Abstract
Alzheimer’s disease (AD) is represented by neuronal loss and this loss is correlated to a constant state of neuronal instability induced by intrinsic and extrinsic factors. In this paper data is presented regarding the possible roles of late phase cell cycle proteins in normal and affected neurons with the goal that understanding the mechanisms involved in the regulation of these proteins may represent a novel strategy for AD treatment. The results demonstrate a relative differential pattern of expression of certain proteins (APC/C, Mad1 and Mad2, Bub R1, Bub1, CDK 11, cohesin subunit Rad 21 and astrin) in the AD brain versus age matched controls, and it is suggested that targeting these proteins might translate into potential treatments for AD. Although the data presented here is of some interest, the ability to translate such information into clinical applications is often a challenge.
Key Words
Alzheimer’s disease, neuronal instability
Introduction
Alzheimer’s disease (AD) in its two forms, FAD and SAD, is represented by accumulated oligomeric fragments of Aβ and Tau phosphorylated proteins. AD is still a highly deadly disease with no cure on the horizon [1]. A shift has been recently introduced i.e. after on only concentrating on Aβ and Tau as the only causative agents for AD, researchers explored the notion that there must be a more fundamental underlining mechanisms, such as inflammatory processes mediated by astrocytes and microglia [2-4] which then in coordination with Aβ and Tau deregulates the neuronal cell, thus leading to cell cycle activation [4,5]. Neuronal responses to such insults in AD brain include increased protein levels and immunoreactivity for kinases known to regulate cell cycle progression and consequently cell death [4,6-10].
Data supports the hypothesis that neurons have evolutionary acquired the ability to use molecular mechanisms primarily developed to control proliferation alternatively to control synaptic plasticity [11,12]. So, protein multifunctionality has gained attention in the neuroscience community, i.e. a number of core cell cycle proteins, essential for cell cycle division has been found to coordinate a number of complex processes in neurogenesis from neuronal migration, axonal elongation, axon pruning, dendrite morphogenesis and synaptic maturation and plasticity [13] to neuronal survival [14] and the maintenance of the post-mitotic state of neurons [15-17]. These core cell cycle proteins have been extensively reviewed by Frank and Tsai, 2009 [13] so here in this review we shall report only findings of how of some of these proteins are expressed in normal versus vulnerable neurons of AD patients and their possible relation to achieving an active state in non-dividing neurons [18]. One feature of altered expression of late phase cell cycle proteins may have been findings of aneuploidy in AD [19,20]. Potter et al,2008 [ 21-23] has reported that in AD models and the AD brain there is constantly a presence of aneuploidy of chromosome 21 in high percentage of neurons leading to pathology. In our view the brain is highly genomicaly heterogeneous meaning that aneuploidy must be more broadly perceived, as researchers have found changes from DNA content variations, LINE 1 retrotransponsons, copy number variations (CNV) to DNA sequence deletion, duplication, microdeletions [24-28].
We have recently proposed a new hypothesis that might incorporate the cell cycle reentry hypothesis with the amyloid hypothesis, trisomy of chromosome 21 and FAD. This hypothesis named ‘’the postmitotic state maintained protein hypothesis‘’ [29] represents a relationship between the ‘’aneuploidy phenotype’’ of neural progenitors (during development and in adult brain) and late phase cell cycle proteins that maintain an balanced proteome.
Aneuploidy increases proteotoxic stress [30] relaying to the state of quality control machinery of the neuronal cell. If the neurons become unbalanced, through the inhibition or excess activation of the late phase cell cycle proteins or the percentage of cells with the aneuploidogenic phenotype, then the cell is unable to cope with an increase in proteotoxic stress which then deregulates APP expression and Aβ accumulation. If the balance is not restored the cell undergoes cell cycle reentry which always leads to apoptosis and cell death [29,31-33].
Here in this review we shall address the Anaphase Promoting Complex /cyclosome (APC/C) and cohesin complex with related proteins, in genetic processes that are unrelated to separation and segregation of centromeres. By emphasing their roles in neurons and their expression levels in AD we shall address the question of target evaluation for AD treatment.
Neurodegeneration: it starts with the APC/C-cdh 1?
Anaphase Promoting Complex /cyclosome (APC/C) is a highly complex protein initially identified more than 15 years ago, a E3 ubiquitin ligase, that has 13 subunits, which triggers the metaphase to anaphase transition by marking select proteins by polyubiquitin chains for degradation. The APC/C requires the binding of a cofactor, Cdc20 or Cdh1, in order to select substrates and perform its activity [34]. APC/C has substrates that regulate the mitotic checkpoint (MCC) and chromosome segregation. They must be degraded for cells to exit mitosis (Figure 1). Securin is a protein that inhibits separase, which in turn inhibits cohesin, a protein that holds sister chromatids together. Therefore, in order for anaphase to progress, securin must be inhibited so that cohesin can be cleaved by separase. Separase activated cleaves Scc 1 or RAD 21 subunit of the multi protein complex cohesin that holds sister chromatides together (Figure 1) [35]. The anaphase-promoting complex (APC) is tethered to cohesion in processes of chromosome segregation and separation in mitotic cells and new data show functional activity in post-mitotic neurons in which alterations of these proteins may lead to aneuploidy and increased proteotoxicity [29,30,36-40].
Figure 1. Late phase cell cycle proteins regulate the metaphase anaphase transition
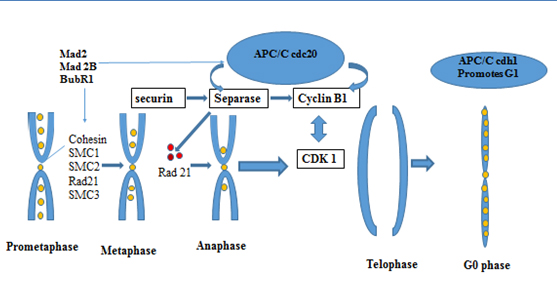
*APC/C cdh1, Anaphase Promoting complex/cyclosome , cdh 1- cadherin type 1
SMC- : structural maintenance of cohesion; Mad 2 and Mad 2B- The Mitotic Arrest Deficient Protein, BubR1 – (budding uninhibited by benzimidazoles) Interaction between BUBR1(budding uninhibited by benzimidazoles) and C-MAD2 (="mitotic arrest deficient") plays a critical role in mitotic control center (MCC)-mediated inhibition of APC/C
Gieffers et al., 1999 [41] were the first to report that the APC/C complex has a role in postmitotic neurons. APC activator subunit cdh1 functions in several major processes in the nervous system, from cdh-1 controlling G1 maintenance [39,42] to axonal growth [36,43] ,cdh-1 also, coordinates neurogenesis [44], synaptic plasticity [45], dendritic morphogenesis, presynaptic differentiation [46] and memory [47]. Aulia, et al. 2006 [18] proposed that APC/cdh1 has a possible role in neurodegenerative diseases as it inhibits cyclin B1. Cyclin B1 has been found to be re expressed in Alzheimer’s disease [48].
Recent finding by T. Fuchsberger, 2016 [48] that Aβ with APC/C mediates excitotoxicity through the glutaminase pathway showed that this protein not only exerts properties for the late phases of the cell cycle but also properties for the maintenance of the postmitotic state through repressing cyclin B activity which suggest that APC/C has an important role in the pathogenesis of Alzheimer’s disease. Glutamate is the most abundant neurotransmitter in the brain, with a wide range of functions. One of the more important functions is its role in activation NMDAR (N-methyl-D aspartate receptors (NMDARs). NMDR activation increases intracellular Ca2+, and if Ca2+ goes unbalanced it may impair mitochondrial oxidation, increase cdk 5-p25 protein levels and lead to hyperphosphorylation of Tau. Interestingly, glutaminase the enzyme that converts glutamine to glutmate and ammonia is a target for APC/C cdh-1. More interesting is that Aβ inhibits cdh-1, depresses APC activity inducing rising levels of glutaminase and increases glutamate levels expressing thus exicitotoxicity in AD. This mediated excitotoxicity was found to be regulated through Cdh-1[48]. Cdh 1 is primarily in a non phosphyrated form found in the nucleus. These processes are also regulated through cdk5 suggesting a crosstalk between these two proteins. Cdk 5 is known to be altered in AD [49]. The process mediated by cdk 5-p25, indicates that cdk 5 is involved in Aβ induced degradation of cdh1 in neurons. Moreover, Maestre, et al. 2008 [15] showed that NMDAR stimulation triggers CDK 5 activation. CDK 5 phosphorylates cdh1, modulates cyclin B1 and through the NMDAR pathway triggers neuronal death [15,50].
Lopes, et al. 2009 [49] found that the pathway of CDK 5 a serin-threonin kinase is involved in axonal guidance, cortical layering and synaptic plasticity. Over activated CDK 5 relocalizes from its protective role in the neuronal nuclei to cytoplasm in AD and prion induced pathologies.
Are the APC/C cdh-1 regulated processes connected through the dysregulation of CDK 5 activity by elevated levels of p25 relative to p35 is a question to be explored? It is well known that cdk 5 dysregulation may cause neuroinflammation and neurodegeneration [51]
One study demonstrated that in response to Aβ, neurons will reenter the cell cycle and transit through the cell cycle until the M phase, and that these processes are mediated by CDK 5 and its downstream effectors. So, the importance of CDK 5 as a neuronal ‘’switch’’ is due to the fact that aberrant hyperactivation of CDK 5 by the production of truncated activator p25 results in the formation of hyperphosphorylated TAU, neuroinflamation, amyloid deposition and neuronal death in vivo and in vitro [52].
To our view this ‘’switch’’ is not regulated solely through CDK 5 but also through the APC/C complex and one or more of its substrates. The data suggest an unsuspected role for Cdk5 during the progression of a normal cell cycle and may offer new pharmaceutical targets for regulating neuronal cell cycling and cell death.
More on the roles of Cdh1
Cdh1 also plays an important role in DNA damage repair [42,53] and cellular metabolism including deoxyribonucleotide synthesis [54] and glycolysis [37]. These findings suggested an emerging role for Cdh1 in controlling developmental processes. Consistent with this notion, recent studies demonstrated that depletion of Cdh1 in post- mitotic neurons enhanced axonal growth [36], which might occur through impaired destruction of the inhibitor of differentiation/DNA binding 2 (Id2) transcription factor [55] and SnoN [56] (Figure 2). APC/C-Cdh1 regulated axonal growth and patterning can be achieved through down regulation of Id2 (by the APC/C –cdh1) in primary neuronal stem cells exposed to retinoic acid [57]. Cdh1 was also found to activate TGF-β signaling by targeting its specific inhibitor, SnoN, for destruction [58] (Figure 2). Also, Stegmuller, et al. 2008 [59] reports that the Cdh1APC/SnoN pathway is regulated by TGF beta-Smad2. A therapeutic potential is suggested by inhibiting Smad signaling thus stimulating axonal growth after injury of the brain [59].
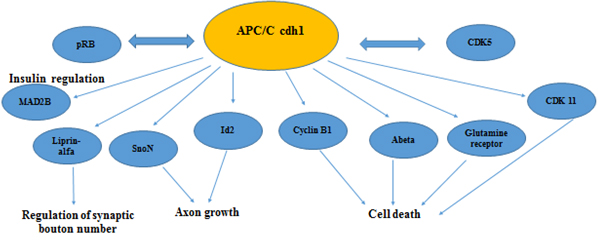
Figure 2. Secondary roles of APC/C in neuronal physiology
In Figure 2 we are showing known substrates of APC/C cdh1 that regulate neuronal development, neurogenesis and have a active role in maintaining the postmitotic state in neurons. A number of these proteins are found to have altered expression in Alzheimer’s disease (CDK 5, CDK11, Abeta, Cyclin B1, pRB, Mad2B)
Liprin-α
In the regulation of synaptic function, the APC target protein Liprin-α, regulates bouton number at neuromuscular junction synapses but also has a post-synaptic role as it has been demonstrated that this protein has a role in mammalian AMPA receptor trafficking, implying that Liprin-α may orchestrate pre- and post-synaptic events [45,60-62].
APC/C-Cdh1 has been found to have a role in the organization of synapses and neuromuscular junctions [63]. How was this acquired in an evolutionary sense is best stated by Arendt, et al. [11] , that the proliferative machinery has in postmitotic neurons evolved new functions such as regulation of synaptic plasticity [12,64]. Interestingly, Cdh1/Hct1-APC is essential for the survival of postmitotic neurons [18], through degrading cycline B1, a cyclin that has been repeatedly found to be reactivated in degenerating neurons of AD patients [65,66-68]. The role of APC/C in AD is reinforced by the observation that Cdh1 overexpression protects neurons against the neurotoxicity of amyloid beta. Also, to our view APC/C cdh-1 clearly plays a major role in the process of cell cycle re-entry of affected neurons in AD.
APC/c cdh 1 interacts with pRB
The retinoblastoma protein has been found to be ectopically expressed in Alzheimer’s disease [69-71]. These findings suggest that entry into the cell cycle requires activation of G1 to S phase cell cycle proteins, among which retinoblastoma protein (pRb) is a key regulator. In healthy cells pRB inhibits transcription of cell cycle proteins and consequently blocking E2F [42,70,71]. Increased levels of phosphorylation is found in affected cells with redistribution (ectopically expression) of pRB protein from the nucleus to the cytoplasm which has been found to be significantly co-localizing with tangles and plaques [70,71]
Fascinatingly, recent data show that pRb physically and genetically interacts with both APC/C Cdh-1 and with S-phase kinase-associated protein 2 (Skp2). Skp2 functions as the receptor component of the Skp–Cullin–F-box complex and is implicated in the degradation of several cell cycle regulators, such as p21Cip1, p27Kip1, p57Kip2, and cyclin E [72]. By contributing to Skp 2 degradation, pRB connects the G0/G1 transcriptional repression machinery directly to the APC/C [72]. Interestingly all degradation products of skp2 have been found to be expressed in AD [73].
DNA repair in postmitotic neurons are accompanied by cell cycle activation. Tomashevski, et al. 2010 [74] have found that cyclin C is directed by a pRb-dependent G0 exit which activates the non-homologous end joining pathway of DNA repair (NHEJ) in postmitotic neurons. Recent reports also link pRB with p53 through skp2. P53 is also an essential protein for DNA repair. The APC/CCdh1 substrate Skp2 can act as an inhibitor of the transcriptional and apoptotic function of p53 [75]. Results from these studies suggest that pRb mutations may influence APC/C activity directly. Impaired function of both pRb and APC/ CCdh1 is expected to give a significant increase in both Cyclin-Cdk production and activity, which would strongly favour S-phase entry, as this is seen in cell cycle re-entry in AD brains. Thus impaired cdh-1 function may feed forward pRB inactivation and promote proliferation of cells through the G 1 phase.
A late G1 phase cell cycle checkpoint has been found [76]. This checkpoint was found to be related to metabolic changes in the proliferating cell, thus monitoring the balance of oxidative stress, and ROS in coordination with the APC/C complex. How does this restriction point play out in postmitotic neurons is a notion needed to be explored as APC/cdh1 has a clearly different in role neurons than in proliferating cells. Also, authors suggested that oxidative stress is a co-founding factor in the emergence of changes to vulnerable neurons in AD [1,77,78].
We have postulated in previous papers that APC/C in AD neurons might be regulated by cohesion and cohesion related proteins such as CDK 11 and MAD 2B, Bub R1, Securin [29,80-84].
MAD2B
MAD 2 B comes from a group of proteins, MAD 2 and MAD 1 that regulate the metaphase cell cycle checkpoint and inhibit APC/C. MAD 2 inhibits the cdc20, activator of APC and MAD2B inhibits, both activators of APC, cdc 20 and cdh1. For the checkpoint to be active, Mad 2 must interact with MAD1 [85] as MAD2B does not react with MAD 1 suggesting that MAD2B transduces the cellular signals other than the checkpoint control. This also emphases the fact that MAD2B may function in a physiological process other than cell cycle regulation [86]. By its regulation of cdh1, it is conceivable that MAD 2B may play role in the control of APC in neuronal tissues through the association of RAN [87]. By the association with RAN, MAD2B may be involved in controlling the transport of Cdh 1 across the nuclear membrane and, by doing so, the transition from the G1 to the S phase of the cell cycle as well [88]. This process might be accompanied by a novel cohesion regulated cyclin, CDK 11 [81,88-90] that is Ran GTP regulated [88]. CDK 11 is differentially expressed in AD [81] and is also known to regulate the apoptotic process [89]. Indirectly we show that Mad2B and CDK 11 might be regulated by the APC/C complex. Interestingly enough MAD 2B has been found to regulate apoptosis through its suppression in the rat diabetic model [90]. This property of MAD 2B has been explained through Mad 2 regulation of cyclin B1 and cdh1.The involvement of neuronal apoptosis has been demonstrated in diabetic animal models, associated with a decrease in neuronal density with a present learning and cognitive decline. Attenuation of apoptosis of neurons in the hippocampus and cerebral cortex ameliorates the cognitive defects in diabetic rats [90]. APC/C is responsible for cyclin B stability in postmitotic neurons, and APC is regulated by MAD 2B [39] suggesting that Meng, et al. 2014 [90] may found a dependent MAD2B apoptotic pathway in a DM animal model. In DM rats cyclin B1 was increased, especially in neurons. MAD 2B modulates cyclin B stability in cortical neurons by downregulating cyclin B1 accumulation, suggesting a regulative role in neuronal stability, i.e. elevated expression of MAD2B induced by hyperglycemia results in cyclin B1 accumulation in neurons. By decreasing MAD 2B expression, the number of apoptotic cells also decreased. Inhibition of MAD 2B expression prevents neurons from entering the S phase. The decreased number of cells in the S phase results in abolishing neuronal injury and apoptosis that have resulted from hyperglycemia. In the AD brain, MAD2B protein expression is decreased [unpublished results]. In AD depletion of MAD2B may result of cells ability to cope with ROS and or DNA damage. These findings do give a way to strategize MAD2B as a possible target for therapy.
BUBR1
Bub R1 is also one of the key proteins in the spindle assembly checkpoint (SAC). Importantly, decreased expression of BubR1 induces aneuploidation but its overexpression protects against aneuploidy [91]. The mitotic checkpoint complex (MCC) is structured from BubR1, Mad2 and Bub3. The MCC dissociates once the chromosomes are properly arranged in the mitotic spindle. This allows cdc20 to activate the APC/C complex, thus degrading cyclin B1 and securin, triggering anaphase onset (Figure 1). Interestingly we have showed in previous paragraphs that the late anaphase machinery is also used to maintain the G1 phase of neurons. The role of BubR1 at this phase is still not understood except for the notion that BubR1 is decreased in brain samples of AD patients [84]. However, little is known about the function of BUBR1 other than mitotic control. Using cell lines from the patients with the mosaic variegated aneuploidy (MVA) syndrome, investigators demonstrated that BubR1 is essential for the formation of primary cilium [92]. MVA syndrome is a novel ciliopathy. Primary cilium is a microtubule-based organelle on the surface of most vertebrate cells found in the G0 phase, and that the primary cilium is regulated by APC/CCDH1 activity through ubiquitin-mediated proteolysis of Disheveled (DVL) [92]. Other than cilium formation, Watanabe Y, 2014 [93] found that the APC/C complex through the BubR1 protein can mediate regulation of dendritic development through the schizophrenia-associated gene FEZ1 protein. Except for APC/C complex as a functional key for postmitotic maintenance, there is another complex, the Cohesin complex and its associated proteins that have been found to be expressed and to regulate numeral processes in neurons [Table 1].
Table 1. Cohesin and cohesin related proteins: their secondary roles in post mitotic neurons
Cohesin and cohesin related proteins |
Secondary roles in Post- mitotic neurons and other cell types |
Cohesin |
Mutations in the cohesion complex are associated with cohesionopathies (Cornelia de Lang and Roberts syndrome) |
Cohesin |
Gene transcription regulation (transcriptional insulators, CTCF) |
Cohesin subunit SMC1 and SA1 |
Essential for axon pruning in Drosophila;Dentritic targeting |
Cohesin subunit
Rad 21 |
Axon pruning in humans; Repair by homologues recombination |
PDSB5 cohesion regulatory protein |
Abnormal projections of the superior cervical ganglia (mouse model CdLs) |
Aurora A |
A centrosomal kinase that directs translation at the synapse; regulates synaptic plasticity; essential for microtubular dynamics in neurons; |
Polo like kinases |
Dendritic spikes; synaptic plasticity and remoulding; synaptic strength |
BUB R1 |
Essential for primary cilium formation; cerebellum development in the medaka fish |
MAD2B |
MAD2B has a protective role by controlling APC/C and cyclin B in insulin sensitive neuronal cells |
SMC 3 |
Axon pruning |
CDK 11 |
Regulates G2/M cohesion, possible role in synaptic regulation |
Cohesin: the postmitoic genetic switch?
The centromere is a complex structure that is fundamental for chromosome separation and segregation. These processes are coordinated with cell division and by the metaphase cell cycle control center (MCC), in which Mad2 and BUbR1 play a pivotal role [94,95]. Fundamental to the centromere complex is the multifaceted protein called Cohesin [34,35,95,96] (Figure 1).
The cohesin protein builds a complex that is contained by two SMC (structural maintenance of cohesion) subunits and two non SMC subunits, designated SMC 1 and 3, Rad 21 (SA1) and SA2, respectively [96] in vertebrate cells. Cohesion is dependent on the working of regulatory factors designated, PDS 5, Scc2, ESCO 1/ 2, NIPBL and MAU-2.
Removal of cohesin arms in the metaphase anaphase transition are regulated by PLK1, Aurora B kinase, condensin and WAPL. Both regulatory factors and factors for cohesin removal have secondary roles (see Table 1). To date, PLK 1 a cohesin removal factor is found to be elevated in AD [97]. During the metaphase-to-anaphase transition, the separase inhibitor securin is degraded by APC (Figure 1). This enables sister chromatide separation. This process has been altered in AD and has been seen as premature centromere division or PCD [98-101]. Moh, et al. 2011 [31] proposed that PCD occurrence expresses genome instability in which cell cycle re-entry takes place. Interestingly, PCD has been found to result in aneuploidy [101], which is again a consequence of altered cohesion complex, especially RAD 21 [102], CDK11 [89], SMC1 [103], Bub1B [104]. Cohesin is essential for chromosome segregation, but also for DNA damage repair, suggesting a link with the APC cdh 1 complex in regulation of the late pages G1/S transition in cell exposed to oxidative stress [76]. This damage repair property is found to be tied to BRCA1 [105], a known DSB repair protein implicated in cancer [106] and AD [107]. The findings of cohesion instability or premature centromere division in peripheral cells [98,108] and in neuronal cells [99] of AD patients suggest that the secondary roles of cohesion are impaired [29,109]. A number of the proteins in the cohesin complex are associated with various diseases named ‘’cohesionapathies’’ [110] suggesting that cohesin has roles in dividing and non-dividing cells other that chromosome regulation (Table 1).
Cohesinopathies such as Cornelia de Lange syndrome (CdLS; caused by mutations in SMC1, SMC3, and SCC2) and Roberts syndrome (caused by mutations in ESCO2; is a rare autosomal recessive developmental disorder) [111] shows us the principle of how these processes in gene regulation of neuronal development are altered. So, mutation in cohesion genes are not related per se to chromosome segregation defects but the problem is in the secondary role of cohesins, i.e. in regulation of gene transcription and or gene regulation [110,112,113]. From these studies and many others, knowledge is emerging in which the necessity of cohesin for chromosome segregation is separated from its role in gene regulation in neuronal and non-neuronal cells suggesting that cohesin and cohesin related protein may be targets for AD treatment (Table 1).
Conclusion
APC or cyclosome is the major ubiqintin ligase involved in cell cycle regulation via cyclins recognition and targeting for destruction. So, G0 of the postmitotic neurons is an active process in which a new hierarchy of CDK and relative proteins has to be established. The other crucial complex to postmitotic regulative hierarchy is the cohesin complex.
In AD there is an increase in cyclin D, cyclin B1, CDk4, Ki67 which suggests exit from Go and these proteins are also ectopically found in the cytoplasm rather than in the nucleus, their typical site of action [65,66-68]. Also, M phase markers: an increase of MPM 2 phosphoepitopes , Cdc 35A and B phosphatases and the findings of cell cycle phenotypes found as binucleation [114] and premature centromere division ‘’PCD’’ [98,99,108] may result in abortive karyokinesis [115-117] found in AD.
The ubiquitation system is also altered in AD, including mutations in the ubiqitin 1 protein complex which may influence cell cycle regulation [118,119] and protein aggregation and accumulation. Moreover, as some ubiqitin ligase, e.g. BRCA 1 , are overexpressed in AD neurons, the ubiquintation substrates, ubiquintation dependent signaling are most likely to be altered in AD [107]. The recent report by Meng et al. 2014 [90] showed that the APC/C complex may be in the epicenter of the cell cycle reentry process, and that these processes are affected by the Aβ peptide. Clearly, the studies described above are just beginning to uncover the importance of late phase cell cycle proteins in neurobiology.
Two protein complexes that are crucial to healthy G1 neurons, the Cdh1-APC complex and the Cohesin complex. To date, from outside of the cell cycle regulators, late phase cell cycle proteins exert a number of important function in neurons, from axon patterning, neuronal death, synaptic function, to processes that lead the transition from a neural stem cell to a differentiated neuron. The substrates of neuronal Cdh1–APC are only beginning to be identified. Many questions remain in the analysis of the functions of these proteins. One of such is the PP2A protein (protein phosphatase 2A). It has been shown that downregulation of PP2A in AD patients is linked to Tau hyper phosphorylation [120]. PP2A is a major tau phosphatase in human brain and recently Cheng, et al. 2015 [121] found that PP2A promotes axonal growth.
Abrogating apoptosis: The way to therapy ?
One strategy for AD treatment is to extend the cell cycle by abrogating the apoptosis pathway. Some experiments have shown that by diminishing apoptosis, neuronal cells do not die, and the cognitive phenotype is preserved [116,117]. We here postulate a hypothesis that targeting late phase cell cycle proteins will increase neuronal survival, thus alleviating the symptoms of AD, dementia and neuronal loss. Here, we evaluate the hypothesis that abortive cell cycle reentry is due to alteration in the switch of core cell cycle proteins in which Late phase cell cycle proteins have a primer role. To consider this view, that APP-cell cycle core proteins signaling processes are the core of the AD neurodegenerative process may be strengthen by new research of Melnikova, et al. 2013 [118 ] in which they found that in the inducible mouse model od AD, the Aβ plaque that exceeds the same in human disease is without cognitive defects in the mouse if the APP gene is switched off. This shows that the Aβ burden can be dissociated from the cognitive decline associated with AD and that by blocking APP did the trick. The study suggests ‘’ that if we could extrapolate these findings to AD patients by reducing expression of ‘’bad’’ APP (and its βC terminal fragment accumulation) then this ‘’action’’ might be sufficient to improve cognitive function’’. In our view APP must have a fundamental role in maintaining the Go phase of neurons and by this notion we may conclude that altered reorganization of the secondary core cell cycle proteins and APP are crucial to the survival/death processes in AD neurons. Concerning this view we than have proposed a number of proteins that might be screened for their altered expression patterns as possible targets for therapy. Our preliminary results showed a relative differential pattern of expression of proteins: Mad1 and Mad2, Bub R1, Bub1, CDK 11, cohesin subunit Rad 21 and astrin in the AD brain versus age matched controls and also in Tg 257 brain (results not published). An alternative approach is to utilize exploration of cohesion and cohesion associated proteins that are not directly involved in the formation of amyloid plaques. These processes intertwined with late phase cell cycle proteins in the pathology of AD are and may thereby contribute to the pathogenesis of AD. These new findings imply us to address late phase cell cycle proteins as new targets for AD treatment.
Acknowledgments
Author thanks the Fulbright Association , German DAAD Association and the Ministry of Education, Science and Technological Development of the Republic of Serbia for financial support (Project No: 173034).
References
- Smith MA (1998) Alzheimer disease. Int Rev Neurobiol 42: 1-54. [crossref]
- Castellani RJ, Lee HG, Siedlak SL, Nunomura A, Hayashi T, et al. (2009) Reexamining Alzheimer's disease: evidence for a protective role for amyloid-beta protein precursor and amyloid-beta. J Alzheimers Dis 18: 447-452. [crossref]
- Zhu X, Lee HG, Perry G, Smith MA (2007) Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta 1772: 494-502. [crossref]
- Krstic D, Knuesel I (2013) Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol 9: 25-34. [crossref]
- Arendt T (2002) Dysregulation of neuronal differentiation and cell cycle control in Alzheimer's disease. J Neural Transm Suppl : 77-85. [crossref]
- Hernandez-Ortega K, Ferrera P, Arias C (2007) Sequential expression of cell-cycle regulators andAlzheimer’s disease-related proteins in entorhinal cortex after hippocampal excitotoxic damage. J Neurosci Res 85: 1744–1751. [crossref]
- Lee HG, Casadesus G, Nunomura A, Zhu X, Castellani RJ, et al. (2009) The neuronal expression of MYC causes a neurodegenerative phenotype in a novel transgenic mouse. Am J Pathol 174: 891-897. [crossref]
- Nguyen MD, Mushynski WE, Julien JP (2002) Cycling at the interface between neurodevelopment and neurodegeneration. Cell Death Differ 9: 1294-1306. [crossref]
- Ding XL, Husseman J, Tomashevski A, Nochlin D, Jin LW, et al. (2000) The cell cycle Cdc25A tyrosine phosphatase is activated in degenerating postmitotic neurons in Alzheimer's disease. Am J Pathol 157: 1983-1990. [crossref]
- Moreira PI, Santos MS, Oliveira CR, Shenk JC, Nunomura A, et al. (2008) Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol Disord Drug Targets 7: 3-10. [crossref]
- Arendt T, Brückner MK (2007) Linking cell-cycle dysfunction in Alzheimer's disease to a failure of synaptic plasticity. Biochim Biophys Acta 1772: 413-421. [crossref]
- Arendt T, Brückner MK (2007) Linking cell-cycle dysfunction in Alzheimer's disease to a failure of synaptic plasticity. Biochim Biophys Acta 1772: 413-421. [crossref]
- Frank CL, Tsai LH (2009) Alternative Functions of Core Cell Cycle Regulators in Neuronal Migration, Neuronal Maturation, and Synaptic Plasticity. Neuron 62: 312-326. [crossref]
- Almeida A, Bolaños JP, Moreno S (2005) Cdh1/Hct1-APC is essential for the survival of postmitotic neurons. J Neurosci 25: 8115-8121. [crossref]
- Maestre C, Delgado-Esteban M, Gomez-Sanchez JC, Bolaños JP, Almeida A (2008) Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J 27: 2736-2745. [crossref]
- Zhang J, Li H, Zhou T, Zhou J, Herrup K (2012) Cdk5 Levels Oscillate during the Neuronal Cell Cycle: Cdh1 ubiquitination triggers proteosome-dependent degradation during S-phase. J Biol Chem 287: 25985–25994. [crossref]
- Van Roessel P, Elliott DA, Robinson IM, Prokop A, Brand AH (2004) Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell 119: 707-718. [crossref]
- Aulia S, Tang BL (2006) Cdh1-APC/C, cyclin B-Cdc2, and Alzheimer's disease pathology. Biochem Biophys Res Commun 339: 1-6. [crossref]
- Iourov IY, Vorsanova SG, Yurov YB (2006) Chromosomal variation in mammalian neuronal cells: known facts and attractive hypotheses. Int Rev Cytol 249: 143-191. [crossref]
- Iourov IY, Vorsanova SG, Yurov YB (2008) Molecular cytogenetics and cytogenomics of brain diseases. Curr Genomics 9: 452-465. [crossref]
- Potter H (1991) Review and hypothesis: Alzheimer disease and Down syndrome--chromosome 21 nondisjunction may underlie both disorders. Am J Hum Genet 48: 1192-1200. [crossref]
- Potter H (2008) Down's syndrome and Alzheimer's disease: two sides of the same coin. Future Neuro l3: 29-37.
- Granic A, Padmanabhan J, Norden M, Potter H (2010) Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol Biol Cell 21: 511-520. [crossref]
- Iourov IY, Vorsanova SG, Yurov YB (2010) Somatic genome variations in health and disease. Curr Genomics 11: 387-396. [crossref]
- Kingsbury MA, Friedman B, McConnell MJ, Rehen SK, Yang AH, et al. (2005) Aneuploid neurons are functionally active and integrated into brain circuitry. Proc Natl Acad Sci U S A 102: 6143-6147. [crossref]
- Bushman DM, Chun J (2013) The genomically mosaic brain: aneuploidy and more in neural diversity and disease. Semin Cell Dev Biol 24: 357-369. [crossref]
- Bushman DM, Kaeser GE, Siddoway B, Westra JW, Rivera RR, et al. (2015) Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer's disease brains. Elife 4: 05116. [crossref]
- Cai X, Evrony GD, Lehmann HS, Elhosary PC, Mehta BK, et al. (2014) Single-Cell, Genome-wide Sequencing Identifies Clonal Somatic Copy-Number Variation in the Human Brain. Cell Rep 8: 1280–1289. [crossref]
- Bajic V, Spremo-Potparevic B, Zivkovic L, Isenovic ER, Arendt T (2015) Cohesion and the aneuploid phenotype in Alzheimer's disease: A tale of genome instability. Neurosci Biobehav Rev 55: 365-374. [crossref]
- Oromendia AB, Amon A (2014) Aneuploidy: implications for protein homeostasis and disease. Dis Model Mech 7: 15-20. [crossref]
- Moh C, Kubiak JZ, Bajic VP, Zhu X, Smith MA, et al. (2011) Cell cycle deregulation in the neurons of Alzheimer's disease. Results Probl Cell Differ 53: 565-576. [crossref]
- Boeras D, Granic A, Padmanabhan J, Crespo NC, Rojiani AM, et al. (2008) Alzheimer's presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging 29: 319-328. [crossref]
- Bonda DJ, Bajić VP, Spremo-Potparevic B, Casadesus G, Zhu X, et al. (2010) Review: cell cycle aberrations and neurodegeneration. Neuropathol Appl Neurobiol 36: 157-163. [crossref]
- Nasmyth K, Haering CH (2009) Cohesin: its roles and mechanisms. Annu Rev Genet 43: 525-558. [crossref]
- Nasmyth K, Peters JM, Uhlmann F (2000) Splitting the chromosome: cutting the ties that bind sister chromatids. Science 288: 1379-1385. [crossref]
- Konishi Y, Stegmüller J, Matsuda T, Bonni S, Bonni A (2004) Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303: 1026-1030. [crossref]
- Herrero-Mendez A, A2021 Copyright OAT. All rights reserv C, Moncada S, et al. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11: 747-752. [crossref]
- Harper JW, Burton JL, Solomon MJ (2002) The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev 16: 2179-2206. [crossref]
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, et al. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11: 747-752. [crossref]
- Puram SV, Kim AH, Bonni A (2010) An old dog learns new tricks: a novel function for Cdc20-APC in dendrite morphogenesis in neurons. Cell Cycle 9: 482-485. [crossref]
- Gieffers C, Peters BH, Kramer ER, Dotti CG, Peters JM (1999) Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc Natl Acad Sci U S A 96: 11317-11322. [crossref]
- Gao D, Inuzuka H, Korenjak M, Tseng A, Wu T, et al. (2009) Cdh1 regulates cell cycle through modulating the claspin/Chk1 and the Rb/E2F1 pathways. Mol Biol Cell 20: 3305-3316. [crossref]
- Stegmüller J, Bonni A (2005) Moving past proliferation: new roles for Cdh1-APC in postmitotic neurons. Trends Neurosci 28: 596-601. [crossref]
- Yao W, Qian W, Zhu C, Gui L, Qiu J, et al. (2010) Cdh1-APC is involved in the differentiation of neural stem cells into neurons. Neuroreport 21: 39-44. [crossref]
- Teng FY, Tang BL (2005) APC/C regulation of axonal growth and synaptic functions in postmitotic neurons: the Liprin-alpha connection. Cell Mol Life Sci 62: 1571-1578. [crossref]
- Zarnescu DC, Moses K (2004) Born again at the synapse: a new function for the anaphase promoting complex/cyclosome. Dev Cell 7: 777-778. [crossref]
- Kuczera T, Stilling RM, Hsia HE, Bahari-Javan S, Irniger S, et al. (2010) The anaphase promoting complex is required for memory function in mice. Learn Mem 18: 49-57. [crossref]
- Fuchsberger T, Martínez-Bellver S, Giraldo E, Teruel-Martí V, Lloret A, et al. (2016) Aβ Induces Excitotoxicity Mediated by APC/C-Cdh1 Depletion That Can Be Prevented by Glutaminase Inhibition Promoting Neuronal Survival. Scientific Reports 6: 31158. [crossref]
- Lopes JP, Oliveira CR, Agostinho P (2009) Cdk5 acts as a mediator of neuronal cell cycle re-entry triggered by amyloid-beta and prion peptides. Cell Cycle 8: 97-104. [crossref]
- Veas-Pérez de Tudela M, Maestre C, Delgado-Esteban M, Bolaños JP, Almeida A (2015) Cdk5-mediated inhibition of APC/C-Cdh1 switches on the cyclin D1-Cdk4-pRb pathway causing aberrant S-phase entry of postmitotic neurons. Sci Rep 5: 18180. [crossref]
- Chang KH, Vincent F, Shah K (2012) Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J Cell Sci 125: 5124-5137. [crossref]
- Shah K, Lahiri DK (2016) A Tale of the Good and Bad: Remodeling of the Microtubule Network in the Brain by Cdk5. Mol Neurobiol [Epub ahead of print]. [crossref]
- Lafranchi L, Sartori AA (2015) The ubiquitin ligase APC/C (Cdh1) puts the brakes on DNA-end resection. Mol Cell Oncol 2: e1000696. [crossref]
- Chabes AL, Pfleger CM, Kirschner MW, Thelander L (2003) Mouse ribonucleotide reductase R2 protein: a new target for anaphase-promoting complex-Cdh1-mediated proteolysis. Proc Natl Acad Sci USA 100: 3925-3929. [crossref]
- Lasorella A, Stegmuller J, Guardavaccaro D, Liu G, Carro MS, et al. (2006) Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 442: 471–474. [crossref]
- Bonni S, Bonni A (2012) SnoN signaling in proliferating cells and postmitotic neurons. FEBS Lett 586: 1977-1983. [crossref]
- Lasorella A, Stegmüller J, Guardavaccaro D, Liu G, Carro MS, et al. (2006) Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 442: 471-474. [crossref]
- Bonni S, Bonni A (2012) SnoN signaling in proliferating cells and postmitotic neurons. FEBS Lett 586: 1977-1983. [crossref]
- Stegmüller J, Huynh MA, Yuan Z, Konishi Y, Bonni A (2008) TGFbeta-Smad2 signaling regulates the Cdh1-APC/SnoN pathway of axonal morphogenesis. J Neurosci 28: 1961-1969. [crossref]
- Chatterjee P, Roy D (2016) Structural insight into GRIP1-PDZ6 in Alzheimer's disease: study from protein expression data to molecular dynamics simulations. J Biomol Struct Dyn 1: 1-13. [crossref]
- LaConte LE, Chavan V, Liang C, Willis J, Schönhense EM, et al. (2016) CASK stabilizes neurexin and links it to liprin-? in a neuronal activity-dependent manner. Cell Mol Life Sci 73: 3599-3621. [crossref]
- Li L, Tian X, Zhu M, Bulgari D, Böhme MA, et al. (2014) Drosophila Syd-1, liprin-α, and protein phosphatase 2A B' subunit Wrd function in a linear pathway to prevent ectopic accumulation of synaptic materials in distal axons. J Neurosci 34: 8474-8787.
- Kowalski JR, Dube H, Touroutine D, Rush KM, Goodwin PR, et al. (2014) The Anaphase-Promoting Complex (APC) ubiquitin ligase regulates GABA transmission at the C. elegans neuromuscular junction. Mol Cell Neurosci 58: 62-75. [crossref]
- Mosch B, Morawski M, Mittag A, Lenz D, Tarnok A, et al. (2007) Aneuploidy and DNA replication in the normal human brain and Alzheimer's disease. J Neurosci 27: 6859-6867. [crossref]
- Vincent I, Pae CI, Hallows JL (2003) The cell cycle and human neurodegenerative disease. Prog Cell Cycle Res 5: 31-41. [crossref]
- Vincent I, Jicha G, Rosado M, Dickson DW (1997) Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci 17: 3588-3598. [crossref]
- Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P (1998) Mitotic phosphoepitopes precede paired helical filaments in Alzheimer's disease. Neurobiol Aging 19: 287-296. [crossref]
- Vincent I, Bu B, Hudson K, Husseman J, Nochlin D, et al. (2001) Constitutive Cdc25B tyrosinephosphatase activity in adult brain neurons with M phase-type alterations in Alzheimer’sdisease. Neuroscience 105: 639–650. [crossref]
- Stone JG, Siedlak SL, Tabaton M, Hirano A, Castellani RJ, et al. (2011) The cell cycle regulator phosphorylated retinoblastoma protein is associated with tau pathology in several tauopathies. J Neuropathol Exp Neurol 70: 578-587. [crossref]
- Thakur A, Siedlak SL, James SL, Bonda DJ, Rao A, et al. (2008) Retinoblastoma protein phosphorylation at multiple sites is associated with neurofibrillary pathology in Alzheimer disease. Int J Clin Exp Pathol 1: 134-146.
- Jordan-Sciutto KL, Malaiyandi LM, Bowser R (2002) Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J Neuropathol Exp Neurol 61: 358-367. [crossref]
- Binné UK, Classon MK, Dick FA, Wei W, Rape M, et al. (2007) Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat Cell Biol 9: 225-232. [crossref]
- Ueberham U, Arendt T (2005) The expression of cell cycle proteins in neurons and its relevance for Alzheimer's disease. Curr Drug Targets CNS Neurol Disord 4: 293-306. [crossref]
- Tomashevski A, Webster DR, Grammas P, Gorospe M, Kruman II (2010) Cyclin-C-dependent cell-cycle entry is required for activation of non-homologous end joining DNA repair in postmitotic neurons. Cell Death Differ 17: 1189-1198. [crossref]
- Gao D, Inuzuka H, Korenjak M, Tseng A, Wu T, et al. (2009) Cdh1 regulates cell cycle through modulating the claspin/Chk1 and the Rb/E2F1 pathways. Mol Biol Cell 20: 3305-3316. [crossref]
- Havens CG, Ho A, Yoshioka N, Dowdy SF (2006) Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol Cell Biol 26: 4701-4711. [crossref]
- Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, et al. (2008) Oxidative stress activates apositive feedback between the gamma- and beta-secretase cleavages of the beta-amyloidprecursor protein. J Neurochem 104: 683–695. [crossref]
- Nakamura M, Shishido N, Nunomura A, Smith MA, Perry G, et al. (2007) Three histidine residues of amyloid-beta peptide control the redox activity of copperand iron. Biochemistry 46: 12737–12743. [crossref]
- Bonda DJ, Wang X, Perry G, Nunomura A, Tabaton M, et al. (2010) Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology 59: 290-294. [crossref]
- Bajic VP, Spremo-Potparevic B, Zivkovic L, Djelic N, Smith MA (2008) Is the time dimension of the cell cycle re-entry in AD regulated by centromere cohesion dynamics? Biosci Hypotheses 1: 156-161. [crossref]
- Bajić VP, Su B, Lee HG, Kudo W, Siedlak SL, et al. (2011) Mislocalization of CDK11/PITSLRE, a regulator of the G2/M phase of the cell cycle, in Alzheimer disease. Cell Mol Biol Lett 16: 359-372. [crossref]
- Bajic V, Spremo-Potparevic B, Zivkovic L, Stanojevic B, Bruckner M, et al. (2015) Proteotoxic control in Alzheimer’s Disease may be attained by BubR1: a possible target for therapy? The Journal of Prevention of Alzheimer’s Disease 2.
- Bonda DJ, Bajić VP, Spremo-Potparevic B, Casadesus G, Zhu X, et al. (2010) Review: cell cycle aberrations and neurodegeneration. Neuropathol Appl Neurobiol 36: 157-163. [crossref]
- Bajic V, Zivkovic L, Spremo-Potparevic B, Siedlak S, Smith MA (2010) RAD 21 cohesion protein shows altered levels of expression in Swedish mutant phenotype of M17 neuroblastoma cells. Alzheimer's & Dementia: The Journal of the Alzheimer's Association 6: S257.
- Chen J, Fang G (2001) MAD2B is an inhibitor of the anaphase-promoting complex. Genes Dev 15: 1765-1770. [crossref]
- Meng X, Tian X, Wang X, Gao P, Zhang C (2012) A novel binding protein of single-minded 2: the mitotic arrest-deficient protein MAD2B. Neurogenetics 13: 251-260. [crossref]
- Medendorp JJ, van Groningen M, Vreede L, Hetterschijt L, Wilhelmina H, et al. (2009) The Mitotic Arrest Deficient Protein MAD2B Interacts with the Small GTPase RAN throughout the Cell Cycle. Plosone. [crossref]
- Yokoyama H, Gruss OJ, Rybina S, Caudron M, Schelder M, et al. (2008) Cdk11 is a RanGTP-dependent microtubule stabilization factor that regulates spindle assembly rate. J Cell Biol 180: 867-875. [crossref]
- Hu D, Valentine M, Kidd VJ, Lahti JM (2007) CDK11(p58) is required for the maintenance of sister chromatid cohesion. J Cell Sci 120: 2424-2434. [crossref]
- Meng X, Wang X, Tian X, Yang Z, Li M, et al. (2014) Protection of neurons from high glucose-induced injury by deletion of MAD2B. J Cell Mol Med 18: 844-851. [crossref]
- Weaver RL, Limzerwala JF1, Naylor RM1, Jeganathan KB2, Baker DJ1, et al. (2016) BubR1 alterations that reinforce mitotic surveillance act against aneuploidy and cancer. Elife 5. [crossref]
- Miyamoto T, Porazinski S, Wang H, Borovina A, Ciruna B, et al. (2011) Insufficiency of BUBR1, a mitotic spindle checkpoint regulator, causes impaired ciliogenesis in vertebrates. Hum Mol Genet 20: 2058-2070. [crossref]
- Watanabe Y, Khodosevich K, Monyer H (2014) Dendrite development regulated by the schizophrenia-associated gene FEZ1 involves the ubiquitin proteasome system. Cell Rep 7: 552-564. [crossref]
- Choi E, Zhang X, Xing C, Yu H (2016) Mitotic Checkpoint Regulators Control Insulin Signaling and Metabolic Homeostasis. Cell 166: 567-581. [crossref]
- Nasmyth K (2005) How do so few control so many? Cell 120: 739-746. [crossref]
- Nasmyth K, Haering CH (2005) The structure and function of SMC and kleisin complexes. Annu Rev Biochem 74: 595-648. [crossref]
- Harris PL, Zhu X, Pamies C, Rottkamp CA, Ghanbari HA, et al. (2000) Neuronal polo-like kinase in Alzheimer disease indicates cell cycle changes. Neurobiol Aging 21: 837-841. [crossref]
- Spremo-Potparevic B, Zivkovic L, Djelic N, Bajic V (2004) Analysis of premature centromere division (PCD) of the X chromosome in Alzheimer patients through the cell cycle. Exp Gerontol 39: 849-854. [crossref]
- Spremo-Potparević B, Zivković L, Djelić N, Plećas-Solarović B, Smith MA, et al. (2008) Premature centromere division of the X chromosome in neurons in Alzheimer's disease. J Neurochem 106: 2218-2223. [crossref]
- Bajic V, Siedlak S, Zivkovic L, Spremo-Potparevic B, Casadeus G, et al. (2009) The X-chromosome instability phenotype in Alzheimer’s disease: A clinical sign of accelerating aging? Medical Hypothesis 73: 917-920. [crossref]
- Spremo-Potparevic B, Bajic V, Perry G, Zivkovic L1 (2015) Alterations of the X Chromosome in Lymphocytes of Alzheimer's Disease Patients. Curr Alzheimer Res 12: 990-996. [crossref]
- Zhang N, Jiang Y, Mao Q, Demeler B, Tao YJ, et al. (2013) Characterization of the interaction between the cohesin subunits Rad21 and SA1/2. PLoS One 8: e69458. [crossref]
- Michaelis C, Ciosk R, Nasmyth K (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91: 35-45. [crossref]
- Matsuura S, Matsumoto Y, Morishima K, Izumi H, Matsumoto H, et al. (2006) Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A 140: 358-367. [crossref]
- Skibbens RV (2008) Cell biology of cancer: BRCA1 and sister chromatid pairing reactions? Cell Cycle 7: 449-452. [crossref]
- Kleibl Z, Kristensen VN (2016) Women at high risk of breast cancer: Molecular characteristics, clinical presentation and management. Breast 28: 136-144. [crossref]
- Evans TA, Raina AK, Delacourte A, Aprelikova O, Lee HG, et al. (2007) BRCA1 may modulate neuronal cell cycle re-entry in Alzheimer disease. Int J Med Sci 4: 140-145. [crossref]
- Zivkovic L, Spremo-Potparevic B, Djelic N, Bajic V (2006) Analysis of premature centromere division (PCD) of the chromosome 18 in peripheral blood lymphocytes in Alzheimer disease patients. Mech Ageing Dev 127: 892-896. [crossref]
- Bajic V, Mandusic V, Stefanova E, Bozovic A, Davidovic R, et al. (2015) Skewed X-chromosome inactivation in women affected by Alzheimer's disease. J Alzheimers Dis 43: 1251-1259. [crossref]
- Bose T, Gerton JL (2010) Cohesinopathies, gene expression, and chromatin organization. J Cell Biol 189: 201-210. [crossref]
- Vega H, Waisfisz Q, Gordillo M, Sakai N, Yanagihara I, et al. (2005) Roberts syndromeis caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sisterchromatid cohesion. Nat Genet 37: 468-470. [crossref]
- Mehta GD, Kumar R, Srivastava S, Ghosh SK (2013) Cohesin: functions beyond sister chromatid cohesion. FEBS Lett 587: 2299-2312. [crossref]
- Mehta GD, Rizvi SM, Ghosh SK (2012) Cohesin: a guardian of genome integrity. Biochim Biophys Acta 1823: 1324-1342. [crossref]
- Zhu X, Siedlak SL, Wang Y, Perry G, Castellani RJ, et al. (2008) Neuronal binucleation in Alzheimer disease hippocampus. Neuropathol Appl Neurobiol 34: 457-465. [crossref]
- Ogawa O, Lee HG, Zhu X, Raina A, Harris PL, et al. (2003) Increased p27, an essential component of cell cycle control, in Alzheimer's disease. Aging Cell 2: 105-110. [crossref]
- Raina AK, Zhu X, Monteiro M, Takeda A, Smith MA (2000) Abortive oncogeny and cell cycle-mediated events in Alzheimer disease. Prog Cell Cycle Res 4: 235-242. [crossref]
- Raina AK, Hochman A, Zhu X, Rottkamp CA, Nunomura A, et al. (2001) Abortive apoptosis in Alzheimer's disease. Acta Neuropathol 101: 305-310. [crossref]
- Melnikova T, Fromholt S, Kim H, Lee D, Xu G, et al. (2013) Reversible pathologic and cognitive phenotypes in an inducible model of Alzheimer-amyloidosis. J Neurosci 33: 3765-3779. [crossref]
- Kubiak J, Smith MA (2010) Ubiquitin/proteasome system in mitotic and mitotic-like regulationduring brain development and pathology. In: Di Napoli M, Wojcik C (eds) The ubiquitinproteasome system in the central nervous system: from physiology to pathology – 2008 update. Nova Science, Hauppauge, NY, 113–130.
- Qian W, Shi J, Yin X, Iqbal K, Grundke-Iqbal I, et al. (2010) PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J Alzheimers Dis 19: 1221-1229. [crossref]
- Cheng P, Chen K, Yu W, Gao S, Hu S, et al. (2015) Protein phosphatase 2A (PP2A) activation promotes axonal growth and recovery in the CNS. J Neurol Sci 359: 48-56. [crossref]