Abstract
We have previously proposed that the G protein-coupled receptor, GPR87, may be an LPA receptor. This study evaluated whether GPR87 was functionally involved in the LPA response using ME180 cervical carcinoma cells. Our in vitro wound healing assay revealed that LPA-induced cell migration was suppressed by the LPA antagonist Ki16425. RT-PCR analyses indicated that ME180 cells expressed LPA1 receptor and GPR87, both of which are antagonized by Ki16425. The ME180 cells pretreated with gpr87-specific siRNAs significantly reduced LPA-induced cell migration. These results suggest that GPR87 is activated by LPA and the activation of GPR87 leads cell migration.
Key words
G protein-coupled receptor, GPR87, lysophosphatidic acid, cell migration, cancer progress
Abbreviations
LPA, lysophosphatidic acid; GPCR, G protein-coupled receptor; MAPK, mitogen-activated protein kinase; PI3-K, phosphoinositide3-kinase; EGFR, epidermal growth factor receptor; EMT, epithelial mesenchymal transition; MMP, matrix metalloprotease; NSCLC, non-small-cell lung cancer.
Introduction
Lysophosphatidic acid (LPA) is one of the lysophospholipids produced by autotoxin enzymatic activity invascular endothelialcells and acts as an extracellular signaling molecule [1,2]. LPA increases the phosphorylation of mitogen-activated protein kinase (MAPK), phosphoinositide3-kinase (PI3-K), and low molecular weight G-proteinsby binding to at least six specific G-protein coupled receptors (GPCRs) known as LPA receptors [1–6 ]. LPA1-3 receptors sharing >50% homology are known to be “endothelial differentiation gene” (EDG) members, while three additional LPA receptors LPA [4-6] are members of the P2Y purinergic family. These receptors have significantly different encoding sequences than LPA receptors [1-3], yet still bind and mediate LPA signaling effects [3]. These receptors are expressed in various mammalian cells and tissues [3]. We previously reported that the intracellular Ca2+ concentration in CHO cells expressing a GPR87–Gα16 fusion protein increased with exposure to a low concentration of LPA and proposed that GPR87, a member of P2Y family, was an LPA receptor [4]. Glatt et al. reported that GPR87 may contribute to the viability and proliferation of several human tumor cells [5]. Recently, Yan et al. predicted that GPR87wasinvolved with the regulation of the proliferation and migration of epithelial cancer cells expressing the cancer stem cell-marker CD133 [6]. Moreover, we previously reported that colony dispersal of human A431 epithelial cells was induced by LPA through the activation of GPR87 and the LPA1 receptor [7]. Cell colony dispersal has been known as the epithelial mesenchymal transition (EMT) that occurs in cancer cell migration and transition [8, 9].
In general, cell migration and EMT are both regarded as important factors in cancer progression and malignancy development. Importantly, patients with bladder cancer cells that expressed GPR87 had shorter survival times than patients whose cancer did not express GPR87 [10]. Similar results were reported for patients with lung cancer [11]. Because induction of the migration ability of cancer cells is thought to be important for cancer cell progression to malignancy, GPR87 may be a key molecule in this process. However, definitive evidence showing that GPR87 is an LPA receptor has been lacking.
It is well documented that various cell lines migrate in response to LPA through the activation of diverse signaling pathways [12-15]. Ullich showed that LPA receptor activation transactivates epidermal growth factor receptor (EGFR) through the activation of a matrix metalloprotease (MMP) and the release of an EGF-like peptide [16-18]. Recently, Muthusami et al. reported that the treatment of human cervical carcinoma ME180 cells with EGF induced the nuclear accumulation of transcription activating factor2 downstream of Erk, increasing cell migration and EMT [19]. Because ME180 cells have been shown to express GPR87 [5], the cells can be expected toinduce cell motility following GPR87 activation depending on GPCR-mediated EGFR transactivation.
GPCRs have attracted a great deal of research interest because of their numerous physiological roles, although more than 120 GPCRs exist as orphan receptors with unknown endogenous stimulating ligands [20]. Deorphanization of orphan GPCRs has been expected to lead to a better understanding of their physiological functions and suggest new therapeutic targets in drug discovery. However, the rate at which orphan GPCRs are deorphanized has recently decreased. Initially some 10 GPCRs were deorphanized each year, whereas very few GPCRs have been deorphanized since 2004 [21]. Thus, matching GPR87 to LPA should impact the current understanding of the physiological roles of this receptor and ligand and advance drug discovery. In the present study, we investigated the pharmacological and physiological features of GPR87 on the basis of ME180 cell motility.
Materials and methods
Materials
L-a-lysophosphatidic acid (oleoyl sodium salt, LPA 18:1) was purchased from Avanti Polar Lipid Inc. (Alabaster, AL, USA) and kept at a stock concentration of 10 mM. U0126, SP600125, and LY294002 were obtained from Millipore (Billerica, MA, USA), and Ki16425 was from Cayman Chemical Co. (Ann Arbor, MI, USA). All primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell culture
ME180 cells were maintained in RPMI1640 medium (Nacalai, Kyoto, Japan) supplemented with 10% heat-inactivated fetal bovine serum (FBS; GIBCO, Carlsbad, CA, USA) and 1% penicillin/streptomycin (WAKO, Osaka, Japan) in a humidified atmosphere of 5% CO2 at 37°C. When cells were approximately 80% confluent, they were treated with 0.05% Trypsin/0.53m MEDTA (Nacalai) for 5 min at 37°C and transferred to a new medium. Cells were visualized using an Olympus IX81 microscope (Olympus, Tokyo, Japan) after staining with crystal violet.
Preparation of GPR87 knockdown cells
GPR87 knockdown cells were prepared with Ac cell SMART Pool siRNAs (Dharmacon, Chicago, IL, USA) as described [11]. To confirm the effect of siRNA on target gene expression, total RNA extracted from treated cells with TRI Reagent (Sigma–Aldrich, St. Louis, MO, USA) was reverse transcribed into cDNA using ReverTra Ace kit (TOYOBO, Osaka, Japan) and the products were analyzed by conventional or real time PCR.
Wound healing assay
The scratch assay was performed on native and siRNA-treated cells to identify cell migration. ME180 cells were incubated until they were 80% confluent in 24-well dishes, and then the medium was replaced with a serum-free medium and incubated overnight. Further, the cell monolayer was scraped with a CELL Scratcher (Iwaki, Tokyo, Japan) in a straight line to create a 2 mm wound. Subsequently, the cells were incubated in a new medium including 1% FBS (GIBCO, charcoal-stripped FBS, USDA-approved regions) in the presence or absence of LPA and/or an inhibitor for 36 h. Each inhibitor was added 30 min prior to addition of LPA. Cells were then stained with crystal violet and observed under an Olympus IX71 microscope.
Cell proliferation assay
ME180 cells (5 × 104 cells/well) were seeded in duplicate onto 24-well plates in RPMI1640 medium containing 10% FBS and incubated at 37°C for 48 h to reach 80% confluency. The culture medium was then replaced with serum-free medium and incubated at 37°C overnight. The following day, the culture medium was replaced with medium containing 0, 1, or 10% FBS. Different doses of LPA were then added and the cells were cultured for 0, 24, and 36 h. Cell growth was determined through cell count using Cell Count Reagent SF (NacalaiTesque, Japan).
MAPK and Akt activities
Phosphorylated and unphosphorylated MAPK or Akt were detected by western blot analyses using specific antibodies as described [11]. All primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).
Statistical analyses
Results are expressed as the mean ± SD of at least 3 independent experiments. Significant differences were determined using Student’s t-test, with P<0.05 accepted as statistically significant.
Results
The effects of LPA on ME180 cell migration were examined using an in vitro wound healing assay. The cell-free wound gaps healed slowly in the absence of any treatment. However, in the presence of LPA, the wound closure was significantly accelerated. We measured the wound areas 36 h after the addition of LPA in the following experiments (Figure 1A). LPA-induced ME180 cell migration was detected at the concentration of 0.1–10 mM. As shown in Figure 1, migration was clearly observed following the addition of 1–10 mM LPA. We compared the migration of ME180 cells treated with LPA in the presence or absence of the LPA receptor antagonist Ki16425 and found that the antagonist significantly suppressed LPA-induced cell migration (Figure 1A and B). To eliminate the effects of LPA on cell proliferation, we compared the number of ME180 cells after treatment with 1 or 10 mM LPA to that of the cells treated with vehicle for 36 h. As a result, there was no significant difference in cell proliferation in the presence or absence of LPA, whenmore than 1% FBS were involved in the medium (Figure 2). Theseproliferation results suggested that the presence of cells in the scratched gap was mainly due to cell migration but not cell proliferation.
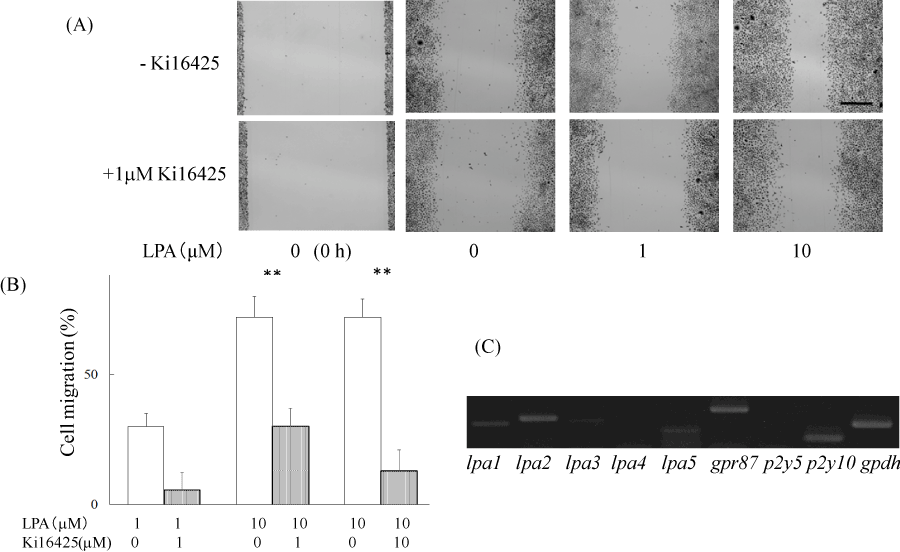
Figure 1. LPA induced cell migration of ME180 cells.
(A) ME180 cell migration was observed 36 h after adding LPA (1 µM and 10 µM) or vehicle in the absence (upper) or presence (lower) of the LPA antagonist, Ki16425. The images shown on the left side were captured at time 0. (B) Quantitative analyses of the results in (A) are resented. The healed areas were quantified with the program ImageJ. The gap area observed 36 h after adding vehicle was estimated at 100%. N=3. **P<0.01. (C) LPA receptor genes were detected by RT-PCR analysis using total RNA extracted from ME180 cells. **P<0.01
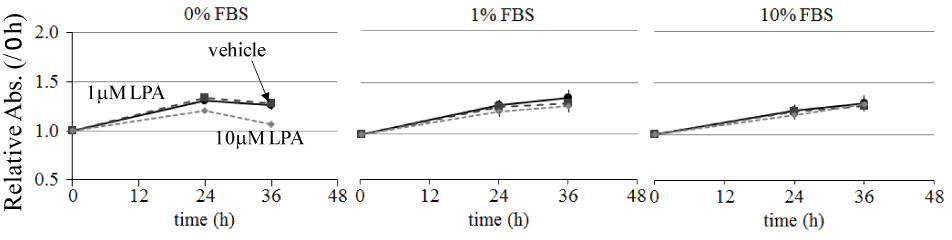
Figure 2. Effect of LPA on ME180 cell proliferation.
ME180 cells (5 × 104 cells/well) were plated on 24-well dishes and incubated in RPMI1640 medium containing 10% FBS until they were 80% confluent. Then, the culture medium was replaced with serum-free medium and incubated overnight. Subsequently, the cell medium was replaced with medium containing 0, 1, or 10% FBS and stimulated with 0, 1, or 10 mM LPA. Cell proliferation was measured at 0, 24, and 36 h after LPA stimulation. Each experiment was repeated 3 times. Data are expressed as the fold increase compared with the level at 0 h.
On the basis of the receptor specificity of Ki16425, LPA1, LPA3 receptor, and GPR87 were expected to be expressed in ME180 cells. We examined the expression levels of LPA receptors in ME180 cells by RT-PCR analysis using total RNAs extracted from ME180 cells (Figure 1C). The analysis showed that the cells expressed LPA receptor genes, such as lpa1, lpa2, lpa3, lpa5 and gpr87. The gpr87 and lpa2 levels were as high as that of the house-keeping enzyme gapdh, but lpa1 and lpa5 levels were relatively low and lpa3 expression was faint.
LPA is known to increase the activities of MAPK and PI3-K in various cell lines and the activation of those kinases induces cell migration [10-14]. We therefore examined the MAPKs and Akt activities in LPA-treated ME180 cells usingwestern blot analyses. As a result, 1 μM LPA increased the phosphorylation of Erk, JNK, p38, and Akt. These phosphorylation wereeffectively inhibited by Ki16425 (Figure 3A).
As we found that LPA activated MAPKs and Akt, we assessed the effects of inhibitors of Erk, JNK, p38, and PI3-K (upstream of Akt) on the LPA-induced cell migration. When ME180 cells were pretreated with5 mM U0126, 3 mM SP600125, or 3 mM PD16931630 min prior to the addition of LPA, the LPA-induced ME180 cell migration was significantly reduced, whereas the treatment with 10 mM LY294002 did not markedly affect migration (Figure 3B and C).

Figure 3. Activation of MAPKs is required for LPA-induced ME180 cell migration.
(A) LPA induced rapid and transient activation of MAPKs and Akt. ME180 cells were stimulated with 1 μM LPA for various time periods. Phosphorylated Erk, p38, JNK, and Akt were detected by Western blot analyses using phospho-specific antibodies against p-Erk, p-JNK, p-p38, and p-Akt. To evaluate the inhibitory effects of Ki16425 on LPA-induced phosphorylation, cells were pretreated with 1 μM Ki1642530 min prior to LPA stimulation. (B) MAPK inhibitors prevented LPA-induced cell migration, but PI3-kinase inhibitor did not. ME180 cells were pretreated with 5 μM U0126, 3 μM SP600125, 3 μM PD169316, or 10 μM LY294002 for 1 h prior to adding LPA. The migration was observed 36 h after LPA addition. Scale bar: 500 μm. (C) Cell migration was quantified using the software ImageJ. Each experiment was repeated 4 times. **P<0.01 vs vehicle-treated cells.
Because we had previously found that LPA activated EGFR via stimulating metalloprotease activity to regulate A431 cell dispersal [7], we investigated the involvement of EGFR in LPA-induced activation of MAPK and Akt in ME180 cells. Incubation with 0.3 mM of PD153035, an EGFR tyrosine kinase inhibitor, inhibited LPA-stimulated phosphorylation of these kinases. BB94 is known to inhibit the metalloproteases that are required to cleave an EGF-like growth factor precursor for EGFR transactivation [22]. 1 mM BB94 also blocked the LPA-stimulated phosphorylation of MAPK and Akt (Figure 4A). Intriguingly, these compounds also suppressed LPA-induced ME180 cell migration (Figure 4B and C).
As mentioned above, we found that the LPA response was mediated through the activation of GPR87 and/or LPA1 receptors. To evaluate the effects of gpr87 knockdown on LPA-induced cell migration, we treated the cells with gpr87-specific siRNA and conducted the scratch assay. As shown in Figure 5A and B, ME180 cells pretreated with non-targeting siRNA responded to 10 mM LPA and more than 73 ± 7% of the wound gap was healed by cell migration. However, cells pretreated with gpr87-specific siRNA reduced cell migration and only 16 ± 9% of the wound area was recovered. Similarly, pretreatment with an lpa1-specific siRNA prevented the LPA-induced cell migration to 22 ± 5%, and double knockdown of gpr87 and lpa1 geneseven more significantlyreduced LPA-induced ME180 cell migration (Figure 5A and B). Real time RT-PCR analyses showed that the expression of gpr87 and lpar1 genes was reduced to 28 ± 4% and 27 ± 10%, respectively, of the original level in each siRNA-treated cells (Figure 5C).

2021 Copyright OAT. All rights reserv
Figure 4. Transactivation of EGFR was involved in the LPA-induced ME180 cell migration.
(A) Effects of EGFR signal transduction inhibitors on LPA-stimulated MAPKs activation. Cells were pretreated with 0.3 μM PD153035 or 1 μM BB941 h prior to LPA stimulation. The phosphorylation levels were analyzed by western blot analyses. (B) Effects of PD153035 and BB94 on LPA-induced ME180 cell migration. Cells were pretreated with 0.3 μM PD153035 or 1 μM BB941 h prior to LPA stimulation. Cell migration was observed 36 h after adding LPA. Scale bar: 500 μm. (C) The migrations shown in B were quantified with ImageJ. Three experiments were repeated. **P<0.01 vs LPA-induced migration of vehicle-treated cells.
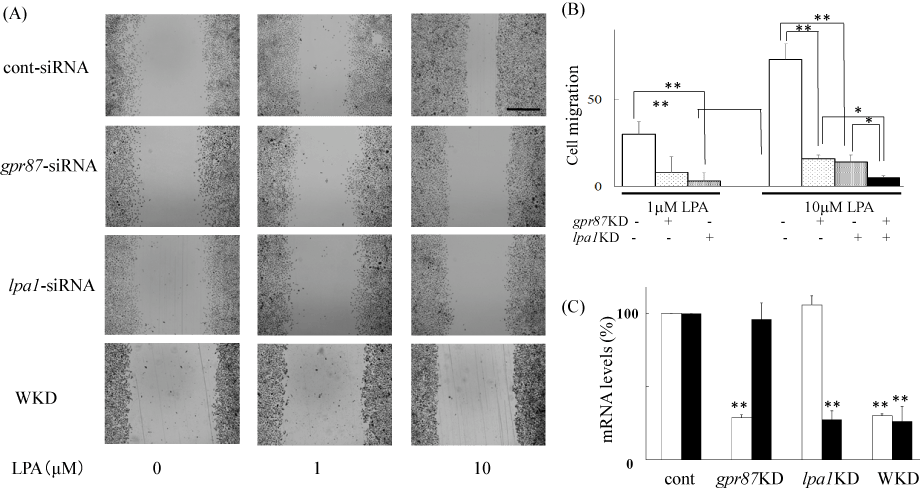
Figure 5. Inhibition of LPA-induced cell migration by silencing gpr87 in ME180 cells.
(A) Inhibition of LPA-induced cell migration by silencing gpr87 or lpa1 genes. gpr87- or lpa1-specific siRNAs or non-targeting siRNAs (control) were transfected into ME180 cells according to the manufacturer’s instructions and as described in the Materials and methods section. Cell migration was observed 36 h after adding 10 μM LPA. Scale bar: 500 μm. WKD represents double knock down. (B) Results shown in (A) were quantified with ImageJ. Values were obtained from 3-4 independent siRNA-experiments. *P<0.05, **P<0.01vs non-targeting siRNA-treated cells. (C) siRNA-induced silencing of gpr87 (open columns) or lpa1 (filled columns) genes. Real time PCR was used to detect the expression of these receptors. The primers used for real time PCR were 5′-AATCGGGATACCATGATGAGTCTT-3′ and 5′-CCAGGAGTCCAGCAGATGATAAA-3′ for GPR87, and 5′-GCACCGTCAAGGCTGAGAAC-3′ and 5′-TGGTGAAGACGCCAGTGGA-3′ for LPA1 receptor. **P<0.01vs non-targeting siRNA-treated cells.
Discussion
Aberrant overexpression of GPCRs can be an essential part of tumor progression [23] and GPCR-mediated signaling can support tumorigenesis by promoting tumor growth, survival, migration, and metastasis [24-26]. In 2008, Glatt et al. and Gugger et al. reported that GPR87 was preferentially overexpressed in squamous cell carcinoma of the lung, cervix, skin, neck, and urinary bladder [5]. Okazoe et al. demonstrated that progression from non-muscle-invasive to muscle-invasive tumor was more frequently observed in patients with GPR87-positive tumors, and they suggested that GPR87 could be a potential prognostic marker for the progression of bladder cancers [11]. Furthermore, Nii et al. demonstrated that in non-small-cell lung cancer (NSCLC), the overexpression of GPR87 is significantly associated with poorer differentiation and higher proliferation. During the progression of NSCLC, GPR87 overexpression may be associated with the acquisition of a more aggressive phenotype [27].
It has been hypothesized that cell migration is critical during many stages of cancer cell metastasis. In the present study, we performed a wound healingassay to investigate the role of GPR87 on LPA-induced cell motility. Because 18:1 oleoyl-LPA (1-acyl-2-hydroxy-sn-glycero-3-phosphate) is a LPA form of LPA that is particularly abundant in human plasma and is also the most commonly used laboratory LPAchemical species, 18:1 LPA was used throughout the present study.
We detected that LPA stimulated wound healing in a dose dependent manner in ME180 cells (Figure 1). The effects of LPA on the closure of the wounded cell monolayers can be attributed to 2 distinct cell processes: cell proliferation and migration [28]. We examined the effects of LPA on the ME180 cell proliferation and found LPA had minor effect on the cell growth under 80% confluent. Thus, the wound healing induced by LPA in ME180 cells was mainly due to cell migration (Figure 2).
As shown in Figure 1, LPA induced ME180 cell migration that was blocked by Ki16425, an efficient antagonist for LPA1, LPA3, and GPR87 [1-3]. Because the LPA3 receptor gene was only faintly identified using RT-PCR in the ME180 cells (Figure 1), LPA1 receptor and GPR87 were primary LPA receptors mediatingthe LPA signaling that leads to cell migration through the activation of MAPKs (Figure 3 and 4).
To determine the relevance of GPR87 to LPA-induced ME180 cell migration, we examined the effects of gpr87-specific siRNA on cell responses. Cell migration was significantly reduced in the GPR87-knockdown cells, consistent with the loss of GPR87 expression (Figure 5). This result strongly suggests that GPR87 acts as an LPA receptor in ME180 cells. However, some LPA-induced cell migration remained. Thus, the LPA1 receptor was predicted to also be important for the LPA-induced cell migration. As expected, it was shown that LPA1 receptor-knockdown cells diminished LPA-induced cell migration. Cell treatment with siRNAs to both gpr87 and lpa1 affected cell migration in an additive manner, highlighting the equal significance of GPR87 and LPA1 receptors in LPA-induced cell migration in ME180 cells.
We showed that Erk, p38, and JNK were the protein kinases required for controlling LPA-induced cell migration; however, we could not distinguish which MAPK was the most critical for these cell motilities (Figure 3). Functional studies have shown that the activation of MAPKs was crucial for cell migration induced by LPA [12-15]. For instance, the migration of prostate cancer PC3 cell was induced via the activation of the LPA receptor with the parallel activation of Erk and p38 [13]. In glioma cells, the p38 and JNK pathways are equally important for LPA1 receptor-mediated migration [14]. Each MAPK is linked to phosphorylation of distinct substrates, such paxillin, FAK, calpain, MLCK, MAP1B, MAP2, and ATF2. These substrates play key roles in cell spreading, lamellipodia formation, and tail retraction during cell migration and invasion. Thus, the three kinases in the MAPK family all seem able to regulate cell migration but by distinct mechanisms [15].
While we identified LPA-activated Akt in ME180 cells, the LPA-induced cell migration was resistant to 10 mM LY294002. Maffucci et al. reported that there are 2 classes of PI3-K; PI3-KC1 and PI3-KC2, both of which are involved in the LPA response [29,30]. However, PI3-KC1 and PI3-KC2 are differentially sensitive to LY294002. PI3-KC2 (b subtype) sensitivity to LY294002 is approximately 6-fold higher than that of the class I enzyme (6.9 mM vs. 1.2M) [30,31]. Thus, further investigation is required to determine whether Akt activation contributes to reduce cell motility at a higher LY294002 concentration. However, since a higher concentration of LY294002 reduced cell migration even in the absence of LPA (data not shown), second generation PI3-K inhibitors may prove more useful, since they exhibit higher selectivity toward PI3-KC2 or PI3-KC1.
As we indicated, the LPA receptor-mediated migration was blocked by both the EGFR-tyrosine kinase inhibitor and the metalloprotease inhibitor, suggesting that the transactivation of EGFR through LPA receptors promotes ME180 cell migration (Figure 4). This hypothesis was also documented in a prior publication; the LPA-induced colony dispersal of A431 cellswas via the transactivation of EGFR [7]. During tumor progression, the colony dispersal allows benign tumor cells to acquire the capacity to infiltrate surrounding tissue and to ultimately metastasize to distant sites. EGFR is expressed in various cervical cancer cells, and its higher expression is associated with an advanced clinical stage and patient poor prognosis [32]. A cascade of signaling events initiated by EGF through EGFR resulted in the induction of EMT in cervical cancer [8]. Moreover, Du et al. reported that EGF treatment activatedMAPKs thatare canonical members of the EGFR and GPCR signaling pathway, and the phosphorylation and activation of MAPK is involved in mediating tumor progression and metastasis [12,16,33]. Thus, these results and our present results demonstratethat the transactivation of EGFR is crucial for LPA-induced motilities of cervical cancer cells.
Hasegawa et al. reported that LPA1–3 receptors play a role in cellular motility, potentially contributing to PC-3 cell invasion and metastases [34]. In the case of A431 cells, the LPA-induced colony dispersal was mediated by GPR87 and LPA1 receptor [7]. Thus, significant receptor redundancies have been reported to stimulate LPA-mediated responses, whereas the physiological meaning of having a system of multiple LPA receptors within a cell line is unknown. Interestingly, the expression of GPR87 was induced in colon carcinoma RKO cells in a p53 dependent manner under genotoxic stress [35]. This may indicate that GPR87 is only expressed at a particular circumstance in the life of the cell to potentiate the LPA response.
In conclusion, our present results on GPR87-mediated ME180 cell migration and our previous report on GPR87-mediated colony dispersal of A431 cells suggest that LPA activates GPR87 and the subsequent development of cancer cells.
Acknowledgments
We thank Prof. Hideo Taniura for discussions and Ms. Ayaka Murashima for technical assistance. This research was supported by MEXT-supported program for the Strategic Research Foundation at Private Universities 2015-2019.
Author contributions
NF conceived and supervised the study; RS, DF, and TF designed experiments; RS, DF, SN, TW, and SO performed experiments; NF and RS wrote the manuscript.
Disclosure of potential conflicts of interest
There are no potential conflicts of interest to disclose.
References
- Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, et al. (2010) LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol 50: 157-186. [Crossref]
- Tigyi G1 (2010) Aiming drug discovery at lysophosphatidic acid targets. Br J Pharmacol 161: 241-270. [Crossref]
- Yung YC, Stoddard NC, Chun J (2014) LPA receptor signaling: pharmacology, physiology, and pathophysiology. J Lipid Res 55: 1192-1214. [Crossref]
- Tabata K, Baba K, Shiraishi A, Ito M, Fujita N (2007) The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem Biophys Res Commun 363: 861-866. [Crossref]
- Glatt S, Halbauer D, Heindl S, Wernitznig A, Kozina D, et al. (2008) hGPR87 contributes to viability of human tumor cells. Int J Cancer 122: 2008-2016. [Crossref]
- Yan M1, Li H, Zhu M, Zhao F, Zhang L, et al. (2013) G protein-coupled receptor 87 (GPR87) promotes the growth and metastasis of CD133⺠cancer stem-like cells in hepatocellular carcinoma. PLoS One 8: e61056. [Crossref]
- Ochiai S, Furuta D, Sugita K, Taniura H, Fujita N (2013) GPR87 mediates lysophosphatidic acid-induced colony dispersal in A431 cells. Eur J Pharmacol 715: 15-20. [Crossref]
- Yilmaz M, Christofori G (2009) EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev 28: 15-33. [Crossref]
- Buonato JM, Lazzara MJ (2014) ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res 74: 309-319. [Crossref]
- Okazoe H, Zhang X, Liu D, Shibuya S, Ueda N, et al. (2013) Expression and role of GPR87 in urothelial carcinoma of the bladder. Int J Mol Sci 14: 12367-12379. [Crossref]
- Gugger M, White R, Song S, Waser B, Cescato R, et al. (2008) GPR87 is an overexpressed G-protein coupled receptor in squamous cell carcinoma of the lung. Dis Markers 24: 41-50. [Crossref]
- Du J, Sun C, Hu Z, Yang Y, Zhu Y, et al. (2010) Lysophosphatidic acid induces MDA-MB-231 breast cancer cells migration through activation of PI3K/PAK1/ERK signaling. PLoS One 5: e15940. [Crossref]
- Hao F, Tan M, Xu X, Han J, Miller DD, et al. (2007) Lysophosphatidic acid induces prostate cancer PC3 cell migration via activation of LPA(1), p42 and p38alpha. Biochim Biophys Acta 1771: 883-892. [Crossref]
- Malchinkhuu E, Sato K, Horiuchi Y, Mogi C, Ohwada S, et al. (2005) Role of p38 mitogen-activated kinase and c-Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine-1-phosphate in glioma cells. Oncogene 24: 6676-6688. [Crossref]
- Huang C, Jacobson K, Schaller M (2004) MAP kinases and cell migration. J Cell Sci 117: 4619-4628. [Crossref]
- Daub H, Weiss FU, Wallasch C, Ullrich A (1996) Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379: 557-560. [Crossref]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, et al. (1997) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402: 884-888. [Crossref]
- Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A (2001) Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for inter receptor signal transmission. Oncogene 20:1594-1600. [Crossref]
- Muthusami S, Prabakaran DS, Yu JR, Park WY (2014) EGF-induced expression of Fused Toes Homolog (FTS) facilitates epithelial-mesenchymal transition and promotes cell migration in ME180 cervical cancer cells. Cancer Lett 351: 252-259. [Crossref]
- Tang XL, Wang Y, Li DL, Luo J, Liu MY (2012) Orphan G protein-coupled receptors (GPCRs): biological functions and potential drug targets. Acta Pharmacol Sin 33: 363-371. [Crossref]
- Chung S, Funakoshi T, Civelli O (2008) Orphan GPCR research. Br J Pharmacol 153 Suppl 1: S339-346. [Crossref]
- Schäfer B, Gschwind A, Ullrich A (2004) Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene 23: 991-999. [Crossref]
- Dorsam RT, Gutkind JS (2007) G-protein-coupled receptors and cancer. Nat Rev Cancer 7: 79–94. [Crossref]
- Lango MN, Dyer KF, Lui VW, Gooding WE, Gubish C, et al. (2002) Gastrin-releasing peptide receptor-mediated autocrine growth in squamous cell carcinoma of the head and neck. J Natl Cancer Inst 94: 375-383. [Crossref]
- Darmoul D, Gratio V, Devaud H, Lehy T, Laburthe M (2003) Aberrant expression and activation of the thrombin receptor protease-activated receptor-1 induces cell proliferation and motility in human colon cancer cells. Am J Pathol 162: 1503–1513. [Crossref]
- Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, et al. (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120: 303-313. [Crossref]
- Nii K, Tokunaga Y, Liu D, Zhang X, Nakano J, et al. (2014) Overexpression of G protein-coupled receptor 87 correlates with poorer tumor differentiation and higher tumor proliferation in non-small-cell lung cancer. Mol Clin Oncol 2: 539-544. [Crossref]
- Teo ST, Yung YC, Herr DR, Chun J (2009) Lysophosphatidic acid in vascular development and disease. IUBMB Life 61: 791-799. [Crossref]
- Maffucci T, Brancaccio A, Piccolo E, Stein RC, Falasca M (2003) Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J 22: 4178-4189. [Crossref]
- Yart A, Chap H, Raynal P (2002) Phosphoinositide 3-kinases in lysophosphatidic acid signaling: regulation and cross-talk with the Ras/mitogen-activated protein kinase pathway. Biochim Biophys Acta 1582: 107-111. [Crossref]
- Maffucci T, Cooke FT, Foster FM, Traer CJ, Fry MJ, et al. (2005) Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J Cell Biol 169: 789-799. [Crossref]
- Yan LX, Huang XF, Shao Q, Huang MY, Deng L, et al. (2008) MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA 14: 2348-2360. [Crossref]
- Xu KP, Yin J, Yu FS (2007) Lysophosphatidic acid promoting corneal epithelial wound healing by transactivation of epidermal growth factor receptor. Invest Ophthalmol Vis Sci 48: 636-643. [Crossref]
- Hasegawa Y, Murph M, YuaS, Tigyi G, Mills GB (2008) Lysophosphatidic acid (LPA) induced vasodilator-stimulated phosphoprotein mediates lamellipodia formation to initiate motility in PC-3 prostate cancer cells. Mol Oncol 2: 54–69. [Crossref]
- Zhang Y, Qian Y, Lu W, Chen X (2009) The G protein-coupled receptor 87 is necessary for p53-dependent cell survival in response to genotoxic stress. Cancer Res 69: 6049-6056. [Crossref]