Abstract
In this review, we comprehensively describe ophthalmic diseases with mitochondrial DNA mutation such as Leber's hereditary optic neuropathy (LHON), progressive external ophthalmoplegia (PEO) and Kearns-Sayre Syndrome (KSS). Ocular involvement is a prominent clinical feature of various mitochondrial diseases as well. The known mitochondrial disorders such as Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Myoclonic epilepsy with ragged-red fibers (MERRF), Mitochondrial neurogastrointestinal encephalopathy (MNGIE) and Neuropathy, ataxia, and retinitis pigmentosa (NARP) exhibiting secondary ophthalmologic manifestations have also been included. In addition, mitochondrial disorders with nuclear gene defects such as Autosomal dominant optic atrophy (ADOA), Autosomal Dominant Optic Atrophy and Cataract (ADOAC), Costeff Optic Atrophy Syndrome (COAS) have also been described. Finally, this review covers ophthalmic diseases with secondary mitochondrial dysfunctions including glaucoma, non-arteritic anterior ischemic optic neuropathy (NAION), diabetic retinopathy (DR) and age-related macular degeneration (AMD). The association of mitochondrial dysfunction in various ophthalmic diseases and ocular manifestations observed in mitochondrial disorders provides a strong evidence for a direct or indirect role of mitochondrial aberrations in ocular diseases and represents a potential therapeutic target. Some of the plausible disease mechanism(s) and therapeutic modalities are also reviewed.
Key words
genetics, mitochondrial DNA, ocular diseases, oxidative stress
Abbreviations
RGCs: Retinal ganglion cells; OXPHOS: Oxidative phosphorylation; ROS: Reactive oxygen species; mtDNA: Mitochondrial DNA; DR: Diabetic retinopathy; AMD: Age-related macular degeneration; LHON: Leber's hereditary optic neuropathy; PEO: Progressive external ophthalmoplegia; COX: Cytochrome c oxidase; CPEO: Chronic Progressive External Opthalmoplegia; KSS: Kearns-Sayre Syndrome; NAION: Non-arteritic anterior ischemic optic neuropathy; MELAS: Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; NGS: Next generation sequencing; MERRF: Myoclonic epilepsy with ragged-red fibers; POLG: Polymerase: DNA directed, gamma; MNGIE: Mitochondrial Neurogastrointestinal encephalomyopathy; NARP: Neurogenic weakness, Ataxia and Retinitis Pigmentosa; LS: Leigh syndrome
Introduction
Mitochondria have been long considered as ‘the powerhouse of the cell’ that plays an important role in energy generation through ATP synthesis necessary to perform all cellular and metabolic functions. Mitochondria are present in nucleated eukaryotic cells and their copy number in a particular cell type varies with energy requirements of the cell. Cells with high metabolic activity including retinal ganglion cells (RGCs) within the papillomacular bundle, the cardiac conduction system, pancreas, kidney, liver, and muscle, have high numbers of mitochondria to meet their high ATP demands. Therefore, such tissues are highly susceptible to ATP disruption and are predominantly affected in mitochondrial disorders [1].
The mitochondrial respiratory chain is composed of five complexes (I–V) through which ATP generation takes place via oxidative phosphorylation (OXPHOS). Complexes I–IV oxidize NADH and FADH2 generated from glycolysis and beta-oxidation of fatty acids; and complex V (also known as ATP synthase), generates an electrochemical gradient that leads to phosphorylation of ADP to ATP. Several other cofactors including ubiquinone facilitate electron transfers between these respiratory chain complexes [2]. While the primary role of mitochondria is ATP generation, these organelles are also involved in other metabolic and biological functions of the cell including detoxification of reactive oxygen species (ROS), iron metabolism, fatty acid oxidation, amino acid biosynthesis, and regulation of cellular apoptosis. Although dysfunction of the OXPHOS pathway is one of the most important factors in the pathogenesis of mitochondrial diseases, defects in cofactors or mitochondrial dynamics and assembly and environmental triggers can also affect the ATP generation and lead to increased oxidative stress, production of ROS and apoptosis which can all have damaging effects on the cells. Genetic defects of OXPHOS caused by mutations in the mitochondrial DNA (mtDNA) may also contribute to energy failure and lead to oxidative stress thereby causing various diseases including those with ocular manifestation [3].
Ocular involvement is a prominent clinical feature of various mitochondrial diseases [4]. This stems from the fact that, the eye is a high energy demand organ and thus is particularly susceptible to the consequences of mitochondrial damage. Mitochondrial damage as a result of oxidative stress and mtDNA instability is implicated in the pathogenesis of ophthalmologic disorders such as diabetic retinopathy (DR), age-related macular degeneration (AMD) and glaucoma [5]. There is also an increased evidence of oxidative stress being involved in the development of many diseases related to aging including Parkinson's disease [6] and Alzheimer's disease [7]. The involvement of mitochondrial abnormalities in ophthalmic diseases can manifest as optic nerve dysfunction specifically involving RGCs as in Leber's hereditary optic neuropathy (LHON), or progressive external ophthalmoplegia (PEO) and ptosis involving the extraocular muscles, which is commonly associated with either primary mtDNA mutations or acquired mtDNA defects secondary to a nuclear genetic disorder of mtDNA maintenance. In this review, we describe the various ophthalmic diseases caused directly by mtDNA mutations, mitochondrial diseases with secondary eye involvement and ophthalmic diseases caused by nuclear genes with implications on mitochondrial function; and the laboratory methods currently used for investigating ophthalmic diseases with direct or indirect relationship with mitochondrial aberration and oxidative stress. In addition, the unique relation of mitochondrial dysfunction and oxidative stress and how they contribute to the development and progression of various ophthalmic diseases as shown in Figure 1 has also been discussed.
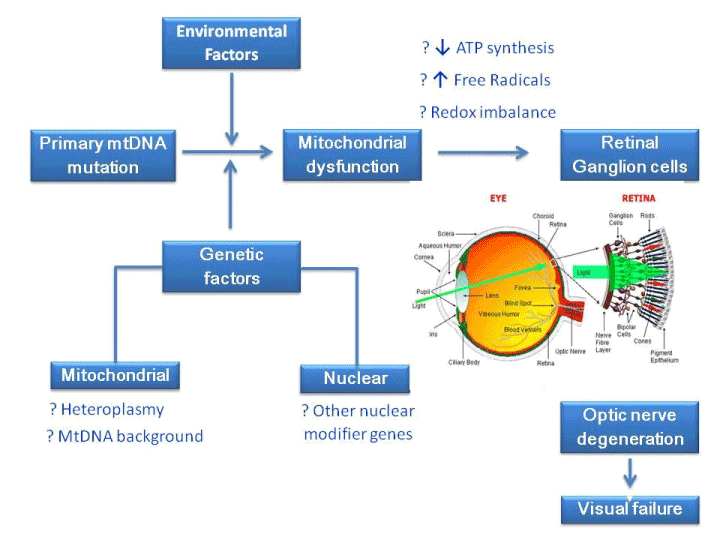
Figure 1. Contribution of various factors to mitochondrial dysfunction and its effect on retinal ganglion cells leading to visual failure.
Mitochondrial genetics
MtDNA is a ~16.6 Kbp circular double stranded genome and encodes 37 genes, all of which are essential for normal mitochondrial function. Thirteen of these genes encode enzymes involved in OXPHOS. The remaining genes code for transfer RNA (tRNA) and ribosomal RNA (rRNA), which help assemble amino acids into functional proteins. MtDNA is a multi-copy DNA, with copy numbers varying between 100 and 10,000 copies of mtDNA depending upon the cellular energy demand. MtDNA is inherited solely from the mother and her male and female offspring's have an equal chance of inheriting a mtDNA mutation causing a mitochondrial disorder [8]. Although mitochondria have their own genome, that however, does not liberate them from being controlled by nuclear genes. The mitochondrial proteome is estimated at ∼3350 proteins and most of the proteins essential for mitochondrial composition and function are encoded by nuclear genes [9,10].
Despite the small size of the mitochondrial genome, mtDNA mutations are an important cause of various inherited disease including ophthalmic diseases. Recent advances have considerably increased our understanding of mitochondrial genetics and the relationship between inherited mutations and disease phenotypes, and in identifying acquired mtDNA mutations related to both normal aging and diseased conditions. However, prevention and treatment of these diseases still remains challenging. This review explores the advances that have been made and the areas in which future progress is likely. According to the MitoMap database (http://www.mitomap.org/bin/view.pl/MITOMAP/MutationsCodingControl), last updated 07/20/2015, there are currently 301 mutations associated with 59 conditions or diseases mainly neurological, metabolic, ophthalmic and even cancer [11] . Similarly, other database such as MitoCarta is also available (http://www.broadinstitute.org/pubs/MitoCarta/human.mitocarta.html). The recent version MitoCarta 2.0 inventory provides a molecular framework for system-level analysis of mammalian mitochondria [12].
Laboratory diagnosis for mitochondrial disorders affecting the eye
The laboratory diagnosis of mitochondrial disease is far from straightforward. Usually, there are several metabolic screening tests which can be performed in blood, urine or spinal fluid (SF). These include but are not limited to: basic chemistries, complete blood count, blood/SF lactate, pyruvate, L/P ratio, amino acid quantifications, liver enzymes and ammonia, creatinine kinase and quantitative urine organic acid. Serum/CSF lactate and lactate stress testing should be included in the diagnostic work-up of suspected mitochondrial disorders.
Histological and biochemical examination of the respiratory function in muscle biopsy were considered as the gold-standard for diagnosing mitochondria disorders (in particular those involving myopathy) by major reference laboratories worldwide [13]. Many patients with mtDNA disease exhibit OXPHOS dysfunction. Histology shows the classical ragged red fibers on Gomori trichrome staining with sub-sarcolemmal accumulation of abnormal mitochondria – and cytochrome c oxidase (COX)-deficient fibers demonstrating mtDNA involvement. Histological and histochemical examination of muscle biopsy are considered gold standard for diagnosis of Chronic Progressive External Opthalmoplegia (CPEO) and Kearns-Sayre Syndrome (KSS). Cultured skin fibroblasts may be particularly helpful to find complex-I defects. In addition, DNA may be extracted from muscle biopsy and investigated for the presence of multiple deletions by southern blotting or long-range PCR, which is the major genetic-underlying cause for CPEO and KSS. Muscle biopsy is an invasive procedure and cannot be justified for diseases such as LHON and non-arteritic anterior ischemic optic neuropathy (NAION) where the diagnosis can be achieved through ophthalmic examination and direct DNA sequencing. This is also true for ophthalmic diseases caused by nuclear gene defect, where diagnosis can be achieved by Sanger sequencing (for single nuclear gene) or exome sequencing (>1 nuclear gene).
Molecular genetic testing can be performed on DNA extracted from the blood, skeletal muscle, hair follicles, buccal mucosa or urinary sediment. MtDNA isolation may require enrichment and few kits from Qiagen®, Abcam® or Biovision® are available for this purpose. These test usually involve Sanger® sequencing to detect single base substitution in the mtDNA extracted from the blood samples of patients with LHON [14] or mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) [15] or a more advanced technique such as exome sequencing [which is part of the next generation sequencing (NGS)] is particularly useful for when the mutation(s) is suspected to be in one of many nuclear genes coding regions [16] contributing to various mitochondrial disorders. NGS involving sequencing of the intronic and exonic regions may also be useful if there is a mutation (known or novel) in the intronic region associated with any of the mitochondrial disorder(s). A good example of this, is mutations in the OPA3 nuclear gene located in the intronic region of the gene.
Mechanisms of mitochondrial diseases
Mitochondrial diseases can be caused by mutations in the genes of mtDNA or nuclear DNA and is not limited to any organ or age group. The inheritance pattern can be autosomal dominant, recessive, X-linked or mitochondrial. In addition to ATP deficiency, mitochondrial disorders can involve various mechanisms: i) defects of structural OXPHOS subunits and assembly factors: Usually presented as a mtDNA mutation in one of the respiratory complexes that can result in decrease in ATP production and defect in ROS detoxification, which ultimately leads to oxidative stress (e.g. MELAS and LHON). In addition, other environmental factor may also plausibly contribute to the development of various ophthalmic diseases as shown in Figure 1; ii) defects in the nuclear-encoded proteins responsible for mtDNA maintenance: This can lead to copy number variations, point mutations and/or multiple deletions in the mtDNA. The disorders disrupting mtDNA maintenance can range from being severe, affecting infants, where mtDNA copy number is severely depleted in one or more tissues (e.g. mtDNA depletion syndrome, MDS), to mild adult-onset autosomal dominant or recessive (e.g. PEO); iii) mitochondrial tRNA and rRNA mutations and single mtDNA deletions: tRNA point mutations are highly common in the human mtDNA (www.mitomap.org) exhibiting varying degree of clinical manifestations. The most commonly studied mitochondrial tRNA point mutations include the A3243G mutation in tRNA-Leu and the A8344G mutation in tRNA-Lys. The former has a population prevalence as high as 5.7 per 100,000 and causes a wide range of phenotypes of which MELAS is prominent, and the latter is associated with myoclonic epilepsy with ragged-red fibers (MERRF); iv) disorders of mitochondrial dynamics: Mitochondria form an interconnected network that is constantly reorganized. Mitochondrial dynamics involving mitochondrial fusion and fission are critical for mitochondrial distribution and function. Defective fusion or fission underlies a number of neurological diseases in humans. OPA1 mutations cause autosomal-dominant optic atrophy [17]. In most cases, this disease specifically involves degeneration of RGCs. However, more diverse OPA1-related phenotypes have been described recently, and they include disorders of mtDNA maintenance [18]. This is discussed in more details in section 2.7.
Ophthalmic diseases caused by mtDNA mutation
Leber's Hereditary optic neuropathy
LHON, a commonly inherited optic neuropathy causing bilateral central vision loss, has a prevalence of 1 in 30,000, 1 in 39,000 and 1 in 50,000 in the North East of England, the Netherlands and Finland, respectively. LHON can affect individuals as early as in childhood to adults over 60 years of age [19]. The age of onset is slightly higher in females ranging from 19-55 years (average 31.3 years) than in males with an age range of 15-53 years (average 24.3 years). For some unclear reasons, LHON shows an increased prevalence among males (over 80%). Early evidence suggested that estrogens play a protective role in females and that these hormones ameliorate mitochondrial dysfunction in LHON through the estrogen receptors [20]. In acute LHON, patients usually present with painless visual blurring, either affecting both eyes simultaneously (25% of cases) or sequentially (75% of cases) within a median time-span of 8 weeks. Clinical ocular examination shows characteristic fundal changes including a circumpapillary, telangiectatic microangiopathy, swelling of the retinal nerve fiber layer, tortuosity of the retinal vasculature and disc hyperemia. The classical field defect in LHON is a steep-sided central or centrocecal scotoma. In chronic LHON, the degeneration of retinal nerve fiber layer is gradual and the loss of RGC axons results in optic atrophy [14]. In the majority of the LHON patients visual loss is permanent and visual recovery is rare [19]. In 90% of LHON patients with family history, one of the three mtDNA mutations, m.3460G>A, m.11778G>A and m.14484T>C, known as primary LHON mutation are observed. The remaining 10% either have other mtDNA mutation or no mutation in the entire mtDNA genome. The total number of mtDNA mutations reported to date in association with LHON are 63 (mitomap website), in which 12 mutations are with confirmed pathogenic status [21]. Primary LHON mutations have been reported to be less common in patients with LHON optic neuropathy selected from a clinical setting than in patients with LHON from multi-generational families and with family history. In other words, the 90% figure only holds if the patient was diagnosed clinically with LHON and a family history of the disease. Screening of the entire mtDNA genome is highly essential in LHON patients lacking one of the three primary LHON mutation(s), in order to identify mtDNA mutation(s) causing the disease. Mutations in the mtDNA clearly leads to mitochondrial dysfunction and disrupted ATP production as demonstrated in cybrid experiments. RGCs are highly susceptible to disrupted ATP production and oxidative stress. Selective degeneration of RGCs as a result of mitochondrial dysfunction and metabolic insult leads to vision loss in LHON [22]. The mechanisms involved in the pathogenesis of LHON continue to be elucidated, paving the way for research into potential therapeutic interventions. It is also plausible, that the primary involvement in LHON is the RGCs and not the optic nerve. The optic nerve involvement is possibly a secondary event in LHON and this may be supported by the fact that the mitochondria are located farther away from the optic nerve as demonstrated in Figure 2.
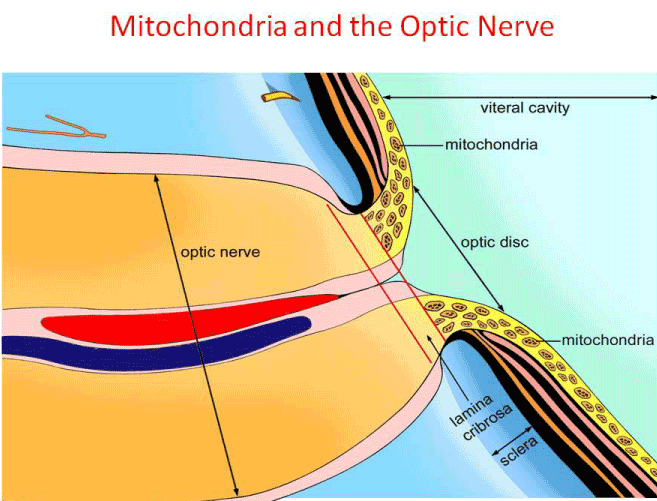
Figure 2. Unmyelinated portion of the optic nerve extending into the lamina cribrosa and halting abruptly in the retrolaminar position. Unmyelinated axons tend to have higher density of mitochondria than myelinated axons. Unmyelinated axons are not very efficient in conducting signals (conduct more slowly and require more energy).
Management of patients is largely supportive with the provision of visual aids and help with occupational rehabilitation. Individuals harboring mtDNA LHON-causing mutations are strongly advised to moderate their alcohol intake and avoid smoking. It also seems reasonable to avoid other putative environmental triggers for visual loss such as exposure to industrial toxins and drugs with mitochondrial-toxic effects. Initial studies have suggested that oral administration of Idebenone (trade name, Raxone®) could benefit early-stage LHON patients at high risk for further visual loss [23-25]. In the absence of any other treatment options available for LHON patients, Idebenone is commonly used by the mitochondrial medicine community. Santhera pharmaceuticals are currently conducting clinical trials of Idebenone for LHON patients. Targeted gene therapy for LHON is also being actively explored [26,27].
Leber's hereditary optic neuropathy-plus
LHON in some instances occur with other symptoms in addition to vision loss. These include dystonia, tremors, and cardiac conduction defects. Some affected individuals develop features similar to multiple sclerosis, which is a chronic, immunological disorder characterized by focal central nervous system demyelination resulting in transient or permanent visual problems, sensory deficits, muscle weakness, poor coordination, and a variety of other health problems. In these patients, the condition is described as LHON-plus [28]. LHON-plus patients also have complex I respiratory defects and mtDNA mutations similar to LHON [11]. In one report, however, cases of LHON-plus demonstrated complex I respiratory defect but showed no mtDNA mutation upon sequencing the entire mitochondrial genome [29].
Progressive external ophthalmoplegia
PEO is a condition characterized by weakness of the external eye muscles affecting adults between ages 18 and 40 years. The most common clinical signs and symptoms include ptosis, which can affect one or both eyelids and ophthalmoplegia (Figure 3A). Some patients may also exhibit myopathy in the neck, arms or legs [30]. The weakness may be especially noticeable during exercise (exercise intolerance.). Muscle weakness may also cause dysphagia. PEO patients often have abnormal looking cells under the microscope presented as ragged-red fibers and show large mtDNA deletions (Figure 3B and 3C) [31]. However, the mtDNA copy number usually remains normal.
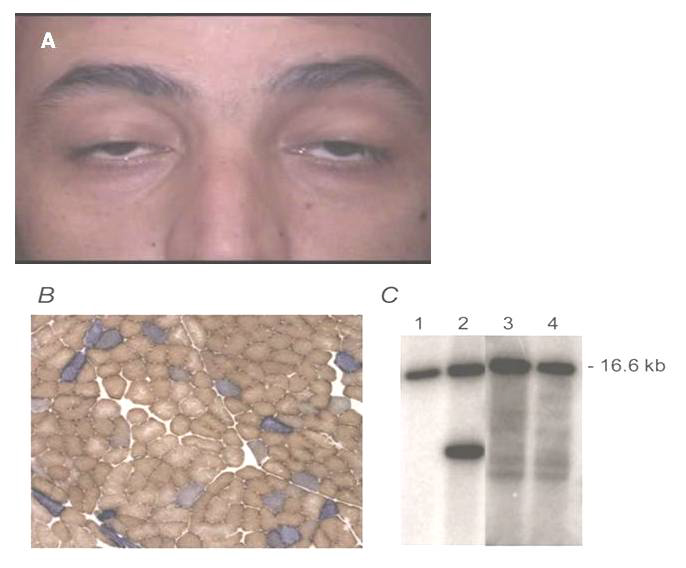
Figure 3. (A) Late onset-PEO is the most common form of extraocular mitochondrial myopathy and often arises spontaneously. (B) COX-deficient fibers in skeletal muscle tissue and (C) heteroplasmic, single large-scale mtDNA rearrangement (predominantly mtDNA deletion, lane 2).
Disorders similar to PEO with overlapping signs and symptoms have also been described. These include conditions caused by polymerase (DNA directed), gamma (POLG) gene mutations [32], such as ataxia neuropathy spectrum, as well as other mtDNA deletion disorders, such as KSS [32]. However, these conditions have many additional features not shared by most individuals with PEO.
Progressive external ophthalmoplegia plus
In certain instances PEO is accompanied by additional signs and symptoms including sensorineural hearing loss, weakness and loss of sensation in the limbs due to neuropathy, impaired muscle and ataxia, a pattern of movement abnormalities known as parkinsonism, or depression. When PEO and ptosis are associated with other neurological or multisystem abnormalities, these may be termed as ophthalmoplegia-plus (PEO-plus) syndromes. Drachman (1968) gave a description of disorders associated with progressive external ophthalmoplegia, which he termed 'ophthalmoplegia plus’ [33]. A myriad of systemic features have been reported including limb, facial and bulbar muscle weakness, short stature, diabetes mellitus, deafness, cardiac conduction defects, central and peripheral respiratory dysfunction and endocrine and sex organ abnormalities [33]. The prevalence of PEO-plus is not known.
Kearns-Sayre Syndrome: KSS is the most common form of a PEO-plus syndrome [34] with prevalence estimated at 1 to 3 per 100,000 individuals. Its cardinal features include early age of PEO onset (before the age of 20 years), degenerative pigmented retinopathy, cardiac conduction block with the risk of potentially life threatening arrhythmias and cerebellar dysfunction or elevated cerebrospinal fluid protein (>100mg/dl) [35]. With disease progression, a proximal myopathy develops in addition to deafness, bulba dysfunction, areflexia and stroke-like episodes. Although reports of sibling pairs exist [35], most cases of KSS are sporadic. Individuals with KSS have a single, large deletion of mtDNA, ranging from 1,000 to 10,000 nucleotides. The cause of the deletion in affected individuals is unknown.
Other mitochondrial diseases associated with PEO
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes: PEO may rarely occur in patients with MELAS. A subset of patients with PEO who harbor the m.3243 A>G point mutation typically associated with MELAS but who manifest no other clinical features of this condition have been reported [36]. During the Imaging may reveal multifocal, predominantly cortical, infarction-like lesions during the stroke-like episodes, suggestive of an embolic or vasculitic disease and calcification within the basal ganglia. It is noteworthy that in contrast to typical ischemic strokes, these lesions are not confined to vascular territories. Additional ocular manifestations may include pigmentary retinopathy and maculopathy. Muscle biopsy shows a classical COX-deficient and ragged-red fibers; and molecular confirmation of the m.3243A>G mutation can be made through mtDNA analysis of blood, muscle or urine sediment [37].
Myoclonic epilepsy, myopathy with ragged-red fibres: MERRF is characterized by myoclonus, myopathy, and spasticity mainly affecting the muscles and nervous system. The clinical features of MERRF are highly variable and may vary widely even among members of the same family. Other clinical features of MERRF include epilepsy, ataxia, peripheral neuropathy and dementia [38]. PEO has also been described with MERRF, in what appears to be an overlapping syndrome [39]. Ragged-red fibers in a muscle biopsy specimen and DNA sequencing for the m.3243 A>G mutation is characteristic for MERRF/PEO [39]. Creatine kinase and serum lactate may be moderately elevated, whilst brain imaging may simply show mild diffuse atrophy.
Mitochondrial neurogastrointestinal encephalomyopathy: Mitochondrial Neurogastrointestinal encephalomyopathy (MNGIE) is a condition primarily affecting the digestive and nervous systems. MNGIE is very commonly associated with gastrointestinal dysmotility. The latter affects the muscles and nerves of the digestive system causing satiety, dysphagia, nausea and vomiting after eating, episodes of abdominal pain, diarrhea, and intestinal blockage which can lead to cachexia. Abnormalities of the nervous system are comparatively mild and may include tingling, numbness and peripheral neuropathy, particularly in the hands and feet. Deafness is common but ataxia and seizures, which are common clinical associates of other mitochondrial myopathies, are typically absent. MNGIE has high morbidity and mortality rates with death occurring in the 3rd or 4th decades of life. MNGIE is a recessively-inherited disorder caused by mutations in the nuclear-encoded TYMP gene encoding thymidine phosphorylase and is associated with PEO and ptosis [40]. Ragged-red and COX-deficient fibers are reported in muscle biopsies also show multiple mtDNA deletions [41].
Neurogenic weakness, ataxia and retinitis pigmentosa: Neurogenic weakness, Ataxia and Retinitis Pigmentosa (NARP) was described in 1990 by Holt and colleagues in a family in which 4 members showed a variable combination of developmental delay, retinitis pigmentosa, dementia, seizures, ataxia, proximal neurogenic muscle weakness, and sensory neuropathy, in a pedigree pattern consistent with maternal transmission. Biochemical assays (pyruvate and lactate) and muscle biopsy findings in affected individuals may be normal. Brain imaging, may show non-specific cerebral or cerebellar atrophy in patients with high disease burden. Although histochemical evidence of mitochondrial myopathy is absent, however, blood and muscle biopsy from the patients show the presence of heteroplasmic (m.8993 T>G) mutation in the mitochondrial ATP6 gene [42].
Leigh syndrome
Leigh syndrome (LS) is a severe neurodegenerative disorder that typically arises in the first year of life affecting roughly 1 in 40,000 live births. The onset of symptoms is often between 3 and 12 months of age, characterized by progressive loss of mental and movement abilities (psychomotor regression) and typically results in death in the first 2 years of life, usually due to respiratory failure following an acute infection. However, adult onset LS has also been described. Several ophthalmological features may occur in people with Leigh syndrome. Many affected individuals develop ophthalmoparesis; nystagmus and optic atrophy. In the biggest study conducted thus far, ophthalmological examination of 44 patients with Leigh syndrome revealed that strabismus (40.9%) was the most frequently observed ophthalmological manifestation. This was followed by pigmentary retinopathy (22.5%), optic atrophy (22.5%), ptosis (15.9%), and nystagmus (13.6%). Thirteen patients were exotropes and five patients were esotropes [43].
Ptosis could be an initial sign in patients with LS and these patients can be easily misdiagnosed as having juvenile myasthenia gravis. Ptosis could be the main presenting sign in patients with LS as in other mitochondrial disorders such as KSS or CPEO. These patients were initially misdiagnosed as having juvenile myasthenia gravis because the presentation of ptosis in LS varied during follow-up. Acquired ptosis in young children has many aetiologies, including trauma and neurologic or systemic disease, and juvenile myasthenia gravis which is not uncommon in the Asian population. The diagnosis of juvenile myasthenia gravis is solely based on clinical features because other tests such as the repetitive nerve stimulation test is not feasible in young adolescents and anti-acetylcholine receptor antibody can be negative. Therefore, LS should be included in the differential diagnosis of patients who develop ptosis or gaze-palsy, and these patients should be followed to check for developmental delay, gait disturbance, or other neurologic symptoms. The most common mtDNA mutation associated with LS and ophthalmological manifestation (including ptosis) is m.13513G>A in the MT-ND5 gene. To-date, pathogenic mutations in more than 75 genes have been reported in the mtDNA as well as nuclear DNA as reviewed elsewhere [44,45].
Autosomal dominant optic atrophy
Autosomal Dominant Optic Atrophy (ADOA) is a genetic disease primarily affecting the RGCs and nerve fiber layer of the retina with an estimated prevalence of 1 in 35,000 in Northern Europe. ADOA is clinically characterized by bilateral and symmetric optic nerve pallor associated with insidious decrease in visual acuity in the first or second decade of life, visual field defects, and color vision defects. Visual impairment varies from being moderate (6/10 to 2/10) to mild or even insignificant to severe (legal blindness with acuity <1/20). The visual field defect is typically centrocecal, central, or paracentral; it is often large in those with severe disease. Spontaneous recovery of vision has not been reported yet. Other findings can include auditory neuropathy resulting in sensorineural hearing loss with variable severity. ADOA is associated with mutations in the nuclear encoded OPA1 gene located on chromosome 3q encoding a mitochondrial-targeted protein implicated in mitochondrial membrane biogenesis and integrity [46]. A recent retrospective study found that OPA1 mutations can be detected in 50% of individuals with family history of visual failure compared to 5.3% in sporadic cases. This rate is comparable to the mutation detection rate reported in other studies [47]. Genetic counseling especially for patients with affected parents and positive OPA1 mutation is highly recommended. Generally, there is no genotype-phenotype correlation of OPA1 mutations with the severity of ADOA, but few reports indicate that in-frame deletions involve loss of visual acuity (1/10 on average) that is statistically slightly more severe than that resulting from truncation mutations or missense substitutions (2/10 on average). Management of ADOA basically involves providing low-vision aids for decreased visual acuity.
OPA1 is related with proteins of the dynamin family [48]. Mutations in this mitochondrial fusion gene causing ADOA highlight the importance of mitochondrial dynamics. Studies have proposed a number of plausible pathogenetic mechanisms. Mutations occurring in the GTPase domain of OPA1 affecting the GTPase activity have been shown to exert a dominant negative effect [49], while the C-terminal truncations caused by removal of the potential GTPase Effector Domain (GED) or deletion of entire OPA1 gene abolish the oligomerization-stimulated activity of the dynamin resulting in haploinsufficiency [50]. Although OPA1 is not exclusively expressed by the RGCs, however, the reason that RGCs are primarily affected by OPA1 mutations remains unknown. Nonetheless, the somewhat specific distribution of the OPA1 mRNA and protein variants [51] indicate a role of a specific variant of this dynamin in the pathogenesis of ADOA. Downregulation of OPA1 induces spontaneous apoptotic cell death [52], so it has been speculated that apoptosis may represent the pathophysiological process leading to the degeneration of the RGC in ADOA. Similarly, downregulated OPA1 results in impaired mitochondrial membrane potential and cellular respiration thereby lowering ATP synthesis and increasing ROS production, a mechanism similar to LHON could occur in ADOA. Studies in Opa1 mutant mouse models (reviewed in [53]) have suggested that defective fusion leads to an increased number of depolarized mitochondria, increased autophagic elimination and dendropathy [54, 55]. It is noteworthy that the phenotype in both models resembles but does not completely match the human disease, which is always characterized by loss of RGCs and optic nerve atrophy. Nevertheless, OPA1, affecting the mitochondrial dynamics and/or structural organization of the cristae, remains the primary cause of ADOA [52]; and future investigation of the functions of this dynamin is crucial to gain further insight into the pathological processes associated with this disease.
Autosomal dominant optic atrophy and cataract
Patients with Autosomal Dominant Optic Atrophy and Cataract (ADOAC) have optic atrophy, cataract, and a neurologic disorder characterized by extrapyramidal signs and ataxia. Inheritance of ADOAC is autosomal dominant; and cataract is often recognized in the first decade. ADOAC is caused by mutations in the OPA3 gene [56]. OPA3 gene is believed to provide instructions for making a protein whose exact function is unknown. The OPA3 protein is found in mitochondria and is involved in regulating the shape of mitochondria. Electron microscope studies had shown that cells without functional OPA3 protein have abnormally shaped mitochondria [57]. There are currently few mutations reported for OPA3 and thus genotype-phenotype correlation is not clear.
This is another disease caused by OPA3 mutation. Truncation mutations in the OPA3 gene are responsible for 3-methylglutaconic aciduria type 3, also called Costeff optic atrophy syndrome (COAS). COAS is a neuro-ophthalmologic syndrome consisting of early-onset bilateral optic atrophy and late-onset spasticity, extrapyramidal dysfunction, and cognitive deficit. Urinary excretion of 3-methylglutaconic acid and of 3-methglutaric acid is increased [58]. Unlike ADOAC, which is also caused by mutations in OPA3 gene, this disease is inherited in autosomal recessive manner.
PEO caused by nuclear gene defect
Autosomal dominant PEO: Autosomal dominant (AD)-PEO can be caused by mutations in one of the following nuclear genes: i) Pol-γ or POLG [59]; functions in mitochondria and DNA repair; ii) Twinkle helicase gene (PEO1) is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication [60]; iii) SLC25A4 encoding adenine nucleotide translocator 1 [61] and iv) RRM2B encoding a subunit of the p53-inducible ribonucleotide reductase protein required for maintaining balanced mitochondrial dNTP pools [62]. Other mutated genes include POLG2, encoding the accessory beta -subunit of pol γ [63] and OPA1 which encodes a dynamin-related GTPase required for mitochondrial dynamics and also mtDNA maintenance [64].
Autosomal recessive PEO: Autosomal recessive (AR)-PEO cases have been reported in pedigrees harbouring mtDNA rearrangements in skeletal muscle, and are particularly associated with POLG mutations [65]. Recessive mutations in the POLG gene also cause the Sensory ataxia neuropathy, dysarthria and ophthalmoparesis (SANDO) phenotype [66] and mitochondrial recessive ataxia syndrome (MIRAS) [67] in addition to AR-PEO. Carrozzo et al. (1998) studied three patients with AD-PEO and three patients with AR-PEO. In particular, the proportions of multiple mtDNA deletions were higher in muscle samples from patients with AD-PEO than in those from AR-PEO [68].
Pigmentary retinopathy
Pigmentary retinopathy is a non-specific finding that may be found in several mitochondrial diseases. The best described primary mtDNA disease in which pigmentary retinopathy may be seen is NARP, which results from a m.8993 T>C mutation in the mitochondrial complex V respiratory complex (ATP6 gene). Pigmentary retinopathy can also occur in a range of other mtDNA cytopathies including Leigh syndrome (degenerative disorder involving the basal ganglia and brainstem), MELAS [69], MERRF, LHON, KSS, and mitochondrial myopathy. A retrospective study identified retinal pigmentary changes in 16 of 59 children and adolescents with definite mitochondrial disease [70]. It was observed that 81% of these patients showed the presence of at least one or more other ophthalmologic findings , including ptosis, reduced eye motility including severe external ophthalmoplegia, strabismus, nystagmus, low visual acuity, refractive error, photophobia, and partial or total optic atrophy [28].These data strongly support the concept for obtaining a dilated ophthalmological examination, including electroretinogram, in individuals of all ages with suspected mitochondrial disease.
Ophthalmic diseases with secondary mitochondrial dysfunction
Glaucoma
Glaucoma is an optic neuropathy that clinically manifests with optic nerve cupping and atrophy similar to what is observed in primary mitochondrial optic neuropathies. The similarities between glaucoma and various mitochondrial optic neuropathies have attracted interest in studying mitochondrial function and abnormalities in glaucoma. The specific loss of RGCs is a common feature of mitochondrial diseases - not only the classic mitochondrial optic neuropathies of LHON, but also occurring together with more severe central nervous system involvement in many other syndromic mitochondrial diseases. The RGC, due to peculiar structural and energetic constraints, appears acutely susceptible to mitochondrial dysfunction. Mitochondrial function is also well known to decline with aging in post-mitotic tissues including neurons [71]. Because age is a risk factor for glaucoma this adds another reason to investigate mitochondria in glaucoma [72]. Various studies have demonstrated oxidative stress in ocular tissues derived from experimental glaucoma animal models and clinical samples collected from glaucoma patients [73]. The first line of evidence was related to the changes in the mitochondrial structure under the electron microscope. These changes includes: i) mitochondria surface area becomes smaller; ii) disrupted cristae or in extreme oxidative stress condition, loss of cristae; iii) fragmentation; iv) swelling; v) membrane permeability changes [74]. Glaucoma is not only a secondary disorder in mitochondrial disorder but also a primary. There is also an accumulating evidence pointing to mitochondrial abnormalities (mtDNA sequence changes, mitochondrial respiratory defects, increased mtDNA copy number or even belonging to a certain mitochondrial haplogroup) in clinical samples from glaucoma patients [72, 75-77]. In 2006, Abu-Amero et al. demonstrated potentially pathological mtDNA sequence changes in a group of patients with primary open angle glaucoma. The group also demonstrated reduction in mitochondrial respiration and increase in mtDNA copy number [72]. This was followed by other investigators reporting potentially pathological mtDNA sequence changes in clinical samples collected from glaucoma patients [78-80]. All the above studies points to the important role mitochondrial abnormalities and oxidative stress plays in glaucoma pathogenesis, however, to date the exact mechanism is not fully understood. A plausible scenario is that decreased ATP production resulting from dysfunctional mitochondria affects the intraocular pressure (as recently demonstrated in the optic nerve of mice with glaucoma [74]) and triggers the glaucoma pathogenesis cascade. Another possible scenario involves the disruption of the trabecular meshwork (TM) during glaucoma development. Oxidative stress and subsequent vascular damage may play a role in initiating the apoptotic cell loss in the TM and the optic nerve head [81] may conceivably be amenable to early initiation of antioxidant therapy.
Non-arteritic anterior ischemic optic neuropathy
Patients with NAION typically show some or all of the signs of an optic neuropathy including decreased visual acuity, dyschromatopsia, a relative afferent papillary defect, a swollen optic nerve with splinter hemorrhages and a visual field defect. Therefore, NAION clinically resemble LHON, but the only difference is that it occurs in old age (>50 years) and has associated optic disc edema. The cause of the optic disc edema is unclear but there is general agreement that the final common pathway leads to a compartment syndrome from axonal edema in a structurally crowded optic disc resulting in apoptosis induced retinal ganglion cell death [82]. Unlike LHON, the genetic underlying cause for NAION is largely unknown. Abu-Amero et al. reported a few potentially pathogenic mtDNA mutations and increased relative mtDNA content, indirect response of mitochondria to oxidative stress, in a group of patients with NAION [21, 83]. In addition, a known pathologically established mtDNA mutation m.9957 T>C was reported in a patient with NAION and seizure [84]. Another mtDNA mutation m.4132 G>A was also reported in one large pedigree of familial NAION [85]. Apart from these studies, to date, the genetic underlying cause of NAION remains largely unknown. The exact mechanism of how mitochondrial DNA mutations (if established) develop NAION remains unknown. Because of similarities between the pathology of LHON and NAION, it is plausible to think that oxidative stress and subsequent degeneration of RGCs may be involved. Because NAION occurs at an old age, it further strengthens the link between NAION and mitochondrial pathology [86].
Diabetic retinopathy
DR is the leading cause of blindness in young adults. Increase in oxidative stress in diabetes and its role in the development of diabetic complications are now well accepted [87, 88]. Molecular mechanisms implicated in the induction of oxidative stress include auto-oxidation of glucose, impaired antioxidant defense enzymes and metabolic abnormalities initiated by hyperglycemia [87]. In addition, since diabetes is a life-long disease, cells are exposed to continuous metabolic stress and increased free radical generation resulting in tissue damage. However, the exact mechanisms leading to oxidative stress and development of DR remains to be elucidated. It is believed that DR involves progressive dysfunction of retinal mitochondria in the setting of hyperglycemia resulting in mtDNA damage and increased apoptosis of the retinal capillary cells [89]. Matrix metalloproteinase-2 (MMP2) is a major protein mediating the pro-apoptotic process in diabetic retinal cells [79]. Activated MMP2 causes mitochondrial membrane degradation and damage to connexin 43, which activates the apoptotic machinery [90]. Antioxidants, such as manganese superoxide dismutase, reduce MMP2-mediated mitochondrial damage inhibiting the development of DR [89]. In a similar study, antioxidant agent, SS31, was found to attenuate glucose-induced injury in human diabetic retinal cells, where significantly decreased mitochondrial ROS generations, decreased cell destruction, and reduced cytochrome-c release (mark the initiation of apoptosis) was seen following treatment of cells in high glucose media with SS31 [53]. These important findings underscore the potential reversibility of oxidative-stress mediated retinal damage in diabetes mellitus.
Age-related macular degeneration
Retinal degeneration, particularly including AMD, is responsible for a large proportion of blindness in the elderly population. Light appears to have a deleterious effect on retinal cells that already have compromised mitochondrial function. Wavelengths of light ranging from 400 to 760 nm appear to specifically affect tissues that are replete with mitochondria by reducing the activity of mitochondrial dehydrogenases and increasing the release of ROS [91]. Since retinal ganglion cells are not protected by macular pigments from short wavelengths of light, they are particularly vulnerable to light-induced damage. Therefore, diseases of the retinal ganglion cells, such as AMD, may be exacerbated by light-induced mitochondrial dysfunction [91]. To determine whether pathogenic mtDNA variants also occur in patients with AMD, Kenney and colleagues compared retinal and blood mtDNA between AMD patients and age matched controls [92]. The authors reported that the retinal cells had more mtDNA rearrangements and deletions than blood, with a greater number of non-synonymous gene variants of potential pathogenic significance occurring in AMD patients. These mtDNA genome alterations seem to accumulate over time in the diseased retinal ganglion cells both as a consequence from, and likely ongoing cause of oxidative stress that exacerbates mitochondrial dysfunction in the retina. With normal aging, mtDNA damage is observed only in the common deletion region of the mitochondrial genome known to be associated with aging. In contrast, mtDNA damage is increased with AMD progression to the entire mitochondrial genome and not limited only to the common deletion region [93]. Previous studies have identified mtDNA as a site of damage that is preferentially affected during the course of AMD and also established that these changes occur by the intermediate stage of the disease. This is consistent with the idea that increased mtDNA damage could be one factor leading to retinal pigment epithelium (RPE) dysfunction [94]. ROS generated under normal conditions, such as light (especially short-wavelength blue light), phagocytosis and metabolism, place the RPE under considerable metabolic demand and oxidative stress. Impaired RPE can cause imbalance in signaling factors (i.e., increased vascular endothelial growth factor) and apoptosis resulting in end-stage AMD. Thus, protecting mtDNA integrity via therapeutics targeted to the mitochondria early in AMD could stop or ameliorate the progression to vision loss [93].
Ocular neoplasm
Ocular cancers, although rare, can be particularly aggressive, with significant morbidity and mortality. While the origin of these cancers is not typically considered to be mitochondrial, some recent investigational therapies have been targeted at disruption of mitochondrial function in tumor cells to specifically induce their apoptosis. Two such cancers studied in the past year were retinoblastoma and uveal melanoma.
Retinoblastoma: Retinoblastoma is the most common form of ocular cancer in young children. Although potentially metastatic if left untreated, retinoblastoma has the highest cure rate of an estimated 95% in the United States [95]. A novel isoquinoline derivative, EDL-155, was recently evaluated as a potential agent to selectively destroy retinoblastoma cells [96]. In vitro treatment resulted in mitochondrial disruption and induction of autophagy. In addition, the authors demonstrated in vivo treatment efficacy using a rat retinoblastoma model in which EDL-155 suggested localized tumor cell destruction after four periocular injections [96] suggesting a role for mitochondria in this tumor. Most retinoblastoma cases are known to be caused by mutations in the RB1 (retinoblastoma-1) gene, which is a tumor-suppressor gene [97]. Mode of inheritance for most retinoblastoma cases is autosomal dominant.
Uveal melanoma: Uveal melanoma, a primary intraocular tumor in adults, is a very rare and aggressive cancer associated with a poor prognosis, where up to half of patients develop liver metastases within 15 years of diagnosis [98]. A recent in vitro study of the effects of curcumin on uveal melanoma cells demonstrated induction of apoptosis [98]. Curcumin is a plant-derived polyphenol antioxidant and anti-inflammatory compound found in the spice turmeric, which prior studies have suggested has an in vitro apoptotic effect on cancers ranging from leukemia to solid tumors such as prostate and ovarian cancers [98, 99]. The viability of uveal melanoma cells was significantly decreased in a dose- and time-dependent manner following curcumin administration, where mitochondrial-induced apoptosis and cell destruction was progressively increased with greater curcumin concentrations. Therapies directed at inhibiting tumor-specific mitochondrial function thus hold promise as a novel means to treat ocular cancers. In addition, germline mutation in the BAP1 (BRCA1 Associated Protein-1) gene has also been associated with uveal melanoma [100].
Gene therapy
Despite the progress made in identifying the genetic causes and diagnosis of mitochondrial diseases, major improvements in the development of treatment are still lacking and required urgently. Currently, there are no effective treatment for mitochondrial group of diseases caused by OXPHOS dysfunction, which are largely restricted to management of symptoms and use of vitamins and antioxidant cocktails adjusted to individual patient needs [101]. Due to the dual genetic origin of the mitochondria, defects in both the nuclear and mitochondrial genome, in principle, could be targeted by directed gene replacement and gene therapy. However, as compared to its nuclear counter-part, development of animal models with mtDNA-based disorders is technically challenging. Notwithstanding these technical challenges, animal models of mtDNA-based optic neuropathy has been developed using the classic complex I inhibitor rotenone or rotenone-loaded microspheres [102]. However, characterization of ocular manifestations in these animals has still been limited. Transgenic studies of the nuclear encoded mitochondrial ATP/ADP translocator (ANT1) knock-out mice demonstrated the feasibility of gene therapy in mitochondrial associated disorders [103]. Recent review by Tischner and Wenz has very elegantly described the numerous gene targeted approaches and their outcomes (reviewed in [104]). The most commonly studied gene therapy approach includes the nuclear allotopic expression of mtDNA-encoded genes in LHON. The delivery of an adeno-associated virus (AAV) vector containing the wild-type ND4 to provide the rescue of RGCs in the ex vivo human eye and mouse model, provides the proof-of-principle of the LHON gene therapy approach [27, 105]. Results of human clinical trial using this approach are pending [106]. In addition, using a different approach, successful direct delivery within mitochondria of the whole mtDNA or of an AAV vector containing the mtDNA-encoded ND4 subunit gene have also been demonstrated [107, 108]. Despite certain critical limitation regarding the unclear mechanism(s) underlying the mitochondrial delivery of mtDNA or AAV construct containing mtDNA-encoded genes, these studies contradict the conventional wisdom demonstrating that mtDNA can be directly delivered to mitochondria and is a very important technical achievement in the field of mitochondrial medicine that needs further investigation. It is noteworthy that a true genetic mouse model of LHON has been established. The model exhibits the presence of a homoplasmic mtDNA complex I mutation in all tissues, biochemical and histological hallmarks of this disease and should provide a platform for further investigations that may shed light on certain unexplained features of human LHON and allow for the proper testing of potential therapies [109]. Replacement gene therapy, an approach successfully used in human inherited retinopathies, may also have future application in human optic nerve disorders caused by nuclear genetic defects such as ADOA with OPA1 mutations. Considering the heterogeneous nature of mitochondrial diseases and the clinical variability observed in these patients with OXPHOS dysfunction it is highly likely that future therapeutic strategies would involve more of a personalized treatment approach.
Conclusion
It is clearly evident that mitochondrial dysfunction is intimately linked to many aspects of ophthalmologic diseases. Besides, primary mitochondrial disorders distinctly exhibit major ophthalmologic sequelae (summarized in Table 1). Similarly, mitochondrial dysfunction, perhaps through induction of oxidative stress mechanism(s) and disruption of mitochondrial function in tumor cells seems to contribute to primary ophthalmic diseases such as glaucoma, DR, AMD and ocular tumors, respectively. As mitochondria play an important role in the pathogenesis of various ophthalmic diseases, multiple strategies targeting mitochondrial protection against oxidative stress and premature apoptosis should be devised to protect RGCs. This might provide a promising way to delay the onset of various ophthalmic diseases, decrease severity or prevent them all together.
Acknowledgments
This Project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia, Award Number (12-MED2621-02)
References
- Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, et al. (2015) Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med 17: 689-701. [Crossref]
- Wallace DC (1999) Mitochondrial diseases in man and mouse. Science 283: 1482-1488. [Crossref]
- Tower J (2015) Programmed cell death in aging. Ageing Res Rev 23: 90-100. [Crossref]
- Finsterer J, Zarrouk-Mahjoub S2, Daruich A3 (2016) The Eye on Mitochondrial Disorders. J Child Neurol 31: 652-662. [Crossref]
- Schrier SA, Falk MJ (2011) Mitochondrial disorders and the eye. Curr Opin Ophthalmol 22: 325-331. [Crossref]
- Sherer TB, Betarbet R, Greenamyre JT (2002) Environment, mitochondria, and Parkinson's disease. Neuroscientist 8: 192-197. [Crossref]
- Wilkins HM, Swerdlow RH1 (2016) Relationships Between Mitochondria and Neuroinflammation: Implications for Alzheimer's Disease. Curr Top Med Chem 16: 849-857. [Crossref]
- Lionaki E, Markaki M, Palikaras K, Tavernarakis N (2015) Mitochondria, autophagy and age-associated neurodegenerative diseases: New insights into a complex interplay. Biochim Biophys Acta 1847: 1412-1423. [Crossref]
- Lotz C, Lin AJ, Black CM, Zhang J, Lau E, et al. (2014) Characterization, design, and function of the mitochondrial proteome: from organs to organisms. J Proteome Res 13: 433-446. [Crossref]
- Chinnery PF, Hudson G (2013) Mitochondrial genetics. Br Med Bull 106: 135-159. [Crossref]
- Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, et al. (2013) mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr Protoc Bioinformatics 44: 1. [Crossref]
- Calvo SE, Clauser KR, Mootha VK (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44: D1251-1257. [Crossref]
- Wolf NI, Smeitink JA (2002) Mitochondrial disorders: a proposal for consensus diagnostic criteria in infants and children. Neurology 59: 1402-1405. [Crossref]
- Abu-Amero KK1, Bosley TM (2006) Mitochondrial abnormalities in patients with LHON-like optic neuropathies. Invest Ophthalmol Vis Sci 47: 4211-4220. [Crossref]
- Abu-Amero KK, Ozand PT, Al-Dhalaan H (2006) Novel mitochondrial DNA transversion mutation in transfer ribonucleic acid for leucine 2 (CUN) in a patient with the clinical features of MELAS. J Child Neurol 21: 971-972. [Crossref]
- Cui H, Dhroso A, Johnson N, Korkin D (2015) The variation game: Cracking complex genetic disorders with NGS and omics data. Methods 79-80: 18-31. [Crossref]
- Thiselton DL, Alexander C, Taanman JW, Brooks S, Rosenberg T, et al. (2002) A comprehensive survey of mutations in the OPA1 gene in patients with autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci 43: 1715-1724. [Crossref]
- Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, et al. (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy 'plus' phenotypes. Brain 131: 338-351. [Crossref]
- Howell N (2003) LHON and other optic nerve atrophies: the mitochondrial connection. Dev Ophthalmol 37: 94-108. [Crossref]
- Pisano A, Preziuso C, Iommarini L, Perli E, Grazioli P, et al. (2015) Targeting estrogen receptor beta as preventive therapeutic strategy for Leber's hereditary optic neuropathy. Hum Mol Genet 24: 6921-6931. [Crossref]
- Bosley TM, Abu-Amero KK, Ozand PT (2004) Mitochondrial DNA nucleotide changes in non-arteritic ischemic optic neuropathy. Neurology 63: 1305-1308. [Crossref]
- Meyerson C, Van Stavern G1, McClelland C1 (2015) Leber hereditary optic neuropathy: current perspectives. Clin Ophthalmol 9: 1165-1176. [Crossref]
- Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, et al. (2011) A randomized placebo-controlled trial of idebenone in Leber's hereditary optic neuropathy. Brain 134: 2677-2686. [Crossref]
- Carelli V, La Morgia C, Valentino ML, Rizzo G, Carbonelli M, et al. (2011) Idebenone treatment in Leber's hereditary optic neuropathy. Brain 134: e188. [Crossref]
- Klopstock T, Metz G, Yu-Wai-Man P, Büchner B, Gallenmüller C, et al. (2013) Persistence of the treatment effect of idebenone in Leber's hereditary optic neuropathy. Brain 136: e230. [Crossref]
- Lam BL, Feuer WJ, Abukhalil F, Porciatti V, Hauswirth WW, et al. (2010) Leber hereditary optic neuropathy gene therapy clinical trial recruitment: year 1. Arch Ophthalmol 128: 1129-1135. [Crossref]
2021 Copyright OAT. All rights reserv
- Ellouze S, Augustin S, Bouaita A, Bonnet C, Simonutti M, et al. (2008) Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet 83: 373-387. [Crossref]
- Nikoskelainen EK, Marttila RJ, Huoponen K, Juvonen V, Lamminen T, et al. (1995) Leber's "plus": neurological abnormalities in patients with Leber's hereditary optic neuropathy. J Neurol Neurosurg Psychiatry 59: 160-164. [Crossref]
- Abu-Amero KK, Bosley TM, Bohlega S, McLean D (2005) Complex I respiratory defect in LHON plus dystonia with no mitochondrial DNA mutation. Br J Ophthalmol 89: 1380-1381. [Crossref]
- Bau V, Deschauer M, Zierz S (2009) [Chronic progressive external ophthalmoplegia--symptom or syndrome?]. Klin Monbl Augenheilkd 226: 822-828. [Crossref]
- DiMauro S, Hirano M (2003) Mitochondrial DNA Deletion Syndromes. [Crossref]
- Hudson G, Chinnery PF (2006) Mitochondrial DNA polymerase-gamma and human disease. Hum Mol Genet 15 Spec No 2: R244-252. [Crossref]
- Drachman DA (1968) Ophthalmoplegia plus. The neurodegenerative disorders associated with progressive external ophthalmoplegia. Arch Neurol 18: 654-674. [Crossref]
- Kearns TP,Sayre GP (1958) Retinitis pigmentosa, external ophthalmophegia, and complete heart block: unusual syndrome with histologic study in one of two cases. AMA Arch Ophthalmol 60: 280-289. [Crossref]
- Rowland LP, Hausmanowa-Petrusewicz I, Bardurska B, Warburton D, Nibroj-Dobosz I, et al. (1988) Kearns-Sayre syndrome in twins: lethal dominant mutation or acquired disease? Neurology 38: 1399-1402. [Crossref]
- Gorman GS, Taylor RW (2011) Mitochondrial DNA abnormalities in ophthalmological disease. Saudi J Ophthalmol 25: 395-404. [Crossref]
- Thambisetty M, Newman NJ, Glass JD, Frankel MR (2002) A practical approach to the diagnosis and management of MELAS: case report and review. Neurologist 8: 302-312. [Crossref]
- Wallace DC1, Zheng XX, Lott MT, Shoffner JM, Hodge JA, et al. (1988) Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 55: 601-610. [Crossref]
- Verma A, Moraes CT, Shebert RT, Bradley WG (1996) A MERRF/PEO overlap syndrome associated with the mitochondrial DNA 3243 mutation. Neurology 46: 1334-1336. [Crossref]
- Bardosi A1, Creutzfeldt W, DiMauro S, Felgenhauer K, Friede RL, et al. (1987) Myo-, neuro-, gastrointestinal encephalopathy (MNGIE syndrome) due to partial deficiency of cytochrome-c-oxidase. A new mitochondrial multisystem disorder. Acta Neuropathol 74: 248-258. [Crossref]
- Nishigaki Y, Marti R, Hirano M (2004) ND5 is a hot-spot for multiple atypical mitochondrial DNA deletions in mitochondrial neurogastrointestinal encephalomyopathy. Hum Mol Genet 13: 91-101. [Crossref]
- Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA (1990) A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet 46: 428-433. [Crossref]
- Han J, Lee YM, Kim SM, Han SY, Lee JB, et al. (2015) Ophthalmological manifestations in patients with Leigh syndrome. Br J Ophthalmol 99: 528-535. [Crossref]
- Lake NJ, Compton AG, Rahman S, Thorburn DR (2016) Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol 79: 190-203. [Crossref]
- Gerards M, Sallevelt SC, Smeets HJ (2016) Leigh syndrome: Resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol Genet Metab 117: 300-312. [Crossref]
- Lenaers G, Hamel C, Delettre C, Amati-Bonneau P, Procaccio V, et al. (2012) Dominant optic atrophy. Orphanet J Rare Dis 7: 46. [Crossref]
- Bonifert T, Karle KN, Tonagel F, Batra M, Wilhelm C, et al. (2014) Pure and syndromic optic atrophy explained by deep intronic OPA1 mutations and an intralocus modifier. Brain 137: 2164-2177. [Crossref]
- Olichon A, Guillou E, Delettre C, Landes T, Arnauné-Pelloquin L, et al. (2006) Mitochondrial dynamics and disease, OPA1. Biochim Biophys Acta 1763: 500-509. [Crossref]
- Marks B, Stowell MH, Vallis Y, Mills IG, Gibson A, et al. (2001) GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 410: 231-235. [Crossref]
- Marchbank NJ, Craig JE, Leek JP, Toohey M, Churchill A, et al. (2002) Deletion of the OPA1 gene in a dominant optic atrophy family: evidence that haploinsufficiency is the cause of disease. J Med Genet 39: e47. [Crossref]
- Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, et al. (2002) The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett 523: 171-176. [Crossref]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, et al. (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278: 7743-7746. [Crossref]
- Williams PA1, Morgan JE, Votruba M (2011) Mouse models of dominant optic atrophy: what do they tell us about the pathophysiology of visual loss? Vision Res 51: 229-234. [Crossref]
- White KE, Davies VJ, Hogan VE, Piechota MJ, Nichols PP, et al. (2009) OPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Invest Ophthalmol Vis Sci 50: 2567-2571. [Crossref]
- Williams PA, Piechota M, von Ruhland C, Taylor E, Morgan JE, et al. (2012) Opa1 is essential for retinal ganglion cell synaptic architecture and connectivity. Brain 135: 493-505. [Crossref]
- Reynier P, Amati-Bonneau P, Verny C, Olichon A, Simard G, et al. (2004) OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. Journal of Medical Genetics 41: e110. [Crossref]
- Ayrignac X, Liauzun C, Lenaers G, Renard D, Amati-Bonneau P, et al. (2012) OPA3--related autosomal dominant optic atrophy and cataract with ataxia and areflexia. Eur Neurol 68: 108-110. [Crossref]
- Ho G, Walter JH, Christodoulou J (2008) Costeff optic atrophy syndrome: new clinical case and novel molecular findings. J Inherit Metab Dis 31 Suppl 2: S419-423. [Crossref]
- Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C (2001) Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 28: 211-212. [Crossref]
- Milenkovic D1, Matic S, Kühl I, Ruzzenente B, Freyer C, et al. (2013) TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum Mol Genet 22: 1983-1993. [Crossref]
- Körver-Keularts IM, de Visser M, Bakker HD, Wanders RJ, Vansenne F, et al. (2015) Two Novel Mutations in the SLC25A4 Gene in a Patient with Mitochondrial Myopathy. JIMD Rep 22: 39-45. [Crossref]
- Tyynismaa H, Ylikallio E, Patel M, Molnar MJ, Haller RG, et al. (2009) A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions. Am J Hum Genet 85: 290-295. [Crossref]
- Graziewicz MA, Longley MJ, Copeland WC (2006) DNA polymerase gamma in mitochondrial DNA replication and repair. Chem Rev 106: 383-405. [Crossref]
- Hudson G, Amati-Bonneau P, Blakely EL, Stewart JD, He L, et al. (2008) Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain: a Journal of Neurology 131: 329-337. [Crossref]
- Chan SS, Copeland WC (2009) DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta 1787: 312-319. [Crossref]
- Umehara F, Kore-Eda Y, Arime T, Kubota R, Arimura K, et al. (1997) Chronic sensory ataxic neuropathy and ophthalmoplegia with oculomotor nerve hypertrophy associated with IgM antibodies against gangliosides containing disialosyl groups. J Neurol Neurosurg Psychiatry 62: 673-674. [Crossref]
- Hudson G, Deschauer M, Busse K, Zierz S, Chinnery PF (2005) Sensory ataxic neuropathy due to a novel C10Orf2 mutation with probable germline mosaicism. Neurology 64: 371-373. [Crossref]
- Carrozzo R, Hirano M, Fromenty B, Casali C, Santorelli FM, et al. (1998) Multiple mtDNA deletions features in autosomal dominant and recessive diseases suggest distinct pathogeneses. Neurology 50: 99-106. [Crossref]
- Latvala T, Mustonen E, Uusitalo R, Majamaa K (2002) Pigmentary retinopathy in patients with the MELAS mutation 3243A-->G in mitochondrial DNA. Graefes Arch Clin Exp Ophthalmol 240: 795-801. [Crossref]
- Grönlund MA, Honarvar AK, Andersson S, Moslemi AR, Oldfors A, et al. (2010) Ophthalmological findings in children and young adults with genetically verified mitochondrial disease. Br J Ophthalmol 94: 121-127. [Crossref]
- Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT (2010) Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 12: 503-535. [Crossref]
- Abu-Amero KK, Morales J, Bosley TM (2006) Mitochondrial abnormalities in patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci 47: 2533-2541. [Crossref]
- McElnea EM, Quill B, Docherty NG, Irnaten M, Siah WF, et al. (2011) Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol Vis 17: 1182-1191. [Crossref]
- Coughlin L, Morrison RS, Horner PJ, Inman DM (2015) Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Invest Ophthalmol Vis Sci 56: 1437-1446. [Crossref]
- Abu-Amero KK, Gonzalez AM, Osman EA, Larruga JM, Cabrera VM, et al. (2011) Mitochondrial DNA lineages of African origin confer susceptibility to primary open-angle glaucoma in Saudi patients. Mol Vis 17: 1468-1472. [Crossref]
- Abu-Amero KK, Morales J, Bosley TM, Mohamed GH, Cabrera VM (2008) The role of mitochondrial haplogroups in glaucoma: a study in an Arab population. Mol Vis 14: 518-522. [Crossref]
- Abu-Amero KK, Bosley TM, Morales J (2008) Analysis of nuclear and mitochondrial genes in patients with pseudoexfoliation glaucoma. Mol Vis 14: 29-36. [Crossref]
- Jeoung JW, Seong MW, Park SS, Kim DM, Kim SH, et al. (2014) Mitochondrial DNA variant discovery in normal-tension glaucoma patients by next-generation sequencing. Invest Ophthalmol Vis Sci 55: 986-992. [Crossref]
- Kumar M, Tanwar M, Faiq MA, Pani J, Shamsi MB, et al. (2013) Mitochondrial DNA nucleotide changes in primary congenital glaucoma patients. Mol Vis 19: 220-230. [Crossref]
- Tanwar M, Dada T, Sihota R, Dada R (2010) Mitochondrial DNA analysis in primary congenital glaucoma. Mol Vis 16: 518-533. [Crossref]
- Saccà SC, Pulliero A, Izzotti A (2015) The dysfunction of the trabecular meshwork during glaucoma course. J Cell Physiol 230: 510-525. [Crossref]
- Beck RW, Servais GE, Hayreh SS (1987) Anterior ischemic optic neuropathy. IX. Cup-to-disc ratio and its role in pathogenesis. Ophthalmology 94: 1503-1508. [Crossref]
- Abu-Amero KK, Bosley TM (2006) Increased relative mitochondrial DNA content in leucocytes of patients with NAION. Br J Ophthalmol 90: 823-825. [Crossref]
- Abu-Amero KK, Bosley TM, Bohlega S, Hansen E (2005) Mitochondrial T9957C mutation in association with NAION and seizures but not MELAS. Ophthalmic Genet 26: 31-36. [Crossref]
- Fingert JH, Grassi MA, Janutka JC, East JS, Howard JG, et al. (2007) Mitochondrial variant G4132A is associated with familial non-arteritic anterior ischemic optic neuropathy in one large pedigree. Ophthalmic Genet 28: 1-7. [Crossref]
- McFarland R, Turnbull DM (2009) Batteries not included: diagnosis and management of mitochondrial disease. J Intern Med 265: 210-228. [Crossref]
- Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54: 1615-1625. [Crossref]
- Kowluru RA, Mishra M (2015) Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim Biophys Acta 1852: 2474-2483. [Crossref]
- Mohammad G, Kowluru RA (2010) Matrix metalloproteinase-2 in the development of diabetic retinopathy and mitochondrial dysfunction. Lab Invest 90: 1365-1372. [Crossref]
- Mohammad G, Kowluru RA (2011) Novel role of mitochondrial matrix metalloproteinase-2 in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci 52: 3832-3841. [Crossref]
- Osborne NN, Kamalden TA, Majid AS, del Olmo-Aguado S, Manso AG, et al. (2010) Light effects on mitochondrial photosensitizers in relation to retinal degeneration. Neurochem Res 35: 2027-2034. [Crossref]
- Kenney MC, Atilano SR, Boyer D, Chwa M, Chak G, et al. (2010) Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Invest Ophthalmol Vis Sci 51: 4289-4297. [Crossref]
- Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA (2010) Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci 51: 5470-5479. [Crossref]
- Lin H, Xu H, Liang FQ, Liang H, Gupta P, et al. (2011) Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci 52: 3521-3529. [Crossref]
- Broaddus E, Topham A, Singh AD (2009) Survival with retinoblastoma in the USA: 1975-2004. Br J Ophthalmol 93: 24-27. [Crossref]
- Nassr M, Wang X, Mitra S, Freeman-Anderson NE, Patil R, et al. (2010) Treating retinoblastoma in tissue culture and in a rat model with a novel isoquinoline derivative. Invest Ophthalmol Vis Sci 51: 3813-3819. [Crossref]
- Zhang K, Nowak I, Rushlow D, Gallie BL, Lohmann DR (2008) Patterns of missplicing caused by RB1 gene mutations in patients with retinoblastoma and association with phenotypic expression. Hum Mutat 29: 475-484. [Crossref]
- Lu C, Song E, Hu DN, Chen M, Xue C, et al. (2010) Curcumin induces cell death in human uveal melanoma cells through mitochondrial pathway. Curr Eye Res 35: 352-360. [Crossref]
- Aggarwal BB1, Kumar A, Bharti AC (2003) Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res 23: 363-398. [Crossref]
- Cebulla CM, Binkley EM, Pilarski R, Massengill JB, Rai K, et al. (2015) Analysis of BAP1 Germline Gene Mutation in Young Uveal Melanoma Patients. Ophthalmic Genet 36: 126-131. [Crossref]
- Avula S, Parikh S, Demarest S, Kurz J, Gropman A (2014) Treatment of mitochondrial disorders. Curr Treat Options Neurol 16: 292. [Crossref]
- Biousse V, Pardue MT, Wallace DC, Newman NJ (2002) The eyes of mito-mouse: mouse models of mitochondrial disease. J Neuroophthalmol 22: 279-285. [Crossref]
- Flierl A, Chen Y, Coskun PE, Samulski RJ, Wallace DC (2005) Adeno-associated virus-mediated gene transfer of the heart/muscle adenine nucleotide translocator (ANT) in mouse. Gene Ther 12: 570-578. [Crossref]
- Tischner C, Wenz T (2015) Keep the fire burning: Current avenues in the quest of treating mitochondrial disorders. Mitochondrion 24: 32-49. [Crossref]
- Koilkonda RD, Yu H, Chou TH, Feuer WJ, Ruggeri M, et al. (2014) Safety and effects of the vector for the Leber hereditary optic neuropathy gene therapy clinical trial. JAMA Ophthalmol 132: 409-420. [Crossref]
- Lam BL, Feuer WJ, Schiffman JC, Porciatti V, Vandenbroucke R, et al. (2014) Trial end points and natural history in patients with G11778A Leber hereditary optic neuropathy: preparation for gene therapy clinical trial. JAMA Ophthalmol 132: 428-436. [Crossref]
- Iyer S, Bergquist K, Young K, Gnaiger E, Rao RR, et al. (2012) Mitochondrial gene therapy improves respiration, biogenesis, and transcription in G11778A Leber's hereditary optic neuropathy and T8993G Leigh's syndrome cells. Hum Gene Ther 23: 647-657. [Crossref]
- Yu H, Koilkonda RD, Chou TH, Porciatti V, Ozdemir SS, et al. (2012) Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber's hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci USA 109: E1238-1247. [Crossref]
- Lin CS, Sharpley MS, Fan W, Waymire KG, Sadun AA, et al. (2012) Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc Natl Acad Sci USA 109: 20065-20070. [Crossref]