, differentiation, plasticity, and the regulation of gene expression [1-3]. However, the various roles of different VGCCs in the expression of neuropsychiatric disorders have yet to be fully characterized.
Neuropsychiatric disorders such as schizophrenia, Huntington's disease, obsessive-compulsive disorder, attention-deficit disorder, and Tourette's syndrome are associated with deficiencies in sensorimotor gating mechanisms [4]. In humans and animals, the integrity of sensorimotor gating mechanisms is often assessed using prepulse inhibition (PPI), which is the suppression of the startle reflex when a startling stimulus is preceded by a weak stimulus at lead times of 20-500 ms [4]. A multitude of evidence suggests that the pathophysiology of schizophrenia involves dysfunction in the dopaminergic, serotonergic, glutamatergic, and g-aminobutyric acid (GABA)-ergic neurotransmitter systems [5] and it has been reported that Cav2.2 channels influence the presynaptic machinery involved in the release of several neurotransmitters, including dopamine [DA; 6-8], serotonin [5-HT; 9], glutamate [10], GABA [11], acetylcholine [12], and norepinephrine [NE; 13], from central neurons. Of clinical relevance, schizophrenia is strongly associated with genes important for presynaptic signaling [14-17]. Thus, the administration of a Cav2.2 blocker should result in PPI deficits, because the precise regulation of neurotransmitter release via Cav2.2 channels plays an important role in the functioning of neuronal circuits.
Therefore, the present study aimed to investigate the relationship between Cav2.2-mediated signaling and sensorimotor gating as a first step towards understanding the mechanisms underlying the expression of schizophrenia. Wild-type mice received intracerebroventricular (i.c.v.) administrations of ω-Conotoxin GVIA, which is an antagonist of the Cav2.2 channel [1], and were then assessed in a PPI test. Additionally, the amounts of monoamines and their metabolites in the brains of the mice were measured to determine the relationship between alterations in monoamine metabolism and behavioral abnormalities.
Mice
All animal procedures were approved by the Animal Experiments Committee of Shanghai Jiao Tong University and RIKEN. The C57BL/6J mice were provided by Charles River Japan (Kanagawa, Japan). The mice were given free access to water and food pellets (CRF-1; Oriental Yeast Co. Ltd., Tokyo, Japan) and were housed under a 12/12-h light/dark cycle (lights on from 08:00 to 20:00) at 23 ± 1°C and 55 ± 5% humidity. Testing was performed during the light phase of the cycle.
Infusion
For the infusion studies, Cav2.2 blocker, ω-conotoxin GVIA (10, 50, or 100 pg/μL, Peptide Institute, Osaka, Japan) were dissolved in saline (vehicle). Under anesthesia and using standard stereotaxic procedures, stainless-steel guide cannulae (22-gauge) were implanted into the lateral ventricle (posterior to bregma, -0.34 mm; lateral to midline, ±0.9 mm; ventral from the dura, −2.3 mm). Mice were allowed to recover for at least 1 week following surgery. The mice were briefly anesthetized with isoflurane to facilitate insertion of the injection cannula (26-gauge). Infusions into the lateral ventricle (0.1 μL/side) are accomplished at a rate of 0.05 μL/min 30 min before behavioral or biochemical tests. The injection cannula was left in place for 2 min following the infusion. The drug doses were determined according to previous report [18,19]. Mice that were not treated with drugs received an equivalent volume of vehicle.
Inhibition of the acoustic startle reflex
The PPI tests were conducted using an SR-LAB system (San Diego Instruments; San Diego CA, USA) constructed of four sound-attenuating chambers that were each equipped with a cylindrical Plexiglas animal enclosure (length: 6.5 cm; inner diameter: 3.8 cm). Ventilation was provided by a small electric fan that also generated a background noise of 70 dB. The tone pulses were presented by a speaker positioned 24 cm directly above the animal enclosure and any motion resulting from the animal’s response was detected by a piezoelectric accelerometer affixed to the animal enclosure frame. The tone pulse parameters were controlled by a microcomputer using a software package (SR-LAB) and interface assembly that also digitized, rectified, and recorded the stabilimeter readings. Measures for both the acoustic startle response (ASR) and PPI were obtained in a single session, as described by Brake et al. [20], with minor modifications.
The mice were placed in the Plexiglas enclosure and allowed to acclimatize to the environment for 5 min before being tested in 37 discrete trials. In the first two trials, the magnitudes of the ASR to a 120 dB tone that lasted 50 ms were measured; these first two startle tones were presented to habituate the animals to the testing procedure and were omitted from the statistical analysis of the mean ASR amplitude. In the subsequent 35 trials, the startle tone was either presented alone or 100 ms after the presentation of a prepulse that lasted 30 ms. The prepulse intensity ranged from 3-15 dB above the background noise and varied randomly between the trials in steps of 3 dB. Measurements of the magnitudes of the ASR were obtained at each of the five prepulse intensities over five trials and the mice were randomly presented with only the startle tone during the other 10 trials.
The same stimulus condition was never presented in more than two consecutive trials, and the intervals between the trials were programmed using a variable interval schedule with an average duration of 15 s (range: 5-30 s). A measure of the ASR amplitude was derived from the mean of 100 digitized data points collected from the stimulus onset at a rate of 1 kHz. The effectiveness of the prepulse to suppress the ASR was expressed as a percentage based on the mean amplitude of responses to the startle tone alone (n = 10) relative to those recorded under the five prepulse conditions (n = 5/condition) and was calculated as follows: % PPI = 100 − [(mean startle amplitude for prepulse + pulse trials/mean startle amplitude for pulse–alone trials) × 100]%.
Biochemical tests
The levels of monoamines and their metabolites in the brain were examined as previously described by Nakagawasai et al. [21] with minor modifications. The mice were sacrificed with focused microwave irradiation 30 min after the i.c.v. injection and the brains were rapidly removed. The striatum and frontal cortex were dissected out onto an ice-cold plate and each brain sample was quickly frozen and stored in a deep freezer at -80°C until assayed. The contents of DA, 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 5-HT, 5-hydroxyindoleacetic acid (5-HIAA), and NE were determined by high-performance liquid chromatography (HPLC) with electrochemical detection (Eicom Corp., Kyoto, Japan).
Data analysis
Data are presented as means ± standard error of the mean (SEM). Statistical analyses for the behavioral and biochemical studies were conducted using Excel Statistics 2006 (SSRI, Tokyo, Japan). Data were analyzed using Dunnett test.
Effects of intracerebroventricular injections of ω-Conotoxin GVIA on the ASR and PPI
The present study examined the effects of i.c.v. injections of ω-Conotoxin GVIA on the ASR and PPI. Four groups of male mice (n = 10 each) were administered i.c.v. injections of either vehicle or ω-Conotoxin GVIA (1, 5, or 10 pg/side). There were no significant differences in the startle amplitude among the groups (Figure 1A). In the PPI paradigm, although there was no significant difference between vehicle-injected and 1 pg/side ω-Conotoxin GVIA-injected mice (Figure 1B), the mice that received either 5 (Figure 1C) or 10 pg/side (Figure 1D) of ω-Conotoxin GVIA showed greater deficits in sensorimotor gating than the mice that received vehicle. Both of these groups exhibited significant deficits at three prepulse intensities (79 dB, 82 dB, and 85dB), which indicates that the blockade of Cav2.2-mediated signaling impairs sensorimotor gating. The correct placements of the guide cannula were verified by ink injections after the experiments were completed (data not shown).
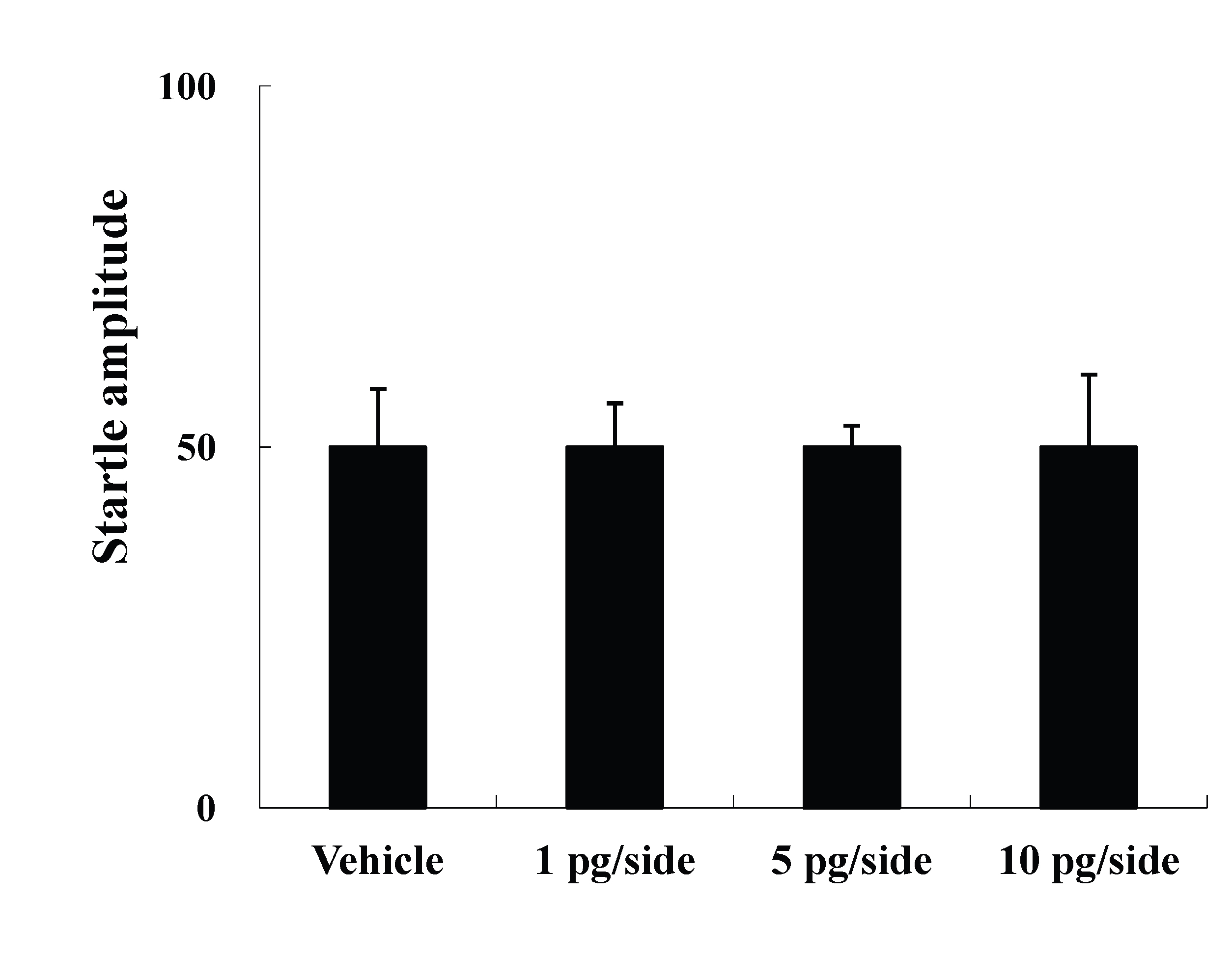
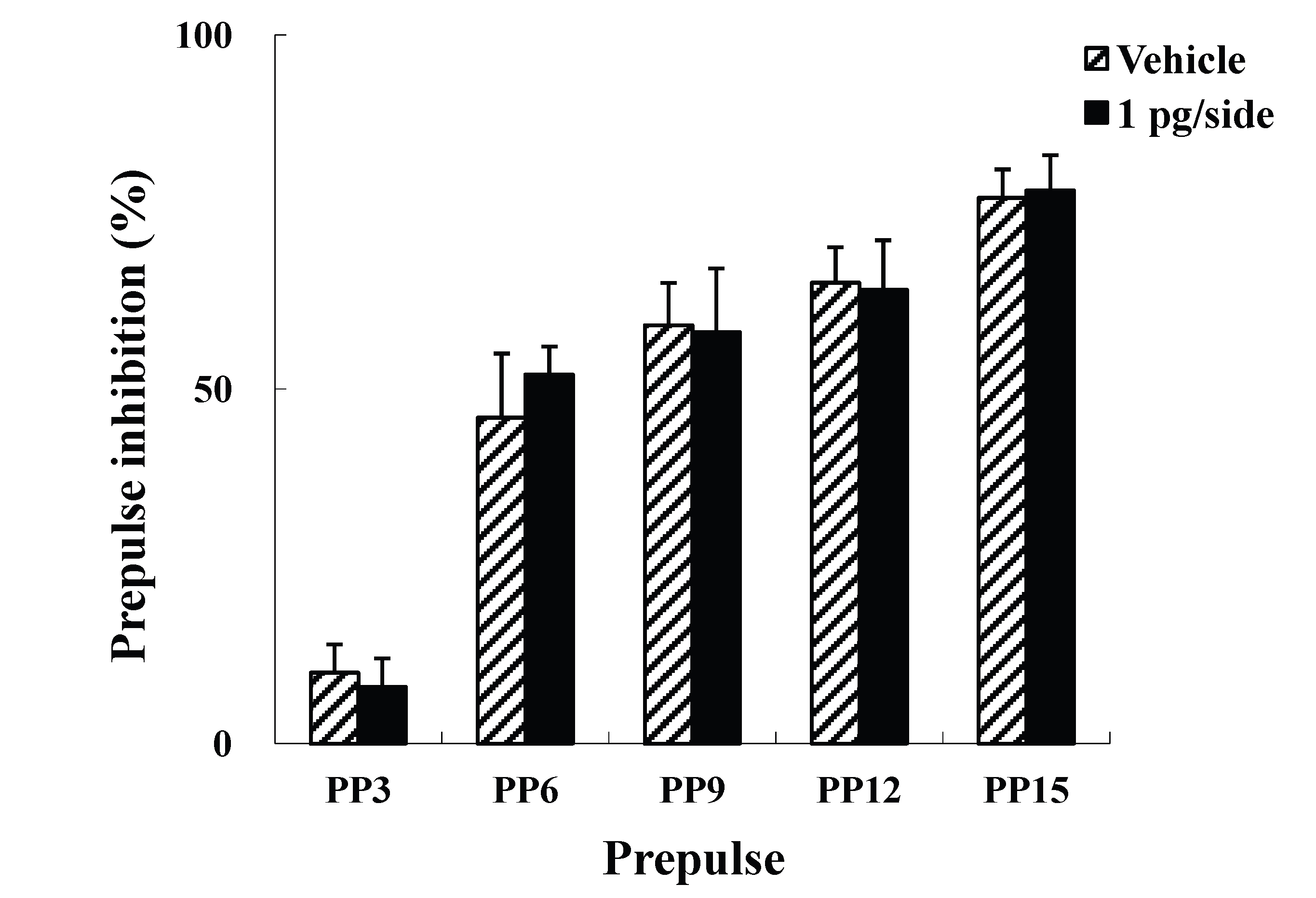
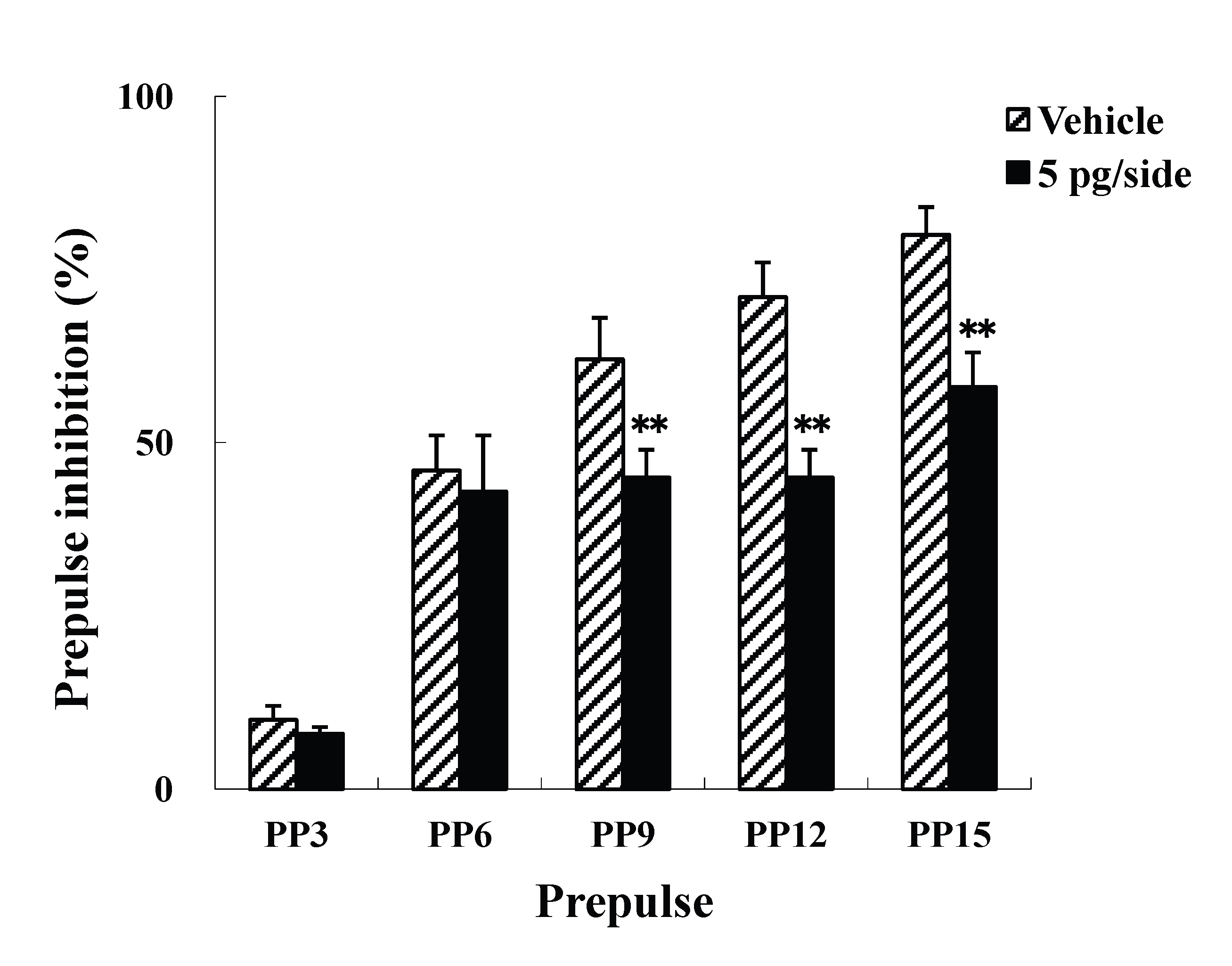
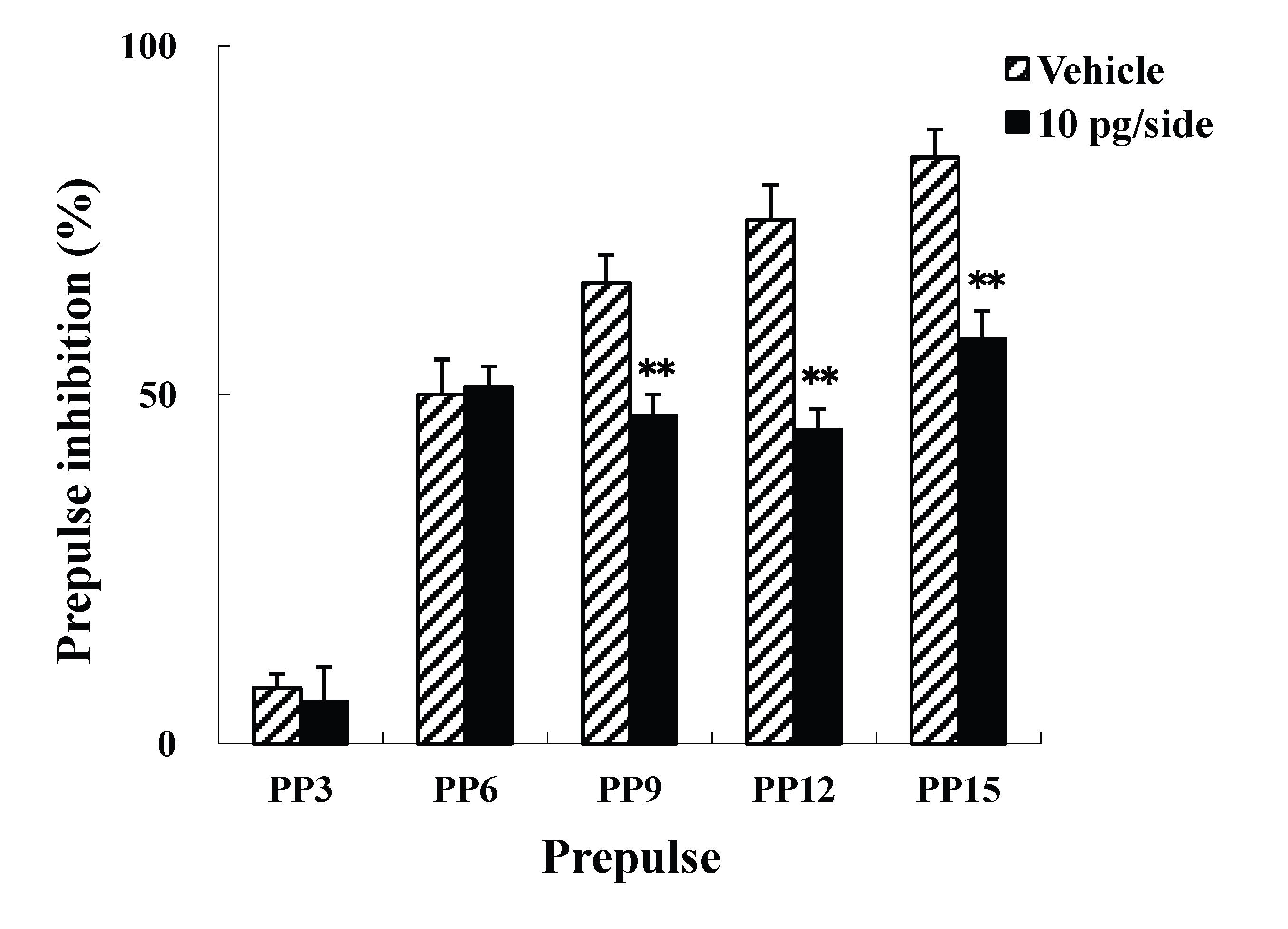
Figure 1. Mean startle amplitudes (A) and percent PPI (B, C, and D) in the vehicle- and ω-Conotoxin GVIA-injected mice. Data are expressed as the means ± SEM. *P<0.05, **P<0.01 compared to vehicle-injected mice.
Effects of intracerebroventricular injections of ω-Conotoxin GVIA on the levels of monoamines and their metabolites
The present study examined the effects of i.c.v. injections of ω-Conotoxin GVIA on the levels of monoamines and their metabolites in the brain. Two groups of male mice (n = 10 each) were administered i.c.v. injections of either vehicle or ω-Conotoxin GVIA (5 pg/side) and the amounts of monoamines and their metabolites in the brain were measured using HPLC analyses (Tables 1 and 2). There were significant decreases in the striatal and frontal cortical DA content of mice that received ω-Conotoxin GVIA compared to vehicle-injected mice (Table 1). Additionally, there was a significant increase in the turnover rate of DA, which was defined as the ratio of DOPAC or HVA to DA, in the frontal cortices of mice that received ω-Conotoxin GVIA. There was no significant difference in NE content between the vehicle-injected mice and the ω-Conotoxin GVIA-injected mice (Table 1). On the other hand, there were significant decreases in the striatal and frontal cortical levels of 5-HT in the mice that received ω-Conotoxin GVIA compared to the vehicle-injected mice (Table 2). The turnover rate of 5-HT, which was defined as the ratio of 5-HIAA to 5-HT, also exhibited a significant increase in the frontal cortices of mice that received ω-Conotoxin GVIA.
Tables 1. Concentrations of monoamines and their metabolites in vehicle – and ɷ -Conotoxin GVIA –injected mice
|
DA |
DOPAC |
DOPAC/DA |
HVA |
HVA/DA |
Striatum Vehicle |
3245 ± 63 |
2135 ± 26 |
0.646 ± 0.056 |
1007 ± 79 |
0.291 ± 0.033 |
ɷ -Conotoxin GVIA |
2751 ± 35* |
2498 ± 17 |
0.892 ± 0.111 |
1101 ± 53 |
0.352 ± 0.045 |
Frontal cortex Vehicle |
333 ± 18 |
441 ± 31 |
1.271 ± 0.142 |
392 ± 22 |
1.075 ± 0.057 |
ɷ -Conotoxin GVIA |
221 ± 21* |
531 ± 83 |
2.419 ± 0.325 |
363 ± 49 |
1.627 ± 0.238 |
Data shown are in ng/g of brain tissue. *p<0.05 and **p<0.01 vs. vehicle-injected mice
Tables 2. Concentrations of 5-hydroxytryptamine and its metabolite in the brain
|
DA |
DOPAC |
DOPAC/DA |
Striatum Vehicle |
2602 ± 39 |
579 ± 63 |
0.231 ± 0.021 |
ɷ -Conotoxin GVIA |
2158 ± 72** |
509 ± 23 |
0.214 ± 0.022 |
Frontal cortex Vehicle |
729 ± 21 |
489 ± 319 |
0.656 ± 0.039 |
ɷ -Conotoxin GVIA |
579 ± 77* |
399 ± 19 |
0.741 ± 0.009** |
Data shown are in ng/g of brain tissue. *p<0.05 and **p<0.01 vs. vehicle-injected mice
Deficiencies in sensorimotor gating contribute to the manifestation of a number of neuropsychiatric disorders. PPI refers to a reduction of the startle reflex following exposure to a weak-intensity prepulse that is administered immediately prior to a startle stimulus; this is a well-established method used to evaluate sensorimotor gating [4]. In clinical populations, there is a disruption of PPI in a majority of schizophrenic patients [5]. Over the past decade, experimental studies using neuroanatomical, pharmacological, behavioral, biochemical, and genetic manipulations have been useful for the elucidation of the neural circuitry underlying PPI [4]. These findings have also provided insight into the pathophysiologies of several forms of mental illness that are characterized by disruptions in sensorimotor gating. As a first step to understanding the role of Cav2.2-mediated signaling during sensorimotor gating, the present study examined this relationship using pharmacological, behavioral, and biochemical approaches.
The present findings demonstrated that mice receiving i.c.v. injections of ω-Conotoxin GVIA exhibited deficits in PPI. It has been shown that PPI is disrupted by the inhibition of dopaminergic [22,23] or serotonergic [24-26] signaling or by manipulations that decrease DA transmission in the prefrontal cortex [27-29]. In the present study, mice that received injections of ω-Conotoxin GVIA exhibited significantly decreased DA levels in conjunction with significantly increased DA turnover in the frontal cortex. It is possible that the increased DA turnover (DOPAC/DA and HVA/DA) was accompanied by decreased DA levels in the mice treated with the Cav2.2 blocker. Regardless, these results indicate that the dopaminergic hypofunctionality detected within the frontal cortical circuitry of ω-Conotoxin GVIA-injected mice might underlie the observed deficits in PPI. The ω-Conotoxin GVIA-injected mice also showed decreased levels of 5-HT in the striatum. This is consistent with a previous report that intraperitoneal injections of the 5-HT synthesis inhibitor para-chlorophenylalanine and microinjections of the 5-HT neurotoxin 5,7-dihydroxytryptamine into the dorsal and median raphe nuclei depleted striatal 5-HT levels and disrupted PPI [30]. Thus, decreased levels of 5-HT in the striatum may also result in PPI deficits. In contrast, there were no significant differences in NE content between the vehicle-injected and ω-Conotoxin GVIA-injected mice, even though the injections of ω-Conotoxin GVIA had a tendency to decrease NE levels relative to vehicle injections.
In conclusion, the present study showed that the inhibition of Cav2.2-mediated signaling by the specific Cav2.2 blocker ω-Conotoxin GVIA induced deficits in sensorimotor gating. The deficits of sensorimotor gating are observed in a majority of schizophrenic patients [4]. The present findings indicated that there were significant relationships between Cav2.2-mediated signal transduction and the dopaminergic and serotonergic systems that likely underlie the expression of various neuropsychiatric disorders. These results are similar to those of a previous study [21] which reported that Cav2.2 knockout mice exhibit deficits in PPI as well as altered DA and 5-HT release. Thus, abnormalities in Cav2.2-mediated neuronal circuits might contribute to the basic pathophysiological mechanisms underlying schizophrenia.
The authors declare no competing interests.
WL and ET designed and supervised the research, and wrote the manuscript. YZ and KN performed the behavioral and biochemical experiments. All authors read and approved the final version of the manuscript.
2021 Copyright OAT. All rights reserv
- Catterall WA, Few AP (2008) Calcium channel regulation and presynaptic plasticity. Neuron 59: 882-901. [Crossref]
- Evans RM, Zamponi GW (2006) Presynaptic Ca2+ channels--integration centers for neuronal signaling pathways. Trends Neurosci 29: 617-624. [Crossref]
- Jarvis SE, Zamponi GW (2007) Trafficking and regulation of neuronal voltage-gated calcium channels. Curr Opin Cell Biol 19: 474-482. [Crossref]
- Braff DL, Geyer MA, Swerdlow NR (2001) Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 156: 234-258. [Crossref]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR (2001) Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 156: 117-154. [Crossref]
- Herdon H, Nahorski SR (1989) Investigations of the roles of dihydropyridine and?-conotoxin-sensitive calcium channels in mediating depolarization-evoked endogenous dopamine release from striatal slices. Naunyn Schmiedebergs Arch Pharmacol 340: 36–40. [Crossref]
- Turner TJ, Adams ME, Dunlap K (1993) Multiple Ca2+ channel types coexist to regulate synaptosomal neurotransmitter release. Proc Natl Acad Sci U S A 90: 9518-9522. [Crossref]
- Woodward JJ, Rezazadeh SM, Leslie SW (1988) Differential sensitivity of synaptosomal calcium entry and endogenous dopamine release to omega-conotoxin. Brain Res 475: 141-145. [Crossref]
- Foehring RC (1996) Serotonin modulates N- and P-type calcium currents in neocortical pyramidal neurons via a membrane-delimited pathway. J Neurophysiol 75: 648-659. [Crossref]
- Luebke JI, Dunlap K, Turner TJ (1993) Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron 11: 895-902. [Crossref]
- Horne AL, Kemp JA (1991) The effect of omega-conotoxin GVIA on synaptic transmission within the nucleus accumbens and hippocampus of the rat in vitro. Br J Pharmacol 103: 1733-1739. [Crossref]
- Wessler I, Dooley DJ, Werhand J, Schlemmer F (1990) Differential effects of calcium channel antagonists (omega-conotoxin GVIA, nifedipine, verapamil) on the electrically-evoked release of [3H] acetylcholine from the myenteric plexus, phrenic nerve and neocortex of rats. Naunyn Schmiedebergs Arch Pharmacol 341: 288–294. [Crossref]
- Dooley DJ, Lupp A, Hertting G, Osswald H (1988) Omega-conotoxin GVIA and pharmacological modulation of hippocampal noradrenaline release. Eur J Pharmacol 148: 261-267. [Crossref]
- Chen Y, Lai M, Maeno-Hikichi Y, Zhang JF (2006) Essential role of the LIM domain in the formation of the PKCepsilon-ENH-N-type Ca2+ channel complex. Cell Signal 18: 215-224. [Crossref]
- Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P (2000) Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 28: 53-67. [Crossref]
- Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, et al. (2004) Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet 13: 2699-2708. [Crossref]
- Talbot K, Eidem WL, Tinsley CL, Benson MA, Thompson EW, et al. (2004) Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest 113: 1353-1363. [Crossref]
- Ogura H, Furuya Y, Teramoto T, Niidome T, Nishizawa Y, et al. (1998) Peptide N- and P/Q-type Ca2+ blockers inhibit stimulant-induced hyperactivity in mice. Peptides 19: 1017-1022. [Crossref]
- Niimi K, Han Y, Zhou Y, Yoshimoto T, Dai F, et al. (2014) Blockade of Cav2.1-mediated NMDA receptor signaling disrupts conditioned fear extinction. Behav Brain Res 259: 45-49. [Crossref]
- Brake WG, Flores G, Francis D, Meaney MJ, Srivastava LK, et al. (2000) Enhanced nucleus accumbens dopamine and plasma corticosterone stress responses in adult rats with neonatal excitotoxic lesions to the medial prefrontal cortex. Neuroscience 96: 687-695. [Crossref]
- Nakagawasai O, Onogi H, Mitazaki S, Sato A, Watanabe K, et al. (2010) Behavioral and neurochemical characterization of mice deficient in the N-type Ca2+ channel alpha1B subunit. Behav Brain Res 208: 224-230. [Crossref]
- Swerdlow NR, Braff DL, Geyer MA, Koob GF (1986) Central dopamine hyperactivity in rats mimics abnormal acoustic startle response in schizophrenics. Biol Psychiatry 21: 23-33. [Crossref]
- Swerdlow NR, Shoemaker JM, Kuczenski R, Bongiovanni MJ, Neary AC, et al. (2006) Forebrain D1 function and sensorimotor gating in rats: effects of D1 blockade, frontal lesions and dopamine denervation. Neurosci Lett 402: 40-45. [Crossref]
- Kehne JH, Padich RA, McCloskey TC, Taylor VL, Schmidt CJ (1996) 5-HT modulation of auditory and visual sensorimotor gating: I. Effects of 5-HT releasers on sound and light prepulse inhibition in Wistar rats. Psychopharmacology (Berl) 124: 95-106. [Crossref]
- Padich RA, McCloskey TC, Kehne JH (1996) 5-HT modulation of auditory and visual sensorimotor gating: II. Effects of the 5-HT2A antagonist MDL 100,907 on disruption of sound and light prepulse inhibition produced by 5-HT agonists in Wistar rats. Psychopharmacology (Berl) 124: 107-116. [Crossref]
- Sipes TA, Geyer MA (1994) Multiple serotonin receptor subtypes modulate prepulse inhibition of the startle response in rats. Neuropharmacology 33: 441-448. [Crossref]
- Bubser M, Koch M (1994) Prepulse inhibition of the acoustic startle response of rats is reduced by 6-hydroxydopamine lesions of the medial prefrontal cortex. Psychopharmacology 113: 487–492. [Crossref]
- Ellenbroek BA, Budde S, Cools AR (1996) Prepulse inhibition and latent inhibition: the role of dopamine in the medial prefrontal cortex. Neuroscience 75: 535-542. [Crossref]
- Koch M, Bubser M (1994) Deficient sensorimotor gating after 6-hydroxydopamine lesion of the rat medial prefrontal cortex is reversed by haloperidol. Eur J Neurosci 6: 1837-1845. [Crossref]
- Fletcher PJ, Selhi ZF, Azampanah A, Sills TL (2001) Reduced brain serotonin activity disrupts prepulse inhibition of the acoustic startle reflex: effects of 5,7-dihydroxytryptamine and p-chlorophenylalanin. Neuropsychopharmacology 24: 399–409. [Crossref]