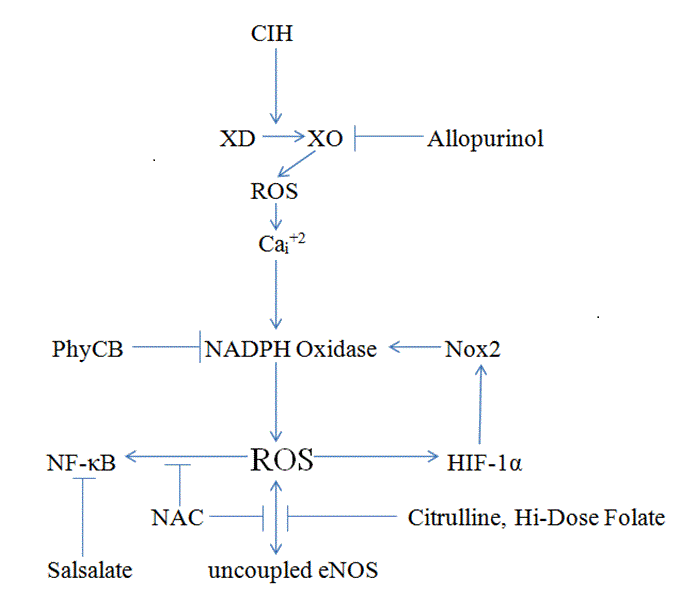
Obstructive sleep apnea (OSA) markedly increases risk for atherosclerosis, systemic and pulmonary hypertension, ventricular hypertrophy, myocardial infarction, stroke, and sudden-death arrhythmias; it also has adverse psychological effects, including daytime somnolence. Clinical and rodent studies indicate that oxidative stress and inflammation underlie the pathogenesis of these complications. NADPH oxidase complexes have emerged as a key source of this oxidative stress, which is amplified in vascular endothelium by uncoupled nitric oxide synthase (eNOS). Proteolytic conversion of xanthine dehydrogenase to xanthine oxidase triggered by hypoxia appears to be upstream from NADPH oxidase activation during intermittent hypoxia. Activation of NF-kappaB downstream from this oxidative stress promotes the inflammation associated with OSA. These considerations suggest therapeutic avenues that, when OSA cannot be fully controlled with CPAP, may lessen the pathogenic impact of this syndrome. The elevated NADPH oxidase activity in OSA might be addressed with spirulina, whose chromophore phycocyanobilin can potently inhibit NADPH oxidases in a manner analogous to bilirubin. Xanthine oxidase inhibitor allopurinol may suppress NADPH oxidase activation in OSA, and has been shown to improve endothelial function in OSA patients. The adverse impact of oxidative stress might be antagonized to some extent by administration of N-acetylcysteine, a glutathione precursor. The uncoupling of eNOS – both a cause and a consequence of oxidative stress – could be combatted with supplemental citrulline and high-dose folate. Docosahexaenoic acid could be of potential benefit by promoting expression of phase 2 antioxidant enzymes. In patients who tolerate it adequately, moderate-dose salsalate could be employed to down-regulate canonical NF-kappaB activation.
Adverse health consequences of obstructive sleep apnea
Epidemiological studies reveal that, independent of associated risk factors, obstructive sleep apnea (OSA) increases risk for heart attack and stroke, systemic hypertension associated with sympathetic activation, pulmonary hypertension, ventricular hypertrophy and heart failure, arrhythmias, erectile dysfunction, and sudden death [1-9]. Patients with OSA have an impairment of flow-mediated vasodilation, and increased circulating markers of endothelial activation, that improve with CPAP (chronic positive airway pressure) therapy [10-13]. When rodents are subjected to chronic intermittent hypoxia, in a manner intended to mimic the episodic hypoxia of OSA, systemic and pulmonary hypertension, ventricular hypertrophy, endothelial dysfunction, and an exacerbation of diet-induced atherosclerosis can be observed [14-18]. Cardiac ischemia-reperfusion produces greater infarct sizes in rats previously subjected to chronic intermittent hypoxia [19]. OSA also has adverse psychological effects; OSA patients tend to experience daytime hypersomnolence and are prone to depression, and rodents subjected to intermittent hypoxia experience a deficit in hippocampus-dependent spatial learning and increases in anxiety responses [20-25].
There is general agreement, based on clinical studies of OSA patients, as well as rodent studies of chronic intermittent hypoxia, that OSA is attended by an increase in oxidative stress and inflammation in a range of organs, including the brain [15,25-28] Clinically, studies comparing OSA patients with matched controls, or comparing patients whose OSA was of greater or lesser severity, have documented increased markers of oxidative stress and/or inflammation associated with significant OSA. The markers found to be higher in OSA included TBARs in LDL particles, lag phase of LDL oxidation ex vivo, paraoxonase level, superoxide production by neutrophils stimulated ex vivo, urinary excretion of 8-hydroxydeoxyguanosine and 8-isoprostane, exhalation of 8-isoprostane, plasma levels of C-reactive protein and adhesion molecules (ICAM-1, VCAM-1, selectin, CD15, CD11c), and diacron reactive oxygen metabolites [29-38]. Conversely, the antioxidant capacity of whole blood (trolox equivalent antioxidant capacity) was found to be decreased relative to controls in OSA patients [39]. In most of these studies, markers of oxidative stress and inflammation were reported to decline significantly following a course of CPAP therapy.
Considerable evidence points to NADPH oxidase complexes as a key source of the oxidative stress underlying OSA, and to increased activation of NF-kappaB as a mediator of the associated inflammation. Additionally, in vascular endothelium, uncoupling of the endothelial nitric oxide synthase (eNOS) contributes both to oxidative stress and a loss of protective NO bioactivity.40, 41
NADPH oxidase and uncoupled eNOS are key mediators of the pathogenic impact of OSA
The central role of NADPH oxidase as a mediator of many pathogenic effects of chronic intermittent hypoxia (CIH) clearly emerges in rodent studies. Mice that are genetically deficient in gp91phox (a.k.a. Nox2) are protected from the pulmonary hypertension, systemic hypertension, ventricular remodeling, hypersomnolence, cognitive deficits, and anxiety produced by CIH in wild-type mice [17,24,42-45]. Analogously, treatment with apocynin, an inhibitor of NADPH oxidase complexes, protects rodents from the systemic hypertension, erectile dysfunction, hypersomnolence, and brain oxidative damage provoked by CIH [16,42,45,46]. Increased expression of various subunits of NADPH oxidase has been observed in the tissues of rodents exposed to CIH, and increased levels of p22phox mRNA in macrophages and neutrophils derived from morning sputum – as contrasted to sputum collected before sleep – were observed in patients with untreated OSA [10,17,19,47,48].
How NADPH oxidase is activated by intermittent hypoxia is not yet entirely clear. The mechanism whereby Nox2-dependent NADPH oxidase activity is increased by intermittent hypoxia in the carotid body – an effect which is key to sympathetic activation and systemic hypertension in CIH – has been studied in PC12 cells [49]. Proteolytic conversion of xanthine dehydrogenase to xanthine oxidase appears to be upstream from NADPH oxidase activation [50]. This is mediated by proteolytic activity, evoked by hypoxia, that is suppressed by a trypsin inhibitor [51]. The resultant initial production of superoxide by xanthine oxidase, provided with purine catabolites owing to hypoxic dephosphorylation of adenosine nucleotides, increases intracellular free calcium, and this in turn promotes activation of protein kinase C activities, leading to membrane assemblage of intact and active NADPH oxidase complexes [50]. The greater level of superoxide production associated with NADPH oxidase activation boosts expression of hypoxia-inducible factor-1alpha (HIF-1alpha) by inhibiting prolyl hydroxylases that induce its proteasomal degradation [52]. One of the effects of increased HIF-1 activity is increased transcription of the Nox2 gene [49] hence, a vicious cycle is established in which HIF-1 activity increases expression of Nox2, leading to greater superoxide production that further promotes HIF-1alpha expression. Notably, in mice that are heterozygous for knockout of HIF-1alpha, the systemic hypertension and brain oxidative stress evoked by CIH are not observed [49]. The extent to which these mechanisms can account for CIH-mediated activation of NADPH in other tissues, such as the vasculature, requires further study.
Other recent research points to uncoupled eNOS as a source of oxidative stress, and a cause of impaired endothelium-dependent vasodilation, in OSA patients and in rats subjected to CIH [40,41]. This likely is a downstream consequence of the oxidative stress generated by NADPH oxidase, and would be expected to further amplify this oxidative stress [53]. Arginase inhibition (in rats) and provision of the eNOS cofactor tetrahydrobiopterin (in humans) have been shown to recouple this enzyme [40,41]. These findings suggest that both an imbalance of arginine and asymmetric dimethylarginine (ADMA), as well as of tetra- and dihydrobiopterin, contribute to the uncoupling associated with OSA. With respect to ADMA, clinical studies have reported elevated ADMA levels in OSA patients, pointing to ADMA as a mediator of eNOS uncoupling in OSA [54,55].
The impairment of endothelium-dependent vasodilation which characterizes OSA appears to be attributable to loss of NO bioactivity, [56-58] and may therefore reflect both the uncoupling of eNOS, and the fact that superoxide generated by either NADPH oxidase or uncoupled eNOS can react spontaneously with NO to form peroxynitrite. The ability of intravenous vitamin C to acutely improve flow-mediated vasodilation in OSA patients likely is attributable to scavenging of superoxide by elevated levels of ascorbate. [59]
NF-KappaB activation drives inflammation in OSA
In untreated patients with OSA, increased NF-kappaB activity has been observed in monocytes, neutrophils, and freshly harvested venous endothelial cells; CPAP therapy reversed this effect [13, 60-62]. In rodents subjected to CIH, activated NF-kappaB has been reported in arterial tissues, cardiomyocytes, vascular endothelium, lung, and liver [14,61,63-66]. Activation of NF-kappaB has also been reported in cultured cells exposed to intermittent hypoxia [67-69]. In some cases, this activation was not accompanied by activation of HIF-1, but was dependent on activation of p38 MAP kinase and IkappaB kinase-beta; hence activation occurred via the canonical pathway [68,69]. The activation of NF-kappaB in cardiomyocytes of mice exposed to CIH was not seen in gp91phox knockout mice similarly treated, suggesting a role for NADPH oxidase as an upstream mediator of NF-kappaB activation [14]. Whereas wild type mice fed a high cholesterol diet developed atherosclerosis when exposed to CIH, no such effect was observed in mice genetically deficient in the p50 subunit of NF-kappaB [18].
These considerations suggest that measures capable of safely down-regulating the activation of NAPDH oxidase, recoupling eNOS, and/or diminishing NF-kappaB activation may have potential for alleviating the pathological consequences of OSA. While CPAP therapy, applied regularly and properly, may provide all of the protection that is needed in this syndrome, compliance with CPAP therapy, which can be unwieldy and uncomfortable for some patients, is less than ideal – and CPAP equipment sometimes malfunctions [70,71]. Hence, it would seem prudent to use safe and convenient adjunctive measures in conjunction with CPAP, if these were indeed effective for mitigating the risks associated with OSA.
Spirulina as an antagonist of NADPH oxidase activity
There is recent evidence that unconjugated bilirubin functions within cells as an inhibitor of NADPH oxidase complexes; this likely explains much of the profound antioxidant activity of the heme oxygenase, which, in response to perceived oxidative stress, cleaves heme to generate carbon monoxide, free iron, and (via biliverdin) bilirubin [72-75]. Moreover, the spirulina chromophore phycocyanobilin (PhyCB), a structural analog and derivative of biliverdin that constitutes about 0.6% of the dry weight of spirulina, has been shown to mimic the ability of biliverdin/bilirubin to inhibit NADPH oxidase complexes – even when administered orally in rodents. [76, 77]. This may account for the manifold anti-inflammatory and antioxidant effects of oral spirulina (or of oral phycocyanin, the spirulina protein which incorporates PhyCB as a chromophore) reported in rodent studies [76-80]. If the metabolism of PhyCB in humans is reasonably analogous to its metabolism in rodents, it should follow that a sufficiently high intake of spirulina, phycocyanin, or free PhyCB will be a clinically effective inhibitor of NADPH oxidase activity. Hence, PhyCB/spirulina may have important potential in the management of OSA. Extrapolations from rodent studies suggest that a spirulina intake of 15-30 g daily might be needed for an optimal antioxidant effect [76].
Inhibiting xanthine oxidase with allopurinol
In light of evidence that conversion of xanthine dehydrogenase to xanthine oxidase is upstream from NADPH oxidase activation in CIH, it is reasonable to suspect that allopurinol treatment might have a favorable clinical impact on OSA. Indeed, in a crossover placebo-controlled clinical study, 2 weeks of allopurinol (300 mg daily) improved flow-mediated vasodilation and lessened plasma markers of oxidative stress in OSA patients [81]. Analogously, in rats exposed to CIH, allopurinol administration boosted acetylcholine-induced vasodilation, and favorably influenced cardiac function and cardiac markers of oxidative stress and apoptosis [82,83].
Supporting glutathione synthesis with N-Acetylcysteine
Some of the downstream effects of excessive superoxide production – whether stemming from NADPH oxidase activation, uncoupling of eNOS, or other sources – can be antagonized by boosting the reduced glutathione content of tissues. Superoxide gives rise to hydrogen peroxide, which oxidizes mildly acidic cysteine groups in proteins, altering their structure and function; reduced glutathione often acts to reverse these effects, while also aiding catabolism of hydrogen peroxide [84-87]. Cysteine supplementation – best achieved with N-acetylcysteine (NAC) – can boost tissue glutathione levels, and there is evidence that this may be particularly beneficial in the elderly, who have a diminished capacity for glutathione synthesis [88-92]. Most clinical trials with NAC have employed daily doses of 1200-1800 mg.
In rodent studies, NAC administration has been reported to alleviate the adverse impact of CIH on erectile dysfunction, and prevent the pro-inflammatory, pro-oxidant effects of CIH on the liver [93,94] But of particular interest is a clinical study in which 20 OSA patients were randomized to receive NAC or placebo for 30 days; sleep function was assessed by polysomnography before and after the supplementation [95]. A number of the parameters measured – including sleep efficiency, oxygen desaturation events per hour, and Epworth sleepiness score – improved markedly and significantly in the NAC group, but not the placebo group. If the findings of this small study prove to be replicable, they suggest that oxidative stress in the central nervous syndrome tends to promote apneic episodes. The authors propose that “long-term treatment with NAC in patients with OSA may reduce their dependency on CPAP.” An attempt to replicate this intriguing clinical study would be warranted.
Citrulline and high-dose folate for recoupling eNOS
eNOS is only fully coupled when it binds its substrate arginine and its cofactor tetrahydrobiopterin (BH4) [53]. The oxidation product of BH4, dihydrobiopterin (BH2), competes with BH4 for binding to eNOS; an elevation of the ratio of BH2 to BH4 therefore is associated with eNOS uncoupling [96-98] Analogously, ADMA competes with arginine for binding to eNOS, and a relatively high ratio of ADMA to arginine likewise promotes uncoupling of eNOS [99]. Endothelial oxidative stress, whether originating from NADPH oxidase activity or other sources, tends to boost levels of both BH2 and ADMA [53]. BH2 increases under oxidative stress owing to direct oxidation of BH4 by oxidants such as peroxynitrite; in addition, oxidative stress may somehow down-regulate endothelial expression of dihydrofolate reductase, an enzyme capable of reconverting BH2 to BH4 [97,98,100]. Oxidative stress also tends to boost endothelial ADMA levels by inhibiting the enzyme responsible for its catabolism, dimethylarginine dimethylaminohydrolase (DDAH) [101-103]. And eNOS can be uncoupled by glutathionylation, which is more likely when oxidants boost cellular levels of diglutathione [106-106] Hence, uncoupling of eNOS may be a consequence of NADPH oxidase activation in OSA – leading to a further increase in oxidant production that would tend to maintain the uncoupling of eNOS in a vicious cycle.
Fortunately, there are practical nutraceutical strategies that can promote the recoupling of eNOS. The adverse impacts of elevated ADMA and of increased arginase activity can be offset by increasing endothelial levels of arginine. This is best achieved by supplementing with the amino acid citrulline, rather than arginine per se [107-109]. The utility of supplemental arginine is compromised by inducible arginase activities in the gastrointestinal tract and liver, such that only a small proportion of supplemental arginine reaches peripheral tissues intact. Citrulline, however, is well absorbed and well distributed to the body’s tissues; within cells, it is efficiently converted to arginine. Hence, supplemental citrulline functions as an efficient delivery form for intracellular arginine. The utility of supplemental citrulline – usually administered in a range of 3-6 g daily, in divided doses – has been documented in clinical circumstances associated with eNOS uncoupling [107,110-114].
With respect to BH2/BH4 imbalance, high-dose folate has the potential to correct this imbalance. This appears to reflect two phenomena. When taken up by cells, folate is rapidly converted to reduced forms, such a 5-methyltetrahydrofolate, which can serve as highly effective scavengers for peroxynitrite and possibly other oxidants which tend to oxidize BH4; this effect is most meaningful when supraphysiological concentrations of folate are employed [115-117]. In addition, there is recent evidence that high-dose folate has an inductive effect on dihydrofolate reductase in endothelial cells, reflecting increased transcription of its gene [118-120]. Since this enzyme plays a key role not only in folate metabolism, but also in mediating the reconversion of BH2 to BH4 [97,98] folate-mediated induction of dihydrofolate reductase may also help to recouple eNOS. Recoupling of eNOS with high-dose folate has been documented in clinical and rodent studies [121-125]. Folate doses in the range of 10-80 mg daily have been employed in the management of vascular disorders, without evident toxicity or side effects; several decades ago, cardiologist Kurt Oster claimed anecdotally that such doses were useful in the management of angina and intermittent claudication [126-128]. Concurrent supplementation with high-dose vitamin B12 – around 1 mg daily – could be used to correct any coexisting B12 deficiency, and hence eliminate the risk that high-dose folate could mask the early symptoms of pernicious anemia [129] (This risk is the FDA’s justification for banning high-dose folate products [130])
Docosahexaenoic acid functions as a sensor of oxidative stress
The long-chain omega-3 fatty acid docosahexaenoic acid (DHA), when incorporated in cellular membranes, is readily oxidized in tissues under oxidative stress. Subsequent metabolism of oxidized DHA leads to generation of the compound 4-hydroxyhexenal (4-HHE) [131] (In contrast, oxidation of linoleic or eicosapentaenoic acids yields 4-hydroxynonenal.) Recent studies show that 4-HHE is an exceptionally potent activator of the nrf2 transcription factor which mediates the transcription of genes coding for a range of phase 2 antioxidant enzymes – including heme oxygenase-1 - as well as for glutamylcysteine synthetase, rate-limiting for glutathione synthesis [131-133]. Hence, DHA can be viewed as a sensor of oxidative stress that promotes feedback induction of natural cellular antioxidant mechanisms.
This recent discovery may rationalize two rodent studies in which DHA supplementation was shown to provide protection from CIH. In atheroma-prone apolipoprotein-E-deficient mice, CIH exacerbates atherosclerosis. Concurrent supplementation with DHA was found to oppose this up-regulatory impact of CIH on atherogenesis, whereas DHA did not influence atherogenesis when these mice were not subjected to CIH [134]. In rats, CIH has been used to promote pulmonary hypertension; concurrent DHA administration was found to have a favorable impact on elevated pulmonary arterial blood pressure, thickening of the pulmonary artery wall, and ventricular hypertrophy [135]. It is reasonable to suspect that DHA’s antioxidant function contributed to these benefits. However, the doses of DHA employed in these rodent studies were high relative to doses that would be feasible for long-term human use; it remains to be seen whether practical clinical doses of DHA could confer benefit in OSA.
A study evaluating OSA patients has observed that DHA levels in erythrocyte membranes tend to be significantly lower in patients with severe OSA, as opposed to less severe OSA [136]. Could DHA be mitigating the severity of OSA – or does severe OSA deplete membrane OSA by oxidative stress or some other mechanism? In any case, clinical evaluation of supplementation with DHA-rich fish oil concentrates in OSA appears to be warranted – as has previously been suggested [136,137].
Addressing NF-KappaB activation with salsalate
With respect to NF-kappaB, clinically tolerable concentrations of salicylic acid can inhibit its activation via the canonical pathway by directly inhibiting the kinase activity of IkappaB kinase-beta [138-140]. Indeed, this effect is currently being employed in the treatment of type 2 diabetes, and is likely responsible for anti-inflammatory impact of salicylate therapy in rheumatoid arthritis (as salicylate per se – as opposed to its derivative aspirin – is only a very weak reversible inhibitor of cyclooxygenase activity) [141-144]. Since salicylate does not produce a physiologically significant inhibition of cyclooxygenase activity, it does not induce the GI bleeds or renal damage that can complicate NSAID therapy [145]. The dose-limiting adverse effect of high-dose salicylate is fully reversible ototoxicity (tinnitus, mild hearing loss); this is seen only in a small minority of patients when a dose of 1.5 g twice daily is employed, a dose sufficient to achieve clinically useful anti-inflammatory or glucose-decreasing effects [142,145,146]. Salicylic acid is best administered as the synthetic dimer salsalate (disalcid), which is cleaved in the alkaline intestinal tract to release free salicylate; salsalate is less prone than salicylate to provoke mild gastric irritation [147-152]. Salsalate, in a dose of 1.5 g twice daily, is usually well tolerated and may have worthwhile potential in the management of OSA.
However, several recent clinical studies have failed to show a favorable effect of salsalate on endothelium-dependent vasodilation – and in some instances a negative effect [153,154]. This suggests that salsalate may have some additional unknown target that compromises its utility for cardiovascular health. A study examining the impact of salsalate on endothelial function in patients with OSA would be of interest.
Figure 1 provides a summary of the molecular mechanisms driving oxidative stress and inflammation in OSA, and of the agents thought to have potential for opposing these mechanisms.
Practical considerations
Daily ingestion of spirulina (or of spirulina extracts enriched in PhyCB), citrulline, high-dose folate, NAC, DHA, allopurinol, and salsalate (for those who tolerate it well) may therefore have potential for favorably impacting the health of OSA patients who fail to achieve good control of their nighttime intermittent apnea. Although the doses of spirulina and of citrulline required for this purpose may be fairly bulky, it would be feasible to incorporate these, along with high-dose folate, into functional foods. Indeed, such foods might have value for managing a range of conditions associated with vascular oxidative stress and eNOS uncoupling [53]. NAC, because of its acrid flavor, and DHA, because of its ready oxidizability, are best administered in capsules.
References
- Bradley TD, Floras JS (2009) Obstructive sleep apnoea and its cardiovascular consequences. Lancet 373: 82-93. [Crossref]
- Drager LF, Polotsky VY, Lorenzi-Filho G (2011) Obstructive sleep apnea: an emerging risk factor for atherosclerosis. Chest 140: 534-542. [Crossref]
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, et al. (2005) Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 353: 2034-2041. [Crossref]
- Lattimore JD, Celermajer DS, Wilcox I (2003) Obstructive sleep apnea and cardiovascular disease. J Am Coll Cardiol 41: 1429-1437. [Crossref]
- Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, et al. (2001) Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med 163: 19-25. [Crossref]
- Narkiewicz K, Somers VK (1997) The sympathetic nervous system and obstructive sleep apnea: implications for hypertension. J Hypertens 15: 1613-1619. [Crossref]
- Peppard PE, Young T, Palta M, Skatrud J (2000) Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 342: 1378-1384. [Crossref]
- Arias MA, Garcia-Rio F, Alonso-Fernandez A, Martinez I, et al. (2006) Pulmonary hypertension in obstructive sleep apnoea: effects of continuous positive airway pressure: a randomized, controlled cross-over study. Eur Heart J 27:1106-13.
- Liu L, Kang R, Zhao S, Zhang T, Zhu W, et al. (2015) Sexual Dysfunction in Patients with Obstructive Sleep Apnea: A Systematic Review and Meta-Analysis. J Sex Med 12: 1992-2003. [Crossref]
- Del BM, Fabiani M, Loffredo L et al. (2012) Oxidative stress mediated arterial dysfunction in patients with obstructive sleep apnoea and the effect of continuous positive airway pressure treatment. BMC Pulm Med 12:36.
- Yoshihisa A, Owada T, Hoshino Y et al. (2010) Flow-mediated dilatation identifies impaired endothelial function in patients with sleep apnea syndrome. Fukushima J Med Sci 56:115-20.
- Ciccone MM, Favale S, Scicchitano P, Mangini F, Mitacchione G, et al. (2012) Reversibility of the endothelial dysfunction after CPAP therapy in OSAS patients. Int J Cardiol 158: 383-386. [Crossref]
- Htoo AK, Greenberg H, Tongia S, Chen G, Henderson T, et al. (2006) Activation of nuclear factor kappaB in obstructive sleep apnea: a pathway leading to systemic inflammation. Sleep Breath 10: 43-50. [Crossref]
- Hayashi T, Yamashita C, Matsumoto C et al. (2008) Role of gp91phox-containing NADPH oxidase in left ventricular remodeling induced by intermittent hypoxic stress. Am J Physiol Heart Circ Physiol 294:H2197-H2203.
- Dumitrascu R, Heitmann J, Seeger W, Weissmann N, Schulz R (2013) Obstructive sleep apnea, oxidative stress and cardiovascular disease: lessons from animal studies. Oxid Med Cell Longev 2013: 234631. [Crossref]
- Liu X, Deng Y, Shang J et al. (2013) Effect of NADPH oxidase inhibitor apocynin on the expression of hypoxia-induced factor-1alpha and endothelin-1 in rat carotid body exposed to chronic intermittent hypoxia. J Huazhong Univ Sci Technolog Med Sci 33:178-84.
- Nisbet RE, Graves AS, Kleinhenz DJ, Rupnow HL, Reed AL, et al. (2009) The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am J Respir Cell Mol Biol 40: 601-609. [Crossref]
- Song D, Fang G, Mao SZ, et al. (1822) Chronic intermittent hypoxia induces atherosclerosis by NF-kappaB-dependent mechanisms. Biochim Biophys Acta 1822: 1650-1659.
- Ramond A, Godin-Ribuot D, Ribuot C et al. (2013) Oxidative stress mediates cardiac infarction aggravation induced by intermittent hypoxia. Fundam Clin Pharmacol 27: 252-261.
- Montplaisir J, Bedard MA, Richer F, Rouleau I (1992) Neurobehavioral manifestations in obstructive sleep apnea syndrome before and after treatment with continuous positive airway pressure. Sleep 15: S17-S19.
- Akashiba T, Kawahara S, Akahoshi T, Omori C, Saito O, et al. (2002) Relationship between quality of life and mood or depression in patients with severe obstructive sleep apnea syndrome. Chest 122: 861-865. [Crossref]
- Ohayon MM (2003) The effects of breathing-related sleep disorders on mood disturbances in the general population. J Clin Psychiatry 64: 1195-1200. [Crossref]
- Bédard MA, Montplaisir J, Richer F, Malo J (1991) Nocturnal hypoxemia as a determinant of vigilance impairment in sleep apnea syndrome. Chest 100: 367-370. [Crossref]
- Nair D, Dayyat EA, Zhang SX, Wang Y, Gozal D (2011). Intermittent hypoxia-induced cognitive deficits are mediated by NADPH oxidase activity in a murine model of sleep apnea. PLoS ONE 6: e19847.
- Wang Y, Zhang SX, Gozal D (2010) Reactive oxygen species and the brain in sleep apnea. Respir Physiol Neurobiol 174: 307-316. [Crossref]
- Ryan S, Taylor CT, McNicholas WT (2010) Systemic inflammation: a key factor in the pathogenesis of cardiovascular complications in obstructive sleep apnoea syndrome? Postgrad Med J 85: 693-698.
- Ramar K, Caples SM (2011) Vascular changes, cardiovascular disease and obstructive sleep apnea. Future Cardiol 7: 241-249. [Crossref]
- Williams A, Scharf SM (2007) Obstructive sleep apnea, cardiovascular disease, and inflammation--is NF-kappaB the key? Sleep Breath 11: 69-76. [Crossref]
- Barceló A, Miralles C, Barbé F, Vila M, Pons S, et al. (2000) Abnormal lipid peroxidation in patients with sleep apnoea. Eur Respir J 16: 644-647. [Crossref]
- Lavie L, Vishnevsky A, Lavie P (2004) Evidence for lipid peroxidation in obstructive sleep apnea. Sleep 27: 123-128. [Crossref]
- Lavie L1 (2003) Obstructive sleep apnoea syndrome--an oxidative stress disorder. Sleep Med Rev 7: 35-51. [Crossref]
- Schulz R, Mahmoudi S, Hattar K et al. (2000) Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med 162: 566-570.
- Yamauchi M, Nakano H, Maekawa J, Okamoto Y, Ohnishi Y, et al. (2005) Oxidative stress in obstructive sleep apnea. Chest 127: 1674-1679. [Crossref]
- Carpagnano GE, Kharitonov SA, Resta O, Foschino-Barbaro MP, Gramiccioni E, Barnes PJ (2003) 8-Isoprostane, a marker of oxidative stress, is increased in exhaled breath condensate of patients with obstructive sleep apnea after night and is reduced by continuous positive airway pressure therapy. Chest 124: 1386-1392.
- Minoguchi K, Yokoe T, Tanaka A, Ohta S, Hirano T, et al. (2006) Association between lipid peroxidation and inflammation in obstructive sleep apnoea. Eur Respir J 28: 378-385. [Crossref]
- Ohga E, Nagase T, Tomita T, Teramoto S, Matsuse T, et al. (1999) Increased levels of circulating ICAM-, VCAM-, and L-selectin in obstructive sleep apnea syndrome. J Appl Physiol (1985) 87: 10-14. [Crossref]
- Dyugovskaya L, Lavie P, Lavie L (2002) Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med 165: 934-939. [Crossref]
- Christou K, Markoulis N, Moulas AN, Pastaka C, Gourgoulianis KI (2003) Reactive oxygen metabolites (ROMs) as an index of oxidative stress in obstructive sleep apnea patients. Sleep Breath 7: 105-110. [Crossref]
- Christou K, Moulas AN, Pastaka C, Gourgoulianis KI (2003) Antioxidant capacity in obstructive sleep apnea patients. Sleep Med 4: 225-228. [Crossref]
- Varadharaj S, Porter K, Pleister A et al. (2015) Endothelial nitric oxide synthase uncoupling: a novel pathway in OSA induced vascular endothelial dysfunction. Respir Physiol Neurobiol 207: 40-47.
- Krause BJ, del RR, Moya EA, Marquez-Gutierrez M, Casanello P, et al. (2015) Arginase-endothelial nitric oxide synthase imbalance contributes to endothelial dysfunction during chronic intermittent hypoxia. J Hypertens 33: 515-524.
- Zhan G, Serrano F, Fenik P, Hsu R, Kong L, et al. (2005) NADPH oxidase mediates hypersomnolence and brain oxidative injury in a murine model of sleep apnea. Am J Respir Crit Care Med 172: 921-929. [Crossref]
- Hayashi T, Yamashita C, Matsumoto C et al. (2008) Role of gp91phox-containing NADPH oxidase in left ventricular remodeling induced by intermittent hypoxic stress. Am J Physiol Heart Circ Physiol 294: H2197-H2203.
- Nair D, Ramesh V, Gozal D (2012) Adverse cognitive effects of high-fat diet in a murine model of sleep apnea are mediated by NADPH oxidase activity. Neuroscience 227: 361-369. [Crossref]
- Schulz R, Murzabekova G, Egemnazarov B, Kraut S, Eisele HJ, et al. (2014) Arterial hypertension in a murine model of sleep apnea: role of NADPH oxidase 2. J Hypertens 32: 300-305. [Crossref]
- Liu K, Liu XS, Xiao L, Shang J, Li MC, et al. (2012) NADPH oxidase activation: a mechanism of erectile dysfunction in a rat model of sleep apnea. J Androl 33: 1186-1198. [Crossref]
- Liu HG, Zhou YN, Liu K, Xu YJ. (2010) Nicotinamide-adenine dinucleotide phosphate oxidase p22phox expression in induced sputum cells for patients with obstructive sleep apnea hypopnea syndrome, 33: 118-122.
- Liu HG, Wu HM, Xu YJ (2011) [Changes of blood pressure and expression of NADPH oxidase in carotid body in rats exposed to intermittent hypoxia]. Zhonghua Jie He He Hu Xi Za Zhi 34: 26-29. [Crossref]
- Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, et al. (2011) Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol 226: 2925-2933. [Crossref]
- Nanduri J, Vaddi DR, Khan SA, Wang N, Makarenko V, et al. (2015) HIF-1α activation by intermittent hypoxia requires NADPH oxidase stimulation by xanthine oxidase. PLoS One 10: e0119762. [Crossref]
- Nanduri J, Vaddi DR, Khan SA, Wang N, Makerenko V, et al. (2013) Xanthine oxidase mediates hypoxia-inducible factor-2α degradation by intermittent hypoxia. PLoS One 8: e75838. [Crossref]
- Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR (2008) Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol 217: 674-685. [Crossref]
- Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A1 (2014) eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr Pharm Des 20: 3579-3594. [Crossref]
- Ozkan Y, Firat H, Simsek B, Torun M, Yardim-Akaydin S (2008) Circulating nitric oxide (NO), asymmetric dimethylarginine (ADMA), homocysteine, and oxidative status in obstructive sleep apnea-hypopnea syndrome (OSAHS). Sleep Breath 12: 149-154.
- Barceló A, de la Peña M, Ayllón O, Vega-Agapito MV, Piérola J, et al. (2009) Increased plasma levels of asymmetric dimethylarginine and soluble CD40 ligand in patients with sleep apnea. Respiration 77: 85-90. [Crossref]
- Ip MS, Tse HF, Lam B, Tsang KW, Lam WK (2004) Endothelial function in obstructive sleep apnea and response to treatment. Am J Respir Crit Care Med 169: 348-353. [Crossref]
- Schulz R, Schmidt D, Blum A, Lopes-Ribeiro X, Lücke C, et al. (2000) Decreased plasma levels of nitric oxide derivatives in obstructive sleep apnoea: response to CPAP therapy. Thorax 55: 1046-1051. [Crossref]
- Duchna HW, Orth M, Schultze-Werninghaus G, Guilleminault C, Stoohs RA (2005) Long-term effects of nasal continuous positive airway pressure on vasodilatory endothelial function in obstructive sleep apnea syndrome. Sleep Breath 9: 97-103.
- Grebe M, Eisele HJ, Weissmann N, Schaefer C, Tillmanns H, et al. (2006) Antioxidant vitamin C improves endothelial function in obstructive sleep apnea. Am J Respir Crit Care Med 173: 897-901. [Crossref]
- Jelic S, Lederer DJ, Adams T, Padeletti M, Colombo PC, et al. (2010) Vascular inflammation in obesity and sleep apnea. Circulation 121: 1014-1021. [Crossref]
- Greenberg H, Ye X, Wilson D, Htoo AK, Hendersen T, Liu SF (2006) Chronic intermittent hypoxia activates nuclear factor-kappaB in cardiovascular tissues in vivo. Biochem Biophys Res Commun 343: 591-596.
- Yamauchi M, Tamaki S, Tomoda K, Yoshikawa M, Fukuoka A, et al. (2006) Evidence for activation of nuclear factor kappaB in obstructive sleep apnea. Sleep Breath 10: 189-193. [Crossref]
- Li S, Qian XH, Zhou W et al. (2011) Time-dependent inflammatory factor production and NFkappaB activation in a rodent model of intermittent hypoxia. Swiss Med Wkly 141: w13309.
- da Rosa DP, Forgiarini LF, Baronio D, Feijó CA, Martinez D, et al. (2012) Simulating sleep apnea by exposure to intermittent hypoxia induces inflammation in the lung and liver. Mediators Inflamm 2012: 879419. [Crossref]
- Wang B, Yan B, Song2021 Copyright OAT. All rights reservmittent hypoxia down-regulates endothelial nitric oxide synthase expression by an NF-kappaB-dependent mechanism. Sleep Med 14: 165-171.
- Feng J, Chen BY, Cui LY et al.(2009) Inflammation status of rabbit carotid artery model endothelium during intermittent hypoxia exposure and its relationship with leptin. Sleep Breath 13: 277-283.
- Han Q, Yeung SC, Ip MS, Mak JC (2013) Intermittent hypoxia-induced NF-κB and HO-1 regulation in human endothelial EA.hy926 cells. Cell Biochem Biophys 66: 431-441. [Crossref]
- Ryan S, Taylor CT, McNicholas WT (2005) Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 112: 2660-2667. [Crossref]
- Ryan S, McNicholas WT, Taylor CT (2007) A critical role for p38 map kinase in NF-kappaB signaling during intermittent hypoxia/reoxygenation. Biochem Biophys Res Commun 355: 728-733.
- Shapiro GK, Shapiro CM (2010) Factors that influence CPAP adherence: an overview. Sleep Breath 14: 323-335. [Crossref]
- Sawyer AM, Gooneratne NS, Marcus CL, Ofer D, Richards KC, et al. (2011) A systematic review of CPAP adherence across age groups: clinical and empiric insights for developing CPAP adherence interventions. Sleep Med Rev15: 343-356. [Crossref]
- Lanone S, Bloc S, Foresti R, Almolki A, Taillé C, et al. (2005) Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J 19: 1890-1892. [Crossref]
- Matsumoto H, Ishikawa K, Itabe H, Maruyama Y (2006) Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol Cell Biochem 291: 21-28.
- Jiang F, Roberts SJ, Datla S, Dusting GJ. (2006) NO modulates NADPH oxidase function via heme oxygenase-1 in human endothelial cells. Hypertension 48: 950-957.
- Datla SR, Dusting GJ, Mori TA, Taylor CJ, Croft KD, et al. (2007) Induction of heme oxygenase-1 in vivo suppresses NADPH oxidase derived oxidative stress. Hypertension 50: 636-642. [Crossref]
- McCarty MF1 (2007) Clinical potential of Spirulina as a source of phycocyanobilin. J Med Food 10: 566-570. [Crossref]
- Zheng J, Inoguchi T, Sasaki S et al. (2012) Phycocyanin and Phycocyanobilin from Spirulina Platensis Protect against Diabetic Nephropathy by Inhibiting Oxidative Stress. Am J Physiol Regul Integr Comp Physiol.
- Romay Ch, González R, Ledón N, Remirez D, Rimbau V (2003) C-phycocyanin: a biliprotein with antioxidant, anti-inflammatory and neuroprotective effects. Curr Protein Pept Sci 4: 207-216. [Crossref]
- Riss J, Decorde K, Sutra T et al. (2007) Phycobiliprotein C-Phycocyanin from Spirulina platensis Is Powerfully Responsible for Reducing Oxidative Stress and NADPH Oxidase Expression Induced by an Atherogenic Diet in Hamsters. J Agric Food Chem 55: 7962-7967.
- Chamorro G, Perez-Albiter M, Serrano-Garcia N, Mares-Samano JJ, Rojas P (2006) Spirulina maxima pretreatment partially protects against 1-methyl-4-phenyl-, 2,3,6-tetrahydropyridine neurotoxicity. Nutr Neurosci 9: 207-212.
- El Solh AA, Saliba R, Bosinski T, Grant BJ, Berbary E, Miller N (2010) Allopurinol improves endothelial function in sleep apnoea: a randomised controlled study. Eur Respir J 27: 997-1002.
- Dopp JM, Philippi NR, Marcus NJ, Olson EB, Bird CE, et al. (2011) Xanthine oxidase inhibition attenuates endothelial dysfunction caused by chronic intermittent hypoxia in rats. Respiration 82: 458-467. [Crossref]
- Williams AL, Chen L, Scharf SM (2010) Effects of allopurinol on cardiac function and oxidant stress in chronic intermittent hypoxia. Sleep Breath 14: 51-57. [Crossref]
- Bindoli A, Rigobello MP (2013) Principles in redox signaling: from chemistry to functional significance. Antioxid Redox Signal 18: 1557-1593. [Crossref]
- Lo Conte M, Carroll KS (2013) The redox biochemistry of protein sulfenylation and sulfinylation. J Biol Chem 288: 26480-26488. [Crossref]
- Dickinson DA, Forman HJ (2002) Glutathione in defense and signaling: lessons from a small thiol. Ann N Y Acad Sci 973: 488-504. [Crossref]
- Parsons ZD, Gates KS (2013) Thiol-dependent recovery of catalytic activity from oxidized protein tyrosine phosphatases. Biochemistry 52: 6412-6423.
- Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA (2007) N-Acetylcysteine--a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 7: 355-359. [Crossref]
- Dodd S, Dean O, Copolov DL, Malhi GS, Berk M (2008) N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther 8: 1955-1962. [Crossref]
- Rebrin I, Sohal RS (2008) Pro-oxidant shift in glutathione redox state during aging. Adv Drug Deliv Rev 60: 1545-1552. [Crossref]
- Dröge W, Kinscherf R, Hildebrandt W, Schmitt T (2006) The deficit in low molecular weight thiols as a target for antiageing therapy. Curr Drug Targets 7: 1505-1512. [Crossref]
- Levine ME, Suarez JA, Brandhorst S et al. (2014) Low protein intake is associated with a major reduction in IGF-, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab 19: 407-417.
- Zhu D, Deng Y, Pan Y et al. (2015) N-acetylcysteine Ameliorates the Erectile Dysfunction Caused by Chronic Intermittent Hypoxia in Rats: Partly Involvement of Endoplasmic Reticulum Stress. Urology 86: 844.
- da Rosa DP, Forgiarini LF, e Silva MB, Fiori CZ, Andrade CF, et al. (2015) Antioxidants inhibit the inflammatory and apoptotic processes in an intermittent hypoxia model of sleep apnea. Inflamm Res 64: 21-29. [Crossref]
- Sadasivam K, Patial K, Vijayan VK, Ravi K (2011) Anti-oxidant treatment in obstructive sleep apnoea syndrome. Indian J Chest Dis Allied Sci 53: 153-162. [Crossref]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, et al. (2003) Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201-1209. [Crossref]
- Crabtree MJ, Tatham AL, Hale AB, Alp NJ, Channon KM. (2009) Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J Biol Chem 284: 28128-28136.
- Crabtree MJ, Channon KM (2011) Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide 25: 81-88. [Crossref]
- Sud N, Wells SM, Sharma S, Wiseman DA, Wilham J, Black SM (2008) Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterial endothelial cells: role of mitochondrial dysfunction. Am J Physiol Cell Physiol 294: C1407-C1418.
- Milstien S, Katusic Z (1999) Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem Biophys Res Commun 263: 681-684. [Crossref]
- Lin KY, Ito A, Asagami T et al. (2002) Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation 106: 987-992.
- Pope AJ, Druhan L, Guzman JE, Forbes SP, Murugesan V, et al. (2007) Role of DDAH-1 in lipid peroxidation product-mediated inhibition of endothelial NO generation. Am J Physiol Cell Physiol 293: C1679-1686. [Crossref]
- Ando R, Ueda S, Yamagishi S et al.(2013) Involvement of advanced glycation end product-induced asymmetric dimethylarginine generation in endothelial dysfunction. Diab Vasc Dis Res 10: 436-441.
- Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, et al. (2010) S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115-1118. [Crossref]
- Zweier JL, Chen CA, Druhan LJ.(2011) S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid Redox Signal 14: 1769-1775.
- Duan DD, Kwan CY (2011) A molecular switch of "yin and yang": S-glutathionylation of eNOS turns off NO synthesis and turns on superoxide generation. Acta Pharmacol Sin 32: 415-416. [Crossref]
- Waugh WH, Daeschner CW 3rd, Files BA, McConnell ME, Strandjord SE (2001) Oral citrulline as arginine precursor may be beneficial in sickle cell disease: early phase two results. J Natl Med Assoc 93: 363-371. [Crossref]
- Schwedhelm E, Maas R, Freese R, Jung D, Lukacs Z, et al. (2008) Pharmacokinetic and pharmacodynamic properties of oral L-citrulline and L-arginine: impact on nitric oxide metabolism. Br J Clin Pharmacol 65: 51-59. [Crossref]
- Wijnands KA, Meesters DM, van Barneveld KW, et al. (2015) Citrulline Supplementation Improves Organ Perfusion and Arginine Availability under Conditions with Enhanced Arginase Activity. Nutrients 7: 5217-5238.
- Figueroa A, Trivino JA, Sanchez-Gonzalez MA, Vicil F(2010) Oral L-citrulline supplementation attenuates blood pressure response to cold pressor test in young men. Am J Hypertens 23: 12-16.
- Balderas-Munoz K, Castillo-Martinez L, Orea-Tejeda A et al. (2012) Improvement of ventricular function in systolic heart failure patients with oral L-citrulline supplementation. Cardiol J 19: 612-617.
- Sureda A, Pons A (2012) Arginine and citrulline supplementation in sports and exercise: ergogenic nutrients? Med Sport Sci 59: 18-28. [Crossref]
- Cormio L, De SM, Lorusso F et al. (2011) Oral L-citrulline supplementation improves erection hardness in men with mild erectile dysfunction. Urology 77: 119-122.
- Ochiai M, Hayashi T, Morita M, Ina K, Maeda M, et al. (2012) Short-term effects of L-citrulline supplementation on arterial stiffness in middle-aged men. Int J Cardiol 155: 257-261. [Crossref]
- Rezk BM, Haenen GR, van der Vijgh WJ, Bast A (2003) Tetrahydrofolate and 5-methyltetrahydrofolate are folates with high antioxidant activity. Identification of the antioxidant pharmacophore. FEBS Lett 555: 601-605. [Crossref]
- Antoniades C, Shirodaria C, Warrick N et al.(2006) 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation 114: 1193-1201.
- McCarty MF, Barroso-Aranda J, Contreras F (2009) High-dose folate and dietary purines promote scavenging of peroxynitrite-derived radicals--clinical potential in inflammatory disorders. Med Hypotheses 73: 824-834.
- Gao L, Chalupsky K, Stefani E, Cai H (2009) Mechanistic insights into folic acid-dependent vascular protection: dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice: a novel HPLC-based fluorescent assay for DHFR activity. J Mol Cell Cardiol 47: 752-760.
- Gao L, Siu KL, Chalupsky K, et al. (2012) Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: treatment with folic acid. Hypertension 59: 158-166.
- Siu KL, Miao XN, Cai H1 (2014) Recoupling of eNOS with folic acid prevents abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E null mice. PLoS One 9: e88899. [Crossref]
- Verhaar MC, Wever RM, Kastelein JJ, van DT, Koomans HA, Rabelink TJ (1998) 5-methyltetrahydrofolate, the active form of folic acid, restores endothelial function in familial hypercholesterolemia. Circulation 97: 237-241.
- Antoniades C, Shirodaria C, Warrick N, et al. (2006) 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation 114: 1193-1201.
- Tawakol A, Migrino RQ, Aziz KS, Waitkowska J, Holmvang G, et al. (2005) High-dose folic acid acutely improves coronary vasodilator function in patients with coronary artery disease. J Am Coll Cardiol 45: 1580-1584. [Crossref]
- Moens AL, Claeys MJ, Wuyts FL, Goovaerts I, Van Hertbruggen E, et al. (2007) Effect of folic acid on endothelial function following acute myocardial infarction. Am J Cardiol 99: 476-481. [Crossref]
- Moens AL, Vrints CJ, Claeys MJ, Timmermans JP, Champion HC, et al. (2008) Mechanisms and potential therapeutic targets for folic acid in cardiovascular disease. Am J Physiol Heart Circ Physiol 294: H1971-1977. [Crossref]
- McCarty MF (2007) Oster rediscovered--mega-dose folate for symptomatic atherosclerosis. Med Hypotheses 69: 325-332. [Crossref]
- Oster KA (1981) Atherosclerosis treated with folic acid. FASEB J 40.
- Oster KA, Ross DJ, Dawkins HHR (1983) The XO Factor. New York: Park City Press.
- Andrès E, Dali-Youcef N, Vogel T, Serraj K, Zimmer J (2009) Oral cobalamin (vitamin B(12)) treatment. An update. Int J Lab Hematol 31: 1-8. [Crossref]
- Sinow RM, Johnson CS, Karnaze DS, Siegel ME, Carmel R (1987) Unsuspected pernicious anemia in a patient with sickle cell disease receiving routine folate supplementation. Arch Intern Med 147: 1828-1829.
- Ishikado A, Morino K, Nishio Y, Nakagawa F, Mukose A, et al. (2013) 4-Hydroxy hexenal derived from docosahexaenoic acid protects endothelial cells via Nrf2 activation. PLoS One 8: e69415. [Crossref]
- Zhang M, Wang S, Mao L, Leak RK, Shi Y, et al. (2014) Omega-3 fatty acids protect the brain against ischemic injury by activating Nrf2 and upregulating heme oxygenase 1. J Neurosci 34: 1903-1915. [Crossref]
- Nakagawa F, Morino K, Ugi S et al. (2014) 4-Hydroxy hexenal derived from dietary n-3 polyunsaturated fatty acids induces anti-oxidative enzyme heme oxygenase-1 in multiple organs. Biochem Biophys Res Commun 443: 991-996.
- Van NL, Back M, Arnaud C et al.(2014) Docosahexaenoic acid supplementation modifies fatty acid incorporation in tissues and prevents hypoxia induced-atherosclerosis progression in apolipoprotein-E deficient mice. Prostaglandins Leukot Essent Fatty Acids 91: 111-117.
- Chen R, Liu P, Yan J2, Gu Y1 (2014) [Effects of docosahexaenoic acid on hypoxia-induced pulmonary arterial hypertension]. Zhonghua Jie He He Hu Xi Za Zhi 37: 109-112. [Crossref]
- Ladesich JB, Pottala JV, Romaker A, Harris WS (2011) Membrane level of omega-3 docosahexaenoic acid is associated with severity of obstructive sleep apnea. J Clin Sleep Med 7: 391-396. [Crossref]
- Scorza FA, Cavalheiro EA, Scorza CA, Galduróz JC2, Tufik S2, et al. (2013) Sleep Apnea and Inflammation - Getting a Good Night's Sleep with Omega-3 Supplementation. Front Neurol 4: 193. [Crossref]
- Yin MJ, Yamamoto Y, Gaynor RB (1998) The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396: 77-80. [Crossref]
- Pierce GL, Lesniewski LA, Lawson BR, Beske SD, Seals DR (2009) Nuclear factor-{kappa}B activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation 119: 1284-1292.
- McCarty MF1 (2010) Salsalate may have broad utility in the prevention and treatment of vascular disorders and the metabolic syndrome. Med Hypotheses 75: 276-281. [Crossref]
- Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, et al. (2008) Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci 1: 36-43. [Crossref]
- Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, et al. (2010) The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med 152: 346-357. [Crossref]
- Desouza CV1 (2010) An overview of salsalate as a potential antidiabetic therapy. Drugs Today (Barc) 46: 847-853. [Crossref]
- Morris HG, Sherman NA, McQuain C, Goldlust MB, Chang SF, et al. (1985) Effects of salsalate (nonacetylated salicylate) and aspirin on serum prostaglandins in humans. Ther Drug Monit 7: 435-438. [Crossref]
- Cryer B, Goldschmiedt M, Redfern JS, Feldman M (1990) Comparison of salsalate and aspirin on mucosal injury and gastroduodenal mucosal prostaglandins. Gastroenterology 99: 1616-1621.
- Goldfine AB, Conlin PR, Halperin F, Koska J, Permana P, et al. (2013) A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia 56: 714-723. [Crossref]
- Singleton PT Jr (1980) Salsalate: its role in the management of rheumatic disease. Clin Ther 3: 80-102. [Crossref]
- Dromgoole SH, Furst DE, Paulus HE (1984) Metabolism of salsalate in normal subjects. J Pharm Sci 73: 1657-1659. [Crossref]
- Estes D, Kaplan K (1980) Lack of platelet effect with the aspirin analog, salsalate. Arthritis Rheum 23: 1303-1307. [Crossref]
- Montrone F, Caruso I, Cazzola M (1989) Salsalate in the treatment of rheumatoid arthritis: a double-blind clinical and gastroscopic trial versus piroxicam. I. Clinical trial. J Int Med Res 17: 316-319. [Crossref]
- Bombardier C, Peloso PM, Goldsmith CH (1995) Salsalate, a nonacetylated salicylate, is as efficacious as diclofenac in patients with rheumatoid arthritis. Salsalate-Diclofenac Study Group. J Rheumatol 22: 617-24. [Crossref]
- Lanza F, Rack MF, Doucette M, Ekholm B, Goldlust B, et al. (1989) An endoscopic comparison of the gastroduodenal injury seen with salsalate and naproxen. J Rheumatol 16: 1570-1574.
- Goldfine AB, Buck JS, Desouza C, et al. (2013) Targeting inflammation using salsalate in patients with type 2 diabetes: effects on flow-mediated dilation (TINSAL-FMD). Diabetes Care 36: 4132-4139.
- Nohria A, Kinlay S, Buck JS, Redline W, Copeland-Halperin R, et al. (2014) The effect of salsalate therapy on endothelial function in a broad range of subjects. J Am Heart Assoc 3: e000609. [Crossref]