Abstract
Recent studies have demonstrated that the cyclic guanosine monophosphate (cGMP) signaling pathway is aberrantly regulated in breast cancer and that activation of this pathway can inhibit growth and induce apoptosis of human breast cancer cells. Little is known about how a cGMP signal elicits such a response in normal or cancerous breast cells, but data suggest that the cGMP dependent protein kinases (PKG) may be involved. Here we studied the expression and importance of PKG isoforms for the anticancer effects of cGMP signaling in human breast cancer cells. We found that PKGIβ is the predominantly expressed isoform of PKG in MDA-MB-231 and SKBr3breast cancer cell lines, while ZR75-1 cells express both PKGIα and PKGIβ. Expression of PKGI was necessary for the anticancer activity of cGMP signaling in the triple negative breast cancer cell line MDA-MB-231, as decreasing expression with PKGI specific siRNA was sufficient to protect the cells from the effects of the cGMP signaling activators sulindac sulfide and NOR-3 on cell viability and apoptosis. Furthermore, the protective effects of PKG knockdown did not appear to involve oncogenic β-catenin signaling because cGMP signaling activation continued to inhibit expression of Tcf/Lef controlled genes despite the lack of PKGI expression. These findings provide further insight into the anticancer mechanism of the cGMP signaling pathway and reiterate the potential utility of this pathway for future breast cancer drug discovery efforts.
Key words
breast cancer, cyclic guanosine monophosphate (cGMP), cGMP dependent protein kinase (PKG), β-catenin
Introduction
Breast cancer remains a major health concern in the United States with more than 40,000 deaths projected to occur from the disease in 2015 [1]. The introduction of targeted therapies and improved screening strategies in the past thirty years has resulted in declining mortality rates and an increase in 5-year survival rates [2]. However, certain molecular subtypes of breast cancer, such as triple negative breast cancers (TNBC), have seen only negligible changes in prognosis [3]. TNBC cells lack appreciable expression of estrogen receptors, progesterone receptors, and HER-2, which limit treatment options; to make matters worse, these types of cancers tend to be highly aggressive with rapid progression and high rates of metastasis and recurrence [4]. Identifying and characterizing previously unrecognized proteins and molecular pathways that are involved in breast cancer cell growth and survival are paramount for developing novel therapies that will further improve breast cancer prognosis irrespective of molecular subtype.
The cyclic guanosine 3’,5’-monophosphate (cGMP) signaling pathway is one such pathway that has recently been identified as a potentially important anticancer pathway [5]. cGMP is a second messenger that is formed in response to the activity of guanylyl cyclase (GC) enzymes [6-9]. Numerous endogenous and exogenous signaling molecules, including nitric oxide (NO) and heat stable enterotoxin, stimulate cGMP formation from guanosine 5’-triphosphate (GTP) by directly activating one or more GC enzymes [6-9]. Once formed, cGMP mediates its effects my modulating the activity of various downstream effector proteins. Three classes of proteins serve as effectors of cGMP signaling; these include cyclic nucleotide gated (CNG) ion channels, phosphodiesterase (PDE) enzymes, and cGMP-dependent serine/threonine protein kinases (PKG) [6,8,10]. cGMP signaling is silenced primarily by cleavage of the cyclic bond by certain PDE isoforms to generate guanosine 5’-monophoshphate (GMP) [6,11,12].
Early studies demonstrated that cancerous breast tissues have lower levels of cGMP signaling compared to normal breast tissue [13]. This was found to be due to higher rates of cGMP degradation due to overexpression of cGMP-degrading PDE isoforms [13-15]. Additionally, cancerous breast tissues have lower levels of PKG activity compared to normal breast tissue [16], which is consistent with lower cGMP signaling activity. These lower levels of cGMP signaling activity may provide breast cancer cells with a survival advantage because numerous studies have shown that activation of cGMP signaling inhibits growth and induces apoptosis of breast cancer cells in culture [14,15,17-24], leading to the proposal that activation of cGMP signaling may be an effective strategy for future drug discovery efforts. What is especially promising about targeting the cGMP pathway is that its anticancer activity appears to be independent of breast cancer subtype, and some studies even suggest that the typically harder to treat triple negative cells are more sensitive to cGMP’s effects than cells with other molecular profiles [14-16,18].
Although the exact mechanism through which cGMP signaling elicits its anticancer response in breast cells has not been established, there are several reasons to suspect that PKG is a key regulator. First, PKG is one of the most widely expressed and commonly activated effector proteins in cGMP signaling [6,10]. Additionally, several investigators have found that the anticancer activity of cGMP signaling in breast cells correlates with PKG activation [14,15,18,21], and more recent findings have demonstrated that direct activation of PKG is sufficient to induce apoptosis of human breast cancer cells [17,18,20]. Should PKG be involved, the means through which it induces apoptosis are up for debate. However, several studies have shown a link between PKG activity and inhibition of oncogenic β-catenin signaling [14,15,21]. Unfortunately, direct involvement of PKG in the anticancer activity of cGMP signaling has yet to be demonstrated and no direct link between PKG and oncogenic β-catenin signaling has been shown.
In this study, we evaluated the importance of PKG for the anticancer activity of cGMP signaling in human breast cancer cells. We found that breast cancer cells predominantly express PKGI isoforms, which are necessary for the reduction in cell viability and activation of apoptosis that is observed in response to cGMP signaling activation in the MDA-MB-231 TNBC cell line. Furthermore, this activity occurs independent of the effects of cGMP signaling on oncogenic β-catenin mediated Tcf/Lef transcriptional activity. This study provides additional insight into the mechanism of the anticancer activity of cGMP signaling in human breast cancer and further establishes this pathway as a promising new target for future drug discovery efforts.
Materials and methods
Cell culture
The human breast cancer cell lines MDA-MB-231, SKBr-3, and ZR75-1 were purchased from ATCC. All cells were grown in RPMI-1640 with L-glutamine (Cellgro) supplemented with an antibiotic antimycotic solution (HyClone) and 10% FBS (Thermo). Cells were incubated at 37°C in 5% CO2 and humidified atmosphere. Cells were passaged at subconfluent density, and only cultures containing >95% viability as determined by trypan blue exclusion were used for experiments. All experiments were performed on cells from passage numbers 5 through 15. Morphology of the cells was consistently monitored.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
RNA was isolated from 300,000 cells using the Axygen Total RNA Miniprep Kit according to the manufacturer’s specifications. Primers specific for β-actin, PKGIα, PKGIβ, and PKGII were designed using the Primer 3 program (MIT) and were synthesized by MWG Operon. Table 1 lists the sequences of all primers. RT-PCR was performed using the QiagenOneStep RT-PCR kit. Each reaction contained 1 µL of RNA and 30 µM of each primer in a total volume of 25 µL. Thermal cycler settings were 50°C for 30 minutes; 95°C for 15 minutes; 35 cycles of 94°C for 30 seconds, 55°C for 1 minute, and 72°C for 1 minute; and 72°C for 10 minutes. Samples were stored at -20°C until analysis. Samples were analyzed by adding 10 µL to a 1% agarose gel containing ethidium bromide. The gels were run for 45 minutes at 100V, then viewed and imaged using a UVP transilluminator.
Table 1. Sequences used for RT-PCR, listed 5’ to 3’
|
Target Name
|
Forward Sequence
|
Reverse Sequence
|
|
PKGIα
|
CGAGTACTTAGCGCCCATTC
|
CTTCTCTGACAGCCGCTTCT
|
|
PKGIβ
|
CACCTTGCGGGATTTACAGT
|
ATCACCGAGCGGTACTTGTC
|
|
PKGII
|
GCCATTGCTGAACTCACAGA
|
GGAAAATTCAGGGGGTTTGT
|
|
β-actin
|
GGACTTCGAGCAAGAGATGG
|
AGCACTGTGTTGGCGTACAG
|
Western blotting
Cells were lysed in SDS lysis buffer (1% SDS, 10mM Tris pH 7.5, 7.5 µg/mL aprotonin, 5 mM benzamidine, 5mM PMSF, 50 mM NaF, and 1.25 mM NaVaO4). Protein concentration was determined using the BCA assay (Thermo Scientific). Samples were diluted in LDS sample buffer (Thermo Scientific) containing β-mercaptoethanol then incubated on a 95°C heat block for 5 minutes. 15 µg of protein was loaded per well of a 12% acrylamide resolving gel with 4% stacking gel. Gels were run at 120V for 1.5 hours in tris/glycine/SDS buffer (Bio-Rad). Proteins were transferred to nitrocellulose membranes at 350 mA for 1 hour in tris/glycine buffer (Bio-Rad) containing 20% methanol. Membranes were blocked in TBST with 5% BSA. Membranes were incubated 2 hours to overnight in 1:1000 dilution of the appropriate primary antibody followed by incubation in 1:5000 dilution of anti-rabbit secondary antibody for 1 to 3 hours. All antibodies were purchased from Cell Signaling Technologies. Membranes were incubated in Immobilin ECL (Millipore) for 60 seconds before exposing to autoradiography film (Thermo Scientific).
PKGI knockdown
PKG siRNA, transfection reagent, and transfection medium were purchased from Santa Cruz Biotechnologies and transient transfection was performed using the manufacturer’s protocol. Cells were plated in growth medium and incubated as described above for at least 24 hours. siRNA was diluted to 7.5 pg per µL of transfection media. 6 µL of transfection reagent was added per 100 µL of transfection media/siRNA mixture and mixture was incubated at room temperature for 45 minutes. Growth media was removed from the cells and the cells were washed with PBS. Transfection mixture was added to the cells and the cells were incubated at 37°C for 5 hours. After incubation, an equal volume of growth media containing 20% FBS was added to the cells and cells were incubated an additional 18-24 hours at which time all media was removed and replaced with normal growth media. For each transfection experiment, one sample was treated with transfection media containing transfection reagent but no siRNA as a negative control.
Drug treatments
Sulindac sulfide and NOR-3 were purchased from Enzo Life Sciences and diluted in DMSO. Cells were treated with transfection media containing transfection reagent alone or with PKGI siRNA as described above. After 24 hours of transfection, media containing the transfection reagent was removed and replaced with normal growth media containing the appropriate concentration of drug or DMSO vehicle control. DMSO concentration was maintained at 0.1% for any experiment in which drug treatments were used. Cells were exposed to drug treatments for 72 hours for the cell viability assay and 6 hours for the Western blotting assays. The time points and dosages used were chosen based on previous studies that demonstrated these conditions produced optimum effects in similar assays [14,15].
Cell viability assay
72 hours after drug treatment was initiated, cell growth was assayed using the CellTiter 96 Assay (Promega). The assay was performed according to the manufacturer’s specifications. Absorbance was measured using a Victor 2 plate reader.
Experimental design and data analysis
Each experiment was performed in either duplicate or triplicate and repeated at least once. All graphs were constructed in GraphPad Prism 5.0 software. Bars represent mean of each replicate and error bars represent standard error of the mean. RT-PCR band density was calculated using ImageJ software (NIH). Statistical analysis was performed using Student’s t test.
Results
Human breast cancer cells predominately express PKGI
Human cells possess two genes for PKG, which give rise to the PKGI and PKGII proteins. mRNA transcribed from the gene for PKGI can be alternatively spliced to yield the distinct PKGIα and PKGIβ protein products [6,10]. We designed primers that were specific to each of the three possible isoform mRNA transcripts (Table 1), and then evaluated PKG mRNA expression by RT-PCR. As shown in Figure 1A, all three breast cancer cell lines – MDA-MB-231, SKBr3, and ZR75-1 – express comparable levels of PKGIβ mRNA. However, the cell lines differed in their expression of other PKG isoforms. MDA-MB-231 cells lacked detectable levels of PKGIα or PKGII mRNA. SKBr-3 cells possessed both PKGIα and PKGII mRNA transcripts. However, the levels of mRNA for the Iα and II isoforms were substantially lower than that of Iβ, producing band densities that were on average just 16% and 3% that of PKGIβ, respectively (Figure 1B). ZR75-1 cells expressed the highest levels of PKGII mRNA of any of the cell lines assayed, yet these levels were still modest compared to the other PKG isoforms.ZR75-1 cells also expressed much higher levels of PKGIα mRNA when compared to the other cell lines. While the level of Iα mRNA was lower than Iβ in this cell line, there was not a significant difference between the amounts of these two transcripts in ZR75-1 cells. Using an antibody specific for PKGI, we confirmed the expression of PKGI at the protein level in each of the three cell lines by western blotting (Figure 1C).
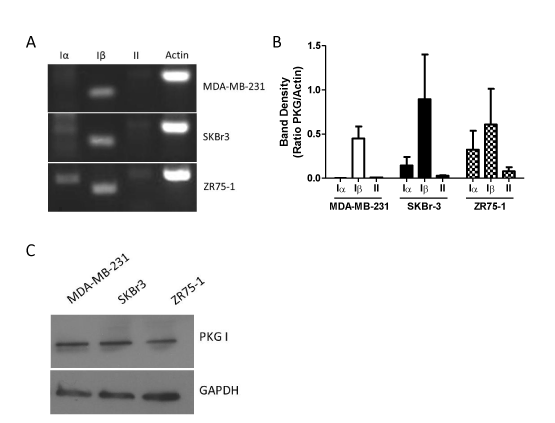
Figure 1. Expression of PKG isoforms in MDA-MB-231, SKBr-3, and ZR75-1 breast cancer cells. A: expression as determined by RT-PCR using primers specific for each PKG isoform. Actin was included as a positive control. B: densitometry of RT-PCR results. Bars represent the average ratio of PKG band density to actin band density from two separate experiments. Error bars are standard error. C: expression of PKGI as determined by western blot. GAPDH was included as a loading control.
PKGI is necessary for the anticancer activity of cGMP signaling
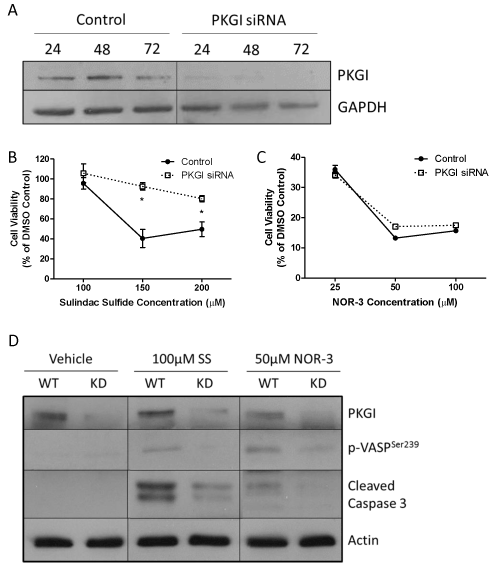
Because of the limited treatment options available for and poor prognosis of TNBC and because previous studies suggest that these cells are more sensitive to the anticancer activity of cGMP signaling, we utilized the MDA-MB-231 TNBC cell line to study the importance of PKGI for mediating the anticancer activity of cGMP signaling. Cells were treated with siRNA specific to PKGI in order to decrease the expression of the protein. As depicted in Figure 2A, as little as 24 hours of transient transfection was sufficient to produce a substantial reduction in PKGI protein levels. 72 hours of transfection resulted in undetectable levels of PKGI protein by western blot.
Control MDA-MB-231 cells that were exposed to transfection reagent but not to PKGI siRNA and cells exposed to both transfection reagent and PKGI siRNA were treated with cGMP signaling activators 24 hours after transfection. 72 hours after drug treatment cell viability was measured. Sulindac sulfide (SS) activates cGMP signaling by inhibiting PDE, while NOR-3 activates cGMP signaling by activating soluble GC enzymes. As shown in Figures 2B and 2C, SS and NOR-3 decreased the viability of both control and PKGI siRNA treated cells. However, PKGI siRNA treated cells were significantly less sensitive to the effects of SS than the control cells. With 150µM SS treatment, only 40.5% of the control cells were viable compared to 92.5% of siRNA treated cells. Although siRNA treated cells were slightly less sensitive to the effects of 50 µM NOR-3, this difference was not statistically significant.
Figure 1. Expression of PKG isoforms in MDA-MB-231, SKBr-3, and ZR75-1 breast cancer cells. A: expression as determined by RT-PCR using primers specific for each PKG isoform. Actin was included as a positive control. B: densitometry of RT-PCR results. Bars represent the average ratio of PKG band density to actin band density from two separate experiments. Error bars are standard error. C: expression of PKGI as determined by western blot. GAPDH was included as a loading control.
By performing a Western blot for PKGI and vasoactivator-stimulated phosphoprotein phosphorylated at the serine 239 residue (p-VASPSer239), a well-documented intracellular marker of PKG activity [25], we confirmed that PKG siRNA reduced the expression of PKGI protein and reduced PKG activity, respectively (Figure 2D). PKGI expression was reduced in all knockdown (KD) cells compared to the wild type (WT) control cells, regardless of drug treatment. As expected, activation of cGMP signaling with 100 µM sulindac sulfide (SS) or 50 µM NOR-3 resulted in activation of PKG in the WT cells as evidenced by increased expression of p-VASPSer239 in drug treated compared to vehicle treated WT cells. Decreased PKGI expression reduced this effect on PKG activity, as KD cells had only negligible levels of p-VASPSer239 expression.
We also evaluated the effects of siRNA treatment on induction of apoptosis in response to drug treatment by measuring the levels of cleaved caspase 3 via Western blotting. As shown in Figure 2D, both 100 µM SS and 50 µM NOR-3 activated apoptosis in the WT cells as indicated by increased expression of cleaved caspase 3 in these cells compared to vehicle treated cells. The effects of SS were more pronounced than those of NOR-3. Both SS and NOR-3 KD cells showed significant reductions in cleaved caspase expression with respect to their WT counterparts, demonstrating that PKGI siRNA treatment reduced the sensitivity of MDA-MB-231 cells to the pro-apoptotic effects of cGMP signaling induced by each drug.
PKGI mediates anticancer activity independent of β-catenin
In order to determine whether oncogenic β-catenin signaling is involved in the PKG-dependent effects of cGMP signaling in MDA-MB-231 cells, we evaluated the expression of two proteins, c-myc and survivin, whose expression is regulated by the β-catenin activated Tcf/Lef family of transcription factors. As shown in Figure 3, neither PKGI siRNA nor 100 µM SS treatment resulted in appreciable changes in β-catenin expression, and KD alone did not alter the expression of c-myc or survivin. 100 µM SS treatment caused significant reduction in the expression of both c-myc and survivin compared to vehicle treatment. PKGI siRNA treatment actually exacerbated this effect as the KD cells demonstrated even lower levels of c-myc and survivin expression despite lacking PKGI expression or activity.
2021 Copyright OAT. All rights reserv
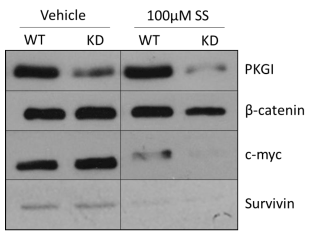
Figure 3. Importance of PKGI for the decrease in β-catenin signaling elicited by cGMP signal activity. Expression of PKGI, pVASPSer239, β-catenin, and c-myc were measured by Western blot after 6 continuous hours of drug treatment. Actin was included as a loading control. “WT” represents control MDA-MB-231 cells and “KD” PKGI siRNA treated cells.
Discussion
Numerous studies have identified the cGMP signaling pathway as a potentially important anticancer pathway in human breast cancer [5,13-24]. However, very little is known about signal transduction through the pathway in human breast cells or the mechanism whereby cGMP elicits its growth inhibitory effects. We found that breast cancer cells predominantly express PKGI isoforms while lacking appreciable expression of PKGII. This is consistent with previous studies that found similar expression patterns in other breast cancer cell lines as well as patient tissue samples [16-18,20]. Previous studies demonstrate a correlation between cGMP signaling activity, PKG activation, and inhibition of breast cancer cell growth [14,15,18,21]. However, the data presented here demonstrate for the first time a direct link between PKGI and the anticancer activity of cGMP signaling in MDA-MB-231 breast cancer cells. Cells that lacked expression of PKGI were significantly less sensitive to the decrease in cell viability and increase in apoptosis that was triggered by cGMP signaling. This observation indicates that PKGI expression is necessary for the growth inhibitory effects of cGMP in these cells. While more work is necessary to determine whether these findings are also true for other types of breast cancer cells or in vivo, other studies that have used pharmacological activators or inhibitors of PKG in other cell lines have had similar results [17,20]. Furthermore, we cannot rule out the involvement of cGMP effector proteins other than PKGI as PKGI knockdown did not completely rescue the cells from the anticancer effects of cGMP signaling activation.
Interestingly, we found that PKG appeared to be more important for the anticancer activity of SS than NOR-3. PKGI knockdown did not rescue cells from the effects of NOR-3 on cell viability and was less effective at protecting the cells from apoptosis induction by NOR-3 when compared to SS. Because these two drugs activate cGMP signaling through different mechanisms (inhibition of PDE for SS and activation of GC for NOR-3), this observation could indicate that signal transduction through the cGMP pathway may differ depending on the manner in which the pathway is activated. Namely, activating cGMP signaling by inhibiting degradation of the second messenger may result in almost exclusive activation of PKG, whereas activating cGMP signaling by promoting production of the second messenger may result in activation of PKG as well as one or more additional effector proteins. This scenario is entirely plausible given the compartmentalization of cyclic nucleotide signaling that has been demonstrated in mammalian cells and the differences in subcellular localization that has been reported for different pathway components [6,26-28]. Another likely explanation for the differences seen with NOR-3 and SS is that the anticancer activity of NOR-3 is at least partially independent of cGMP signaling. This is consistent with the fact that NOR-3 is a NO donor, and NO causes free radical damage that is known to be toxic to breast cancer cells [29]. This idea is supported by the data here that showed NOR-3 to be significantly more potent for reducing cell viability than for activating cGMP signaling or inducing apoptosis. Additional studies are needed to better characterize the differences between these two compounds.
There is relatively little information in the literature about how activation of PKG could induce apoptosis in human breast cells, but several studies have suggested the involvement of oncogenic β-catenin signaling since activation of cGMP signaling reduces the activity of β-catenin activated Tcf/Lef transcription factors and PKG can phosphorylate β-catenin in vitro in a manner that promotes its proteasomal degradation [14,15,21,30,31]. We confirmed previous findings by demonstrating that activation of cGMP signaling with sulindac sulfide inhibits expression of β-catenin regulated proteins like c-myc and survivin. However, this activity appears to be independent of PKG because treating cells with PKGI siRNA did not rescue them from these effects. In fact, PKGI knockdown made the cells even more sensitive to the effects of cGMP signaling on the β-catenin pathway. These findings also indicate that inhibition of β-catenin signaling is not necessary for the anticancer activity of the cGMP pathway since cells remained viable and evaded apoptosis even though β-catenin signaling was reduced. Future studies should explore β-catenin independent mechanisms such as the recently proposed mechanisms involving death associated protein kinase, ERK, and/or JNK [20,24,32,33].
While more work is necessary to fully characterize the anticancer activity of cGMP signaling, our findings demonstrate that PKGI is playing a critical role. Because PKGI is widely expressed throughout the body and is known to play an important role in smooth muscle cell and neuron function, this particular protein is unlikely to be a realistic target for future drug discovery efforts. However, better characterization of the cGMP-PKG pathway in breast cancer still has a strong potential to unveil novel anticancer targets as well as to enhance our understanding of cellular dysfunction within breast cancer.
Our findings presented here further validate that the cGMP signaling pathway is independent of the estrogen receptor, progesterone receptor, and HER2 signaling pathways, indicating the broad therapeutic utility of this pathway for a range of molecular subtypes. Moreover, the sensitivity of a triple negative cell line like MDA-MB-231 to the anticancer activity of cGMP signaling makes this an especially interesting pathway to target for treating these highly aggressive types of breast cancers.
References
- Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, et al. (2015) SEER Cancer Statistics Review, 1975-2012, National Cancer Institute. Bethesda. [http://seer.cancer.gov/csr/1975_2012/]. Based on November 2014 SEER data submission, posted to the SEER web site, April 2015.
- Berry DA, Cronin KA, Plevritis SK, Fryback DG, Clarke L, et al. (2005) Effect of screening and adjuvant therapy on mortality from breast cancer. N Engl J Med 353: 1784-1792. [Crossref]
- Santa-Maria CA, Gradishar WJ (2015) Changing Treatment Paradigms in Metastatic Breast Cancer: Lessons Learned. JAMA Oncol 1: 528-534. [Crossref]
- Tomao F, Papa A, Zaccarelli E, Rossi L, Caruso D, et al. (2015) Triple-negative breast cancer: new perspectives for targeted therapies. Onco Targets Ther 8: 177-193. [Crossref]
- Windham PF, Tinsley HN (2015) cGMP signaling as a target for the prevention and treatment of breast cancer. Semin Cancer Biol 31: 106-110. [Crossref]
- Schmidt HH, Hofmann F, Stasch JP (2009) Handbook of Experimental Pharmacology 191. cGMP: generators, effectors and therapeutic implications. Preface. Handb Exp Pharmacol: v-vi. [Crossref]
- Schulz S, Chinkers M, Garbers DL (1989) The guanylate cyclase/receptor family of proteins. FASEB J 3: 2026-2035. [Crossref]
- Rehmann H, Wittinghofer A, Bos JL (2007) Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat Rev Mol Cell Biol 8: 63-73. [Crossref]
- Feil R, Kemp-Harper B (2006) cGMP signalling: from bench to bedside. Conference on cGMP generators, effectors and therapeutic implications. EMBO Rep 7: 149-153. [Crossref]
- Lincoln TM, Cornwell TL (1993) Intracellular cyclic GMP receptor proteins. FASEB J 7: 328-338. [Crossref]
- Conti M, Beavo J (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76: 481-511. [Crossref]
- Omori K, Kotera J (2007) Overview of PDEs and their regulation. Circ Res 100: 309-327. [Crossref]
- Singer AL, Sherwin RP, Dunn AS, Appleman MM (1976) Cyclic nucleotide phosphodiesterases in neoplastic and nonneoplastic human mammary tissues. Cancer Res 36: 60-66. [Crossref]
- Tinsley HN, Gary BD, Keeton AB, Zhang W, Abadi AH, et al. (2009) Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol Cancer Ther 8: 3331-3340. [Crossref]
- Tinsley HN, Gary BD, Keeton AB, Lu W, Li Y, et al. (2011) Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/ß-catenin-mediated transcription in human breast tumor cells. Cancer Prev Res (Phila) 4: 1275-1284. [Crossref]
- Karami-Tehrani F, Fallahian F, Atri M (2012) Expression of cGMP-dependent protein kinase, PKGIα, PKGIβ, and PKGII in malignant and benign breast tumors. Tumour Biol 33: 1927-1932. [Crossref]
- Fallahian F, Karami-Tehrani F, Salami S (2012) Induction of apoptosis by type Iβ protein kinase G in the human breast cancer cell lines MCF-7 and MDA-MB-468. Cell Biochem Funct 30: 183-190. [Crossref]
- Fallahian F, Karami-Tehrani F, Salami S, Aghaei M (2011) Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS J 278: 3360-3369. [Crossref]
- Kumazoe M, Sugihara K, Tsukamoto S, Huang Y, Tsurudome Y, et al. (2013) 67-kDa laminin receptor increases cGMP to induce cancer-selective apoptosis. J Clin Invest 123: 787-799. [Crossref]
- Isshiki K, Matsuda S, Tsuji A, Yuasa K (2012) cGMP-dependent protein kinase I promotes cell apoptosis through hyperactivation of death-associated protein kinase 2. Biochem Biophys Res Commun 422: 280-284. [Crossref]
- Thompson WJ, Piazza GA, Li H, Liu L, Fetter J, et al. (2000) Exisulind induction of apoptosis involves guanosine 3',5'-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res 60: 3338-3342. [Crossref]
- Wen HC, Chuu CP, Chen CY, Shiah SG, Kung HJ, et al. (2015) Elevation of soluble guanylate cyclase suppresses proliferation and survival of human breast cancer cells. PLoS One 10: e0125518. [Crossref]
- Saravani R, Karami-Tehrani F, Hashemi M, Aghaei M, Edalat R (2012) Inhibition of phosphodiestrase 9 induces cGMP accumulation and apoptosis in human breast cancer cell lines, MCF-7 and MDA-MB-468. Cell Prolif 45: 199-206. [Crossref]
- Mujoo K, Sharin VG, Martin E, Choi BK, Sloan C, et al. (2010) Role of soluble guanylyl cyclase-cyclic GMP signaling in tumor cell proliferation. Nitric Oxide 22: 43-50. [Crossref]
- Deguchi A, Soh JW, Li H, Pamukcu R, Thompson WJ, et al. (2002) Vasodilator-stimulated phosphoprotein (VASP) phosphorylation provides a biomarker for the action of exisulind and related agents that activate protein kinase G. Mol Cancer Ther 1: 803-809. [Crossref]
- Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, et al. (2014) Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov 13: 290-314. [Crossref]
- Lomas O, Zaccolo M (2014) Phosphodiesterases maintain signaling fidelity via compartmentalization of cyclic nucleotides. Physiology (Bethesda) 29: 141-149. [Crossref]
- Fischmeister R, Castro LR, Abi-Gerges A, Rochais F, Jurevicius J, et al. (2006) Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res 99: 816-828. [Crossref]
- Cheng H, Wang L, Mollica M, Re AT, Wu S, et al. (2014) Nitric oxide in cancer metastasis. Cancer Lett 353: 1-7. [Crossref]
- Rice PL, Kelloff J, Sullivan H, Driggers LJ, Beard KS, et al. (2003) Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells. Mol Cancer Ther 2: 885-892. [Crossref]
- Kwon IK, Wang R, Thangaraju M, Shuang H, Liu K, et al. (2010) PKG inhibits TCF signaling in colon cancer cells by blocking beta-catenin expression and activating FOXO4. Oncogene 29: 3423-3434. [Crossref]
- Lan T, Chen Y, Sang J, Wu Y, Wang Y, et al. (2012) Type II cGMP-dependent protein kinase inhibits EGF-induced MAPK/JNK signal transduction in breast cancer cells. Oncol Rep 27: 2039-2044. [Crossref]
- Wang R, Kwon IK, Singh N, Islam B, Liu K, et al. (2014) Type 2 cGMP-dependent protein kinase regulates homeostasis by blocking c-Jun N-terminal kinase in the colon epithelium. Cell Death Differ 21: 427-437. [Crossref]