Nuclear factor–kappa B (NF-κB) has numerous pathophysiologic functions and is therefore considered a suitable target in the search for drugs to treat many diseases, including cancers and inflammatory diseases. We previously reported a novel triazine-based NF-κB inhibitor, 4,6-dichloro-N-phenyl-1,3,5-triazin-2-amine (NI241), which directly inhibits binding of NF-κB to DNA. We predicted the mode of binding between NI241 and p50 (a member of NF-κB family) using in silico docking simulations. Here, we searched for new NF-κB inhibitors using structure-based virtual screening (SBVS), a computational method widely used to identify candidate compounds in drug discovery. In the present study, we performed SBVS with the docking simulation program Glide and selected 33 compounds from our virtual chemical library. Subsequently, we investigated the NF-κB inhibitory effects of these compounds using electrophoretic mobility shift assays. We identified two compounds that inhibited the DNA binding of p50. Although these compounds were less effective than NI241 as NF-κB inhibitors, they have new scaffold structures and provide clues that will aid future efforts to identify novel compounds that specifically inhibit the binding of NF-κB to DNA.
Nuclear factor- kappa B (NF-κB), inhibitor, DNA binding, Structure-based virtual screening (SBVS)
Nuclear factor–kappa B (NF-κB) is a transcription factor that regulates gene expression by binding to regions of DNA known as κB elements, which contain the consensus sequence 5'-GGGRNYYYCC-3' [1–5]. The wide variety of genes regulated by NF-κB are involved in diverse and key cellular and systemic processes, including cell proliferation, cell survival and death, innate immunity, and inflammation [6–8]. Aberrant activation of NF-κB is a characteristic of cancers and inflammatory diseases. NF-κB is constitutively activated in many types of cancers. Chronic inflammation, which is a risk factor for various types of cancers, may also involve aberrant NF-κB activation [9–11]. Therefore, selective NF-κB inhibitors would be useful in both cancer research and treatment.
Two distinct signaling pathways, the canonical and non-canonical pathways, are linked to activation of NF-κB [8,12,13]. Many inhibitors of NF-κB have been reported, including IκB kinase inhibitors, proteasome inhibitors, and antioxidants [14,15]. Though these inhibitors can block the activation of NF-κB at various upstream and midstream steps of NF-κB signaling pathways, they can also affect other targets and pathways. However, compounds that directly inhibit the DNA binding of NF-κB may be highly effective in regulating the expression of target genes, because NF-κB signaling pathways converge at the level of DNA binding. In previous work aimed at identifying new compounds from our chemical library that directly inhibit the DNA binding of NF-κB, we identified the unique compound 4,6-dichloro-N-phenyl-1,3,5-triazin-2-amine, which we designated NI241 (Figure 1). We compared the inhibitory effects of NI241 analogues and predicted in silico the mode of NI241 binding with p50 (a member of NF-κB family) [16]. Subsequently, we began searching for new NF-κB inhibitors using an in silico structure-based virtual screening (SBVS) approach, referencing the mode of NI241 binding with p50 in our previous study.
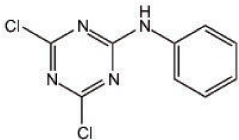
Figure 1. Chemical structure of NI241; 4,6-dichloro-N-phenyl-1,3,5-triazin-2-amine.
SBVS is a powerful computational technique widely used in the drug discovery process to identify candidate compounds by compiling relevant chemical structures from large chemical libraries. SBVS allows researchers to screen large numbers of candidate compounds using docking simulations with an active pocket of target proteins, scoring the compound-protein interaction to estimate the binding energy of the compound [17–19]. In the present study, we first searched for novel inhibitors for NF-κB using SBVS. For this purpose, we prepared a virtual chemical library. We then investigated the inhibitory effect of the high-scoring candidate compounds in vitro. Using this approach, we identified two compounds that inhibited the DNA binding of p50.
Materials
The 2D structure data for the compounds contained in the virtual chemical library were provided by Namiki Shoji (Tokyo, Japan). Compounds selected by SBVS were purchased from Namiki Shoji and dissolved in dimethyl sulfoxide (DMSO). 5'-Alexa680-labeled κB single-stranded oligonucleotide, which corresponds to the binding site of NF-κB, was purchased from Japan Bio Services (Saitama, Japan).
Virtual chemical library preparation and SBVS
The virtual chemical library was prepared with the compound 2D structure data using LigPrep 2.0 [20] and QikProp 2.5 [21]. LigPrep produces a single, low-energy, 3D structure with correct chirality for each successfully processed input structure. From each input structure, the software can also produce a number of structures with various ionization states, tautomers, stereochemistries, and ring conformations and eliminate molecules based on various criteria, including molecular weight, or specified numbers and types of functional groups. Minimization was performed using OPLS_2005 force field, and ionizer was used to generate all possible ionization states at pH 7.0 ± 2.0 [20,22,23]. QikProp enables rapid, accurate, and simple predictions of the absorption, distribution, metabolism, and excretion (ADME) of a compound and predicts physically significant descriptors and pharmaceutically relevant properties of organic molecules, either individually or in batches [21–24].
The specificity of binding between compounds in our virtual chemical library and p50 was evaluated using docking simulations with the docking-simulation program Grid-based Ligand Docking with Energetics (Glide) 4.0 [23,25–29], as reported previously [16]. The 3D structure of p50 was obtained from the protein data bank (PDB code: 1NFK). All water molecules were removed, and just one p50 subunit was used for modeling. The compounds in our virtual chemical library were docked in the crystal structure of p50 in silico using Glide 4.0. This program searches for favorable interactions between one or more typically small-ligand molecules and a typically larger receptor molecule, usually a protein, and provides three mode of docking: high-throughput virtual screening (HTVS), standard precision (SP), and extra precision (XP). The HTVS mode is intended for the rapid screening of very large numbers of ligands, whereas the SP mode is appropriate for screening ligands of unknown quality in large numbers. The XP mode is a more powerful and discriminating procedure that combines a powerful sampling protocol with a custom scoring function designed to identify ligand poses expected to have unfavorable energies based on common principles of physical chemistry [23,25,29]. The docking results are interpreted based on this fitness function in the form of a ‘Glidescore.’ First, the compounds in our virtual chemical library were screened using the HTVS mode of Glide. Compounds with a high Glidescore were selected in the HTVS mode and docked in p50 using the SP mode. The top-ranked compounds were filtered and re-docked using the XP mode. Finally, we selected and purchased 33 candidate compounds for further analysis.
Electrophoretic mobility shift assay (EMSA)
An Alexa680-labeled NF-κB probe was prepared by annealing 5'-Alexa680-labeled single-stranded oligonucleotide with an unlabeled complementary single-stranded oligonucleotide: 5′-Alexa680-AGTTGAGGGGACTTTCCCAGGC-3′ (sense) and 5′-GCCTGGGAAAGTCCCCTCAACT-3′ (antisense), where the sequence of the κB binding site is underlined. His-tagged p50 recombinant was produced in E. coli and purified via HisTrap HP (GE Healthcare, Buckinghamshire, UK) using a Profinia Protein Purification System (Bio-Rad Laboratories, Hercules, CA). Reaction mixtures containing binding buffer (15 mM Tris–HCl [pH 7.5], 180 mM NaCl, 1.5 mM ethylenediamine tetraacetic acid, 1.5 mM dithiothreitol, 4% glycerol, 1 mg/mL bovine serum albumin, and 0.3% Nonidet P-40), 0.5 µg of poly(dI-dC)·(dI-dC), and 62.5 ng of His-tagged p50 recombinant were left to stand on ice for 10 min. The candidate compounds were then added and the mixtures were incubated at room temperature. After 30 min, 20 nM Alexa680-labeled NF-κB probe was added and incubation was continued at room temperature for 30 min. The samples (in a volume of 20 µL) were loaded on native 5% polyacrylamide gels prepared in 0.5× TBE and electrophoresed at 170 CV for 150 min. Gels were then scanned and analyzed using an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE).
SBVS
We performed SBVS to search for candidate inhibitors of NF-κB DNA binding, using 2D structure data for 6,149,846 compounds provided by Namiki Shoji. The data were analyzed with LigPrep and QikProp using optimized parameters. LigPrep produces a single, low-energy, 3D structure with correct chirality for each successfully processed input structure [20,22,23], and QikProp predicts physically significant descriptors and pharmaceutically relevant properties of organic molecules [21–24]. Following optimization, we obtained a virtual chemical library containing 5,469,185 compounds.
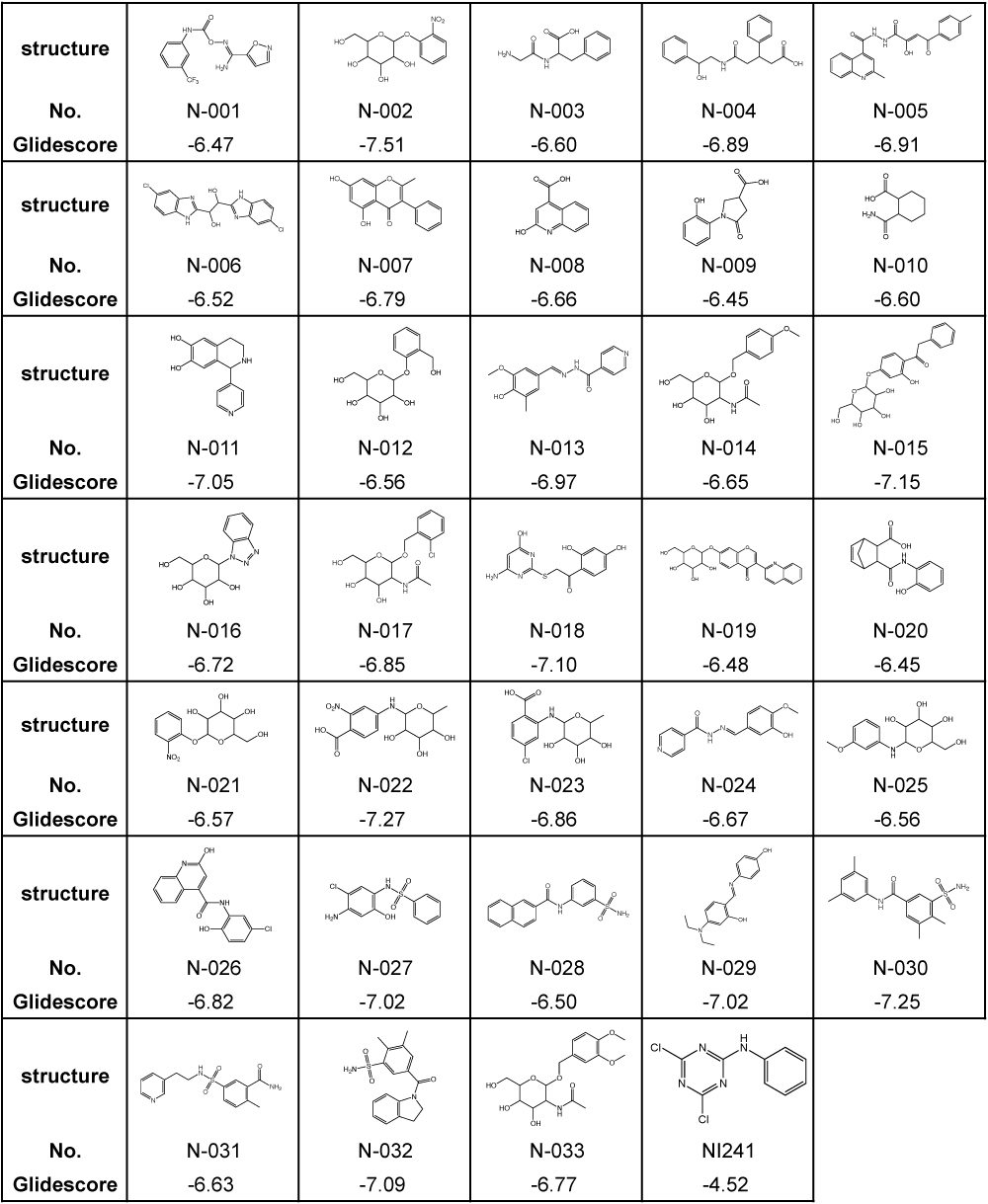
Previously, we identified NI241 as an inhibitor of NF-κB [16], having predicted the mode of NI241 and p50 binding using the docking-simulation program Glide 4.0. Glide searches for favorable and complementary interactions between a given ligand and receptor [23,25,29]. As in our previous study, we evaluated our virtual chemical library via docking simulations using Glide 4.0. First, we selected 4,007 compounds with a Glidescore of less than −6.0 using the HTVS mode. These compounds were then docked in the crystal structure of p50 using the SP mode. Docking simulations using the XP mode were then performed for the top 30% (1,202) of the compounds. A total of 33 compounds were finally selected from our virtual chemical library. The structures and Glidescores of these 33 compounds in comparison with those of NI241 are summarized in Table 1.
Table 1. Structures and Glidescores of NI241 and 33 compounds selected from our virtual chemical library.
Inhibitory effects of selected compounds in vitro
We purchased the 33 compounds that had been selected from our virtual chemical library based on SBVS. These compounds were dissolved in DMSO, and their inhibition of the DNA binding of NF-κB was verified by EMSA using an Alexa680-labeled NF-κB probe and recombinant His-tagged p50. EMSA is a conventional method for assaying sequence-specific DNA binding in vitro. The compounds were first tested at a concentration of 1 mM. Inhibition of DNA binding of NF-κB was observed only for N-018 and N-019 (Figure 2). EMSAs with N-018 and N-019 were then performed at concentrations ranging from 0 to 1 mM to investigate the dose dependence of the inhibitory effects (Figure 3; the inhibitory effect of NI241 is shown for reference). Both N-018 and N-019 inhibited the DNA binding of NF-κB at 1 mM, but they had little or no effect on NF-κB binding at less than 500 μM. Moreover, the inhibitory effects of these compounds were weaker than that of NI241. Although N-018 and N-019 were less effective than NI241 as NF-κB inhibitors, we did identify two new scaffolds of NF-κB inhibitors from our virtual chemical library.
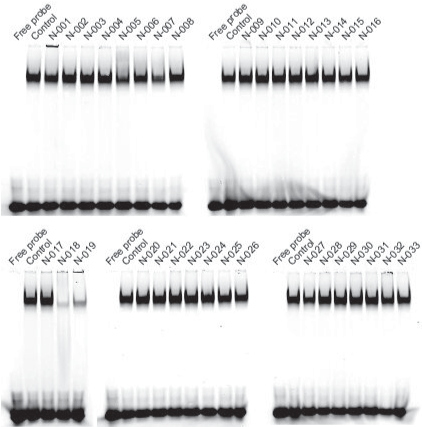
Figure 2. EMSA analysis of the inhibitory effect of 33 compounds selected from our virtual chemical library, at a concentration of 1 mM.
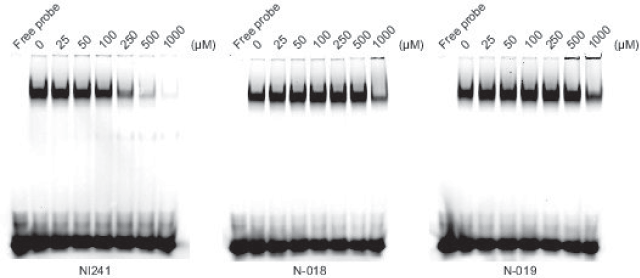
Figure 3. EMSA analysis of the dose dependence of the inhibitory effect of N-018, N-019, and NI241.
NF-κB affects a multitude of biological processes at the cellular and systemic levels and has been considered a potential target in the search for drugs to treat a variety of diseases [14,30,31]. Computational methods involving SBVS are widely used in the drug discovery process, as they enable rapid analysis of enormous amounts of data. The integration of virtual and experimental data has become increasingly important in the identification and development of novel promising compounds [19,32–34]. In our previous study, we searched our chemical library to identify compounds that directly inhibit the DNA binding of NF-κB and identified a single inhibitory molecule, NI241 (Figure 1). In addition, the mode of NI241-p50 binding was predicted using in silico docking simulations [16]. In this study, we prepared a virtual chemical library and searched for additional novel compounds using an SBVS approach that inhibit the DNA binding of p50 of NF-κB. Based on the Glidescore, in our initial screening we selected 33 compounds from our virtual chemical library containing 5,469,185 compounds (Table 1) and then investigated the inhibitory effect of these compounds by EMSA (Figure 2). Two compounds, N-018 and N-019, inhibited the DNA binding of p50 in the EMSA at a concentration of 1 mM (Figures 2 and 3).
When the docking simulations were performed according to the methods of our previous study, the Glidescores of the 33 selected compounds were lower than that of NI241 (Table 1). Although two compounds, N-018 and N-019, actually inhibited the DNA binding of p50 in vitro (Figure 2), the inhibitory effect of these two compounds was weaker than that of NI241 (Figure 3). These results suggest that the SBVS simulations were incomplete. In another previous study, we conducted structure–activity relationship (SAR) analyses of the NF-κB DNA-binding inhibitor NI241 [35]. SAR analyses provide clues that can be used to predict a model for virtual screening in silico. The 2,4-dichloro-1,3,5-triazine substructure in NI241 was identified as the essential pharmacophore responsible for inhibition of NF-κB binding [35]. This was consistent with predictions based on computational analyses indicating that hydrogen bonding between the 2,4-dichloro-1,3,5-triazine substructure in NI241 and p50 is important for inhibition of NF-κB binding [14]. However, this information was not sufficient to enable us to identify NF-κB binding inhibitors more effective than NI241. In order to identify such inhibitors via in silico SBVS, it will be necessary to predict more plausible binding modes of potential inhibitors using p50. More data regarding compounds that inhibit the DNA binding of NF-κB, such as pharmacophores, are also needed.
Two compounds, N-018 and N-019, were identified as inhibitors of the DNA binding of NF-κB in the present study. It is anticipated that the results of SAR analyses of these compounds and their analogues will provide additional useful data. Detailed analyses of these compounds containing of NI241 and their analogues will hopefully provide clues that will enable us to predict the most plausible mode of inhibitors of NF-κB binding using p50 in silico. Further works are underway to identify additional novel compounds that inhibit potently the DNA binding of NF-κB.
This work was supported in part by a Grant-in Aid from The Tokyo Biochemical Research Foundation.
- Sen R, Baltimore D (1986) Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46: 705–716.
- Müller CW, Rey FA, Sodeoka M, Verdine GL, Harrison SC (1995) Structure of the NF-kappa B p50 homodimer bound to DNA. Nature 373: 311-317. [Crossref]
- Chen FE, Huang DB, Chen YQ, Ghosh G (1998) Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature 391: 410-413. [Crossref]
- Huang DB, Phelps CB, FusCo AJ, Ghosh G (2005) Crystal structure of a free kappaB DNA: insights into DNA recognition by transcription factor NF-kappaB. J Mol Biol 346: 147-160. [Crossref]
- Ghosh G, Wang VY, Huang DB, Fusco A (2012) NF-kB regulation: lessons from structures. Immunol Rev 246: 36-58. [Crossref]
- Pahl HL1 (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18: 6853-6866. [Crossref]
2021 Copyright OAT. All rights reserv
- Karin M, Lin A (2002) NF-kappaB at the Crossroads of life and death. Nat Immunol 3: 221-227. [Crossref]
- Hayden MS, Ghosh S (2008) Shared principles in NF-kappaB signaling. Cell 132: 344-362. [Crossref]
- Karin M1 (2006) NuClear factor-kappaB in Cancer development and progression. Nature 441: 431-436. [Crossref]
- Mantovani A, Allavena P, SiCa A, Balkwill F (2008) Cancer-related inflammation. Nature 454: 436-444. [Crossref]
- Hoesel B, SChmid JA (2013) The Complexity of NF-kB signaling in inflammation and cancer. Mol Cancer 12: 86. [Crossref]
- Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18: 2195-2224. [Crossref]
- Perkins ND (2007) Integrating Cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 8: 49-62. [Crossref]
- Gilmore TD, HersCovitCh M (2006) Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 25: 6887-6899. [Crossref]
- Prabhu L, Mundade R, Korc M, Loehrer PJ, Lu T (2014) Critical role of NF- κB in pancreatic cancer. Oncotarget 5: 10969-10975. [Crossref]
- Kobayashi T, Yoshimori A, Kino K, Komori R, Miyazawa H, et al. (2009) A new small molecule that directly inhibits the DNA binding of NF-kappaB. Bioorg Med Chem 17: 5293-5297. [Crossref]
- Chen H, Yao K, Nadas J, Bode AM, Malakhova M, et al. (2012) Prediction of molecular targets of cancer preventing flavonoid compounds using computational methods. PLoS One 7: e38261. [Crossref]
- Xu L, Russu WA (2013) Molecular docking and synthesis of novel quinazoline analogues as inhibitors of transcription factors NF- κB activation and their anti-cancer activities. Bioorg Med Chem 21: 540–546. [Crossref]
- Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD (2015) Molecular docking and structure-based drug design strategies. Molecules 20: 13384-13421. [Crossref]
- LigPrep 2.0, Schrödinger, LLC, NY.
- QikProp 2.5, Schrödinger, LLC, NY.
- Meraj K, Mahto MK, Christina NB, Desai N, Shahbazi S, et al. (2012) Molecular modeling, docking and ADMET studies towards development of novel Disopyramide analogs for potential inhibition of human voltage gated sodium channel proteins. Bioinformation 8: 1139–1146. [Crossref]
- Mahto MK, Yellapu NK, Kilaru RB, Chamarthi NR, Bhaskar M (2014) Molecular designing and in silico evaluation of darunavir derivatives as anticancer agents. Bioinformation 10: 221-226. [Crossref]
- Jorgensen WL, Duffy EM (2002) Prediction of drug solubility from structure. Adv Drug Deliv Rev 54: 355-366. [Crossref]
- Glide 4.0, Schrödinger, LLC, NY.
- Friesner RA, Banks JL, Murphy RB, Halgren TA, KliCiC JJ, et al. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47: 1739-1749. [Crossref]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, et al. (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47: 1750-1759. [Crossref]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, et al. (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49: 6177-6196. [Crossref]
- Koushik Kumar G, Prasanna G, Marimuthu T, Saraswathi NT (2015) Structural basis for complementary and alternative medicine: phytochemical interaction with non-structural protein 2 protease-a reverse engineering strategy. Chin J Integr Med 21: 445–452. [Crossref]
- Epinat JC, Gilmore TD (1999) Diverse agents act at multiple levels to inhibit the Rel/NF-kappaB signal transduction pathway. Oncogene 18: 6896-6909. [Crossref]
- Leban J, Baierl M, Mies J, Trentinaglia V, Rath S, et al. (2007) A novel class of potent NF-kappaB signaling inhibitors. Bioorg Med Chem Lett 17: 5858-5862. [Crossref]
- Sakai J, Yoshimori A, Nose Y, Mizoroki A, Okita N, et al. (2008) Structure-based discovery of a novel non-peptidic small molecular inhibitor of Caspase-3. Bioorg Med Chem 16: 4854-4859. [Crossref]
- Okamoto M, Kojima H, Saito N, Okabe T, Masuda Y, et al. (2011) Virtual screening and further development of novel ALK inhibitors. Bioorg Med Chem 19: 3086-3095. [Crossref]
- Kaserer T, Beck KR, Akram M, Odermatt A, Schuster D (2015) Pharmacophore models and pharmacophore-based virtual screening: concepts and applications exemplified on hydroxysteroid dehydrogenases. Molecules 20: 22799–22832. [Crossref]
- Fujii S, Kobayashi T, Nakatsu A, Miyazawa H, Kagechika H (2014) Synthesis and structure-activity relationship study of triazine-based inhibitors of the DNA binding of NF-κB. Chem Pharm Bull (Tokyo) 62: 700-708. [Crossref]