We present a single-cell application to determine PIK3CA mutations in CTCs, which uncovered the degree of intra-patient heterogeneity in patients with metastatic hormone receptor-positive breast cancer (HR+ MBC) and high CTC count (>10 CTCs/7.5mL). Using CellSearch and DEPArray we isolated circulating tumor cells (CTCs) and white blood cells (WBCs) from peripheral blood and sequenced PIK3CA exons 9 and 20 by targeted amplicon sequencing. Comparative analysis between the primary tumor (PT, n=27 patients), circulating cell-free DNA (cfDNA, n=31 patients), single (n=146 CTCs) and pools (n=70 CTC suspensions, ranging 5-120 cells/suspension) of CTCs from 26 patients and metastases/DTCs (n=11 patients) was performed. Mutations were frequent in PT (15/27 (55.5%)) and showed slight and substantial agreement with cfDNA (n=21; kappa=0.14) and CTCs (n=22; kappa=0.6733), respectively. A wild-type genotype in WBCs indicates a high specificity. Inter-compartmental concordance was observed in 13/18 (72.2%) patients and temporal heterogeneity in 4/18 patients (22.2%). CTC analysis reveals both mutational homo- and heterogeneity with cases showing the presence of different mutant and wild-type CTC subpopulations. Additionally, unique double-mutated CTCs were detected in 8/26 patients (30.7%). The developed liquid biopsy provides an insight into inter and intra-patient PIK3CA mutational heterogeneity in patients with HR+ MBC, paving the way towards the application of a more personalized medicine.
PIK3CA, circulating tumor cells, heterogeneity, breast cancer, liquid biopsy
Breast cancer is a heterogeneous disease, where specific genetic alterations can be associated with response or resistance to a given therapy [1,2]. The PI3K-Akt-mTor pathway is a validated target in the treatment of breast cancer [3,4]. The catalytic subunit of the phosphoinositide-3 kinase (PIK3CA) is mutated in up to 25% of patients with breast cancer, with mutation frequencies rising to 40% in the hormone receptor-positive (HR+) subgroups [5,6]. Hotspot mutations in helical and kinase domain results in constitutively enzymatic activity, enhanced tumorigenicity and the capability to form heterogeneous tumors [7-9].
Genetic discordances between PTs and the metastasis have been repeatedly observed, emphasizing the need for real-time cancer geno- and phenotyping. Since the sequential assessment of biomarkers in metastases is not routinely done, a real-time ‘liquid biopsy’ with the molecular characterization of circulating tumor cells (CTCs) may constitute an alternative approach [10,11]. In both localized and advanced breast cancer the enumeration of CTCs has demonstrated clinical validity with respect to progression-free and overall survival [12]. In addition, the molecular characterization of these cells may assist in the identification of relevant biomarkers [13-15].
Assessment of the inter-patient heterogeneity allows for a useful stratification of patients in histological, molecular and functional subtypes, but is not able to predict the prevailing driving mechanisms of disease progression in an individual patient at any particular point in time. The heterogeneous nature of cancer can be demonstrated by divergence and subtle differences of the molecular profiles between early and advanced disease [16,17]. These observations may be in part explained by the emergence of different tumor cell subpopulations within the primary and/or metastatic sites [18,19]. Clinically, this subclonal development whilst receiving treatment was translated into disease progression and is defined as drug resistance. Therefore, the molecular characterization of the progressing tumor burden and the longitudinal follow-up are an essential prerequisite for personalized medicine to become a reality. This emphasizes the attractiveness of a liquid biopsy using CTCs as real-time input material, which may infer the underlying PT and/or metastases [20,21]. Performing single cell sequencing allows the detection of rare subpopulations, the assessment of the level of genetic heterogeneity and its clonal evolution during therapy [22-24].
Within this study we utilized the previously reported methodology to obtain single CTCs by di-electrophoresis [25] and subjected these samples to a PIK3CA mutational analysis by massive parallel sequencing (MPS). We have assessed the degree of mutational heterogeneity in PT, cfDNA, CTCs and metastases from patients with HR+ MBC. Additionally, this represents a feasibility study to perform CTC mutational analysis by liquid biopsy in a routine clinical setting.
Cell culture and blood spiking experiments
Quality control and performance characteristics of the PIK3CA assay were assessed by profiling and spiking experiments with breast cancer cell lines (MCF-7, BT-20, MDA-MB-361, MDA-MB-231 and SK-BR-3) [26]. Cell lines were cultured as a monolayer at 37°C and 5% CO2 atmosphere in supplemented RPMI-1640 medium (10% FBS, 1% Glu and 1% Anti-Anti) (Invitrogen, BE). MCF-7, BT-20 and SK-BR-3 cells were spiked into 7.5mL blood samples collected from healthy donors (HDs) and processed on the CellSearch system (Janssen Diagnostics, LLC, USA). From the same batch of harvested cells for spiking, DNA was purified, serving as a positive control.
Patient accrual and sample collection
We enrolled total number of 60 patients with HR+ MBC between September 2011 and June 2014 at the Oncology Centre of the GZA Hospitals Sint-Augustinus (Antwerp, BE). Formalin-fixed paraffin-embedded (FFPE) tissue sections from the PT and/or metastases was retrieved from the pathology department. Temporal matched blood samples for CTC enumeration and plasma was collected before a new line of therapy. From two patients we additionally obtained pleural fluid (PF), ascites (A) and/or bone marrow (BM) biopsies for analysis of disseminated tumor cells (DTCs). Blood samples were collected in CellSave (Janssen Diagnostics) and Cell Preparation Tubes (CPT, Becton Dickinson). Study participants (both patients and HDs) gave written informed consent. The Institutional Review Board/Independent Ethics Committee/Research Ethics Board of GZA Hospitals Sint-Augustinus approved the clinical protocol.
DNA extraction from FFPE tissue sections and plasma
Tumor-rich areas on FFPE tissue sections (5 µm) were marked by the pathologist, manually macrodissected and collected in a 1,5 mL Protein LoBind microcentrifuge tube (Eppendorf, DE). DNA was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, USA), according to the manufacturer’s instructions. All blood samples were processed within 1 hour after collection. Blood collected in Cell Preparation Tubes (CPT, BD Biosciences) allows the separation of plasma and peripheral blood mononuclear cells (PBMCs). cfDNA was extracted from 400 μL of plasma using the QIAamp Micro kit (Qiagen, USA). Upon purification, DNA samples were quantitated with the Nanodrop Spectrophotometer System (NanoDrop Technologies, USA) and stored at -20°C until use.
DEPArray purification and whole genome amplification of single cells from CellSearch-enumerated CTC cartridges
CellSave-collected samples were subjected to EpCAM-based immunomagnetical CTC enrichment and enumeration with the CellSearch CTC kit (Janssen Diagnostics, USA). Enumerated CellSearch cartridges were stored at 4°C until CTC cell suspensions were aspirated and transferred into an A300K cartridge for single and groups of cells isolation by di-electrophoresis using the DEPArray system (Silicon Biosystems, Italy), as described previously [25]. Following recovery and volume reduction, single or pools of CTCs or white blood cells (WBCs), 1 ng of unamplified genomic DNA (gDNA) as positive control and reaction mix water (negative control) underwent a whole genome amplification (WGA) using the Ampli1 kit (Silicon Biosystems, IT) [27]. Success of WGA was evaluated using the Ampli1 QC4 kit (Silicon Biosystems, Bologna, Italy).
Primer design and exon-targeted PCR
The MseI restriction, which is proper to the Ampli1 whole genome amplification, was taken into account during the fusionprimer design (Figure1) for downstream PIK3CA amplification and pyrosequencing. Additionally, we assessed the discriminating power of the exon 9 primer pair between the PIK3CA gene and a pseudo-gene on chromosome 22 (NCBI Reference Sequence: NG_027450.1) that has >95% sequence homology [29]. As shown in Table 1, existing target-specific sequences were used for PIK3CA Ex9 [28,29]. Fifty nanograms of extracted DNA from FFPE tissue sections, 2 μL cfDNA from plasma and 1 μL of the WGA product from single and groups of DEPArray-purified cells were subjected to PCR in a final volume of 25 µl containing 1xFastStart High Fidelity Reaction Buffer, 1.6 and 1.4 mM MgCl2 (for Ex9 and 20, respectively), 400 nM of HPLC-purified fusionprimers (IDT Technologies, BE), 200μM dNTPs, 1.25U of FastStart High Fidelity Polymerase Enzyme (All Roche Molecular Systems Inc., USA) and PCR grade water. Program conditions were: 4 min at 95°C; followed by 30 cycles of 30s at 95°C, 45s at 61.8 or 65.6°C (for Ex9 and 20, respectively) and 1 min at 72°C; and 8 min at 72°C. PCR amplicons were purified with Agencourt AMPure XP beads (Beckman Coulter, Spain) at a ratio of 1:1.6 and stored at -20°C until use.
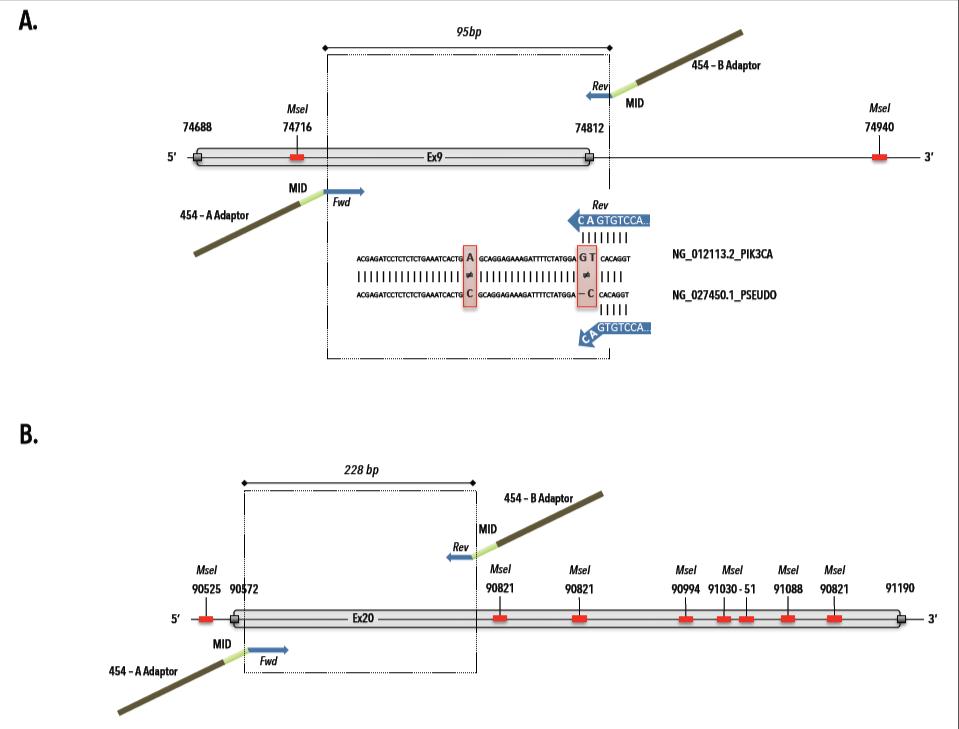
Figure 1. MseI restriction map and 454 fusionprimer design for PIK3CA exon 9 and 20. A) Primer design for PIK3CA exon 9 (gi|383087749|ref|NG_012113.2|:74685-74946 Homo sapiens phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA), RefSeqGene (LRG_310) on chromosome 3) with selection of target-specific reverse primer, which avoids pseudogene (ref|NG_027450.1|: Homo sapiens phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha pseudogene (LOC100422375) on chromosome 22) amplification.
Table 1. Primer design and sequences for PIK3CA mutational analysis by 454 MPS. The presented sequences provide an overview of the fusionprimer design for targeted amplicon sequencing and consist of a universal (i.e. sequencing adaptor), multiplex identifier (MID) and target-specific segment
|
Exon
|
Strand
|
Universal Segment
|
MID Segment
|
Target Specific Segment
|
Reference
|
|
9
|
Fwd
|
CGTATCGCCTCCCTCGCGCCATCAG
|
MID 1-6
|
AACAGCTCAAAGCAATTTCTACACG
|
[29]
|
| |
Rev
|
CTATGCGCCTTGCCAGCCCGCTCAG
|
MID 1-8*
|
CCATTTTAGCACTTACCTGTGAC
|
[30]
|
|
20
|
Fwd
|
CGTATCGCCTCCCTCGCGCCATCAG
|
MID 1-6
|
AGGAGATGTGTTACAAGGCTTATCTA
|
454 FusionPrimers (IDT)
|
| |
Rev
|
CTATGCGCCTTGCCAGCCCGCTCAG
|
MID 1-8*
|
AATCCATTTTTGTTGTCCAGCC
|
454 FusionPrimers (IDT)
|
|
|
|
|
MID Segment
|
|
|
|
|
|
MID 1
|
ACGAGTGCGT
|
|
|
|
|
|
MID 2
|
ACGCTCGACA
|
|
|
|
|
|
MID 3
|
AGACGCACTC
|
|
|
|
|
|
MID 4
|
AGCACTGTAG
|
|
|
|
|
|
MID 5
|
ATCAGACACG
|
|
|
|
|
|
MID 6
|
ATATCGCGAG
|
|
|
|
|
|
MID 7
|
CGTGTCTCTA
|
|
|
|
|
|
MID 8
|
CTCGCGTGTC
|
|
|
* MID 4 was excluded in the reverse primers, due to inability to generate PCR amplicon.
MID: Multiplex identifier, Fwd: forward, Rev: reverse
TCAG: Roche 454 sequencing key
PIK3CA mutation screening by 454 pyrosequencing
Purified PCR products were measured on the BioAnalyzer (Agilent Technologies, USA), diluted to 1 × 109 molecules/µl in 1xTE buffer and equimolarly pooled to create a PIK3CA amplicon library at 1 × 107 molecules/µl. Amplicon libraries of the spiked and patient samples were subjected to targeted pyrosequencing in fifteen independent runs. Emulsion PCR of the libraries was carried out using the Lib-A Titanium emulsion PCR (emPCR) Kit, with an input of 0.5 molecules of library DNA per capture bead. In total 500.000 enriched beads were loaded on a Titanium PicoTiterPlate (PTP) and placed in the GS Junior instrument. The libraries were sequenced with Titanium reagents, and base calling was performed with the ‘FullProcessingAmplicons’ run processor settings in the GS Amplicon Variant Analyzer software (All Roche Molecular Systems Inc., USA). Reads were filtered for mixed and dot reads, adaptor trimmed and de-multiplexed using the ‘Both’ encoding multiplexer. Filter-passed reads were aligned against the PIK3CA reference sequence (NCBI NG_012113.2) and compared to COSMIC and dbSNP databases.
Statistics and performance analysis
During blood spiking experiments with the reference cell lines models, the level of true/false positives and negatives allowed for the calculation of performance parameters of the assay for PIK3CA mutation detection in single CTCs. Optimal sensitivity and specificity of the variant allele frequency (VAF) stringency were assessed by diagnostic odd ratios [30]. To study concordances and correlations between the occurrence of PIK3CA mutations in PT, plasma and CTC samples, standard statistical analysis techniques were used including Cohen’s kappa test (concordance) and Pearson correlation coefficients. A two-sided P<0.05 was considered to be statistically significant. All statistical analyses and graphical representations were performed with SPSS and Microsoft Excel.
PIK3CA PCR assay validation and quality assurance for PIK3CA mutational analysis
Prior to pyrosequencing, PCR assays were validated by direct sequencing of MCF7, BT20 and healthy donor WBC genomic DNA on a 3730XL ABI DNA sequencer (Applied Biosystems, Foster City, CA) using the Big Dye terminator V1.1 DNA sequencing kit. PCR optimization for exons 9 and 20 resulted in 165bp and 301bp amplicons, respectively. Sanger sequencing revealed on-target specificity and identified heterozygously mutant (MT) and wild-type (WT) sequences in genomic DNA from tumor cell lines (MCF7 and BT20) and healthy donor white blood cells, respectively (Figure 2). We validated our 454 pyrosequencing assay on replicate samples from fresh gDNA, obtained from cell lines (i.e., BT-20, MDA-MB-361, MDA-MB-231, MCF-7, SK-BR-3) and gDNA from externally validated FFPE-patient tissue sections (n=3). All inter-run technical replicates from cell line samples generated expected PIK3CA mutational profiles with a coefficient of variations ranging between 0–5,17%. Duplicate analysis of FFPE tissue sections revealed inter-run reproducibility and delivered concordant PIK3CA genotypes for both exons (Figure 3).
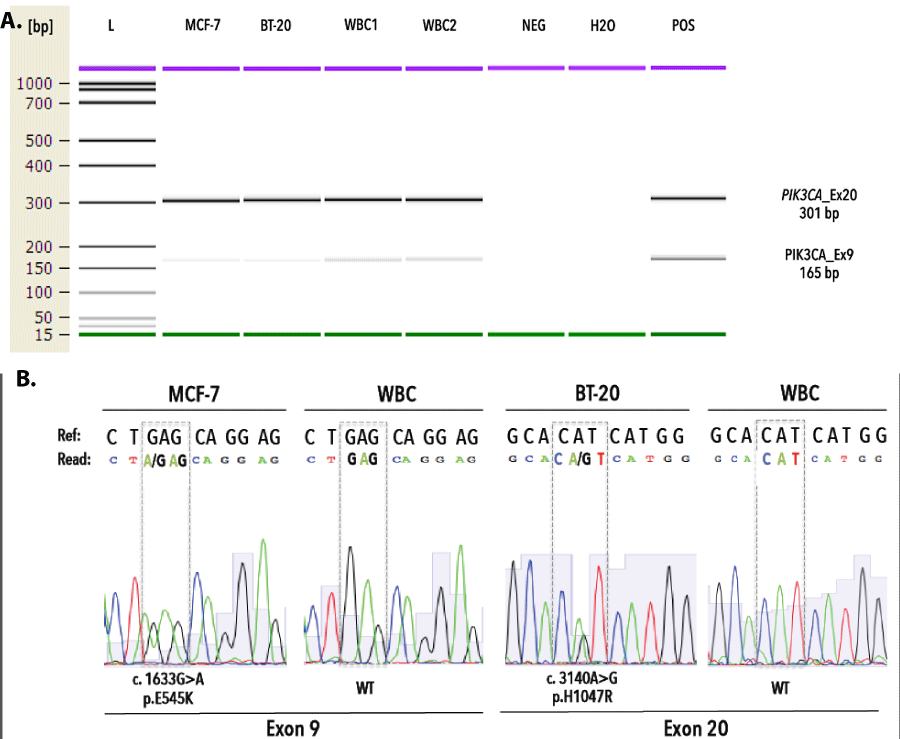
Figure 2. Validation of PCR assay targeting PIK3CA exon 9 and 20. A) Fragment analysis of PIK3CA assays demonstrate the generation of monoclonal PCR fragments, measuring 165bp and 301bp for exon 9 and exon 20, respectively. B) Targeted Sanger sequencing of gDNA from MCF-7, BT-20 and WBC reveals expected MT and WT sequences for exon 9 and 20. L denotes DNA ladder, WBC denotes white blood cells, bp denotes base pairs, Ref denotes reference sequence.
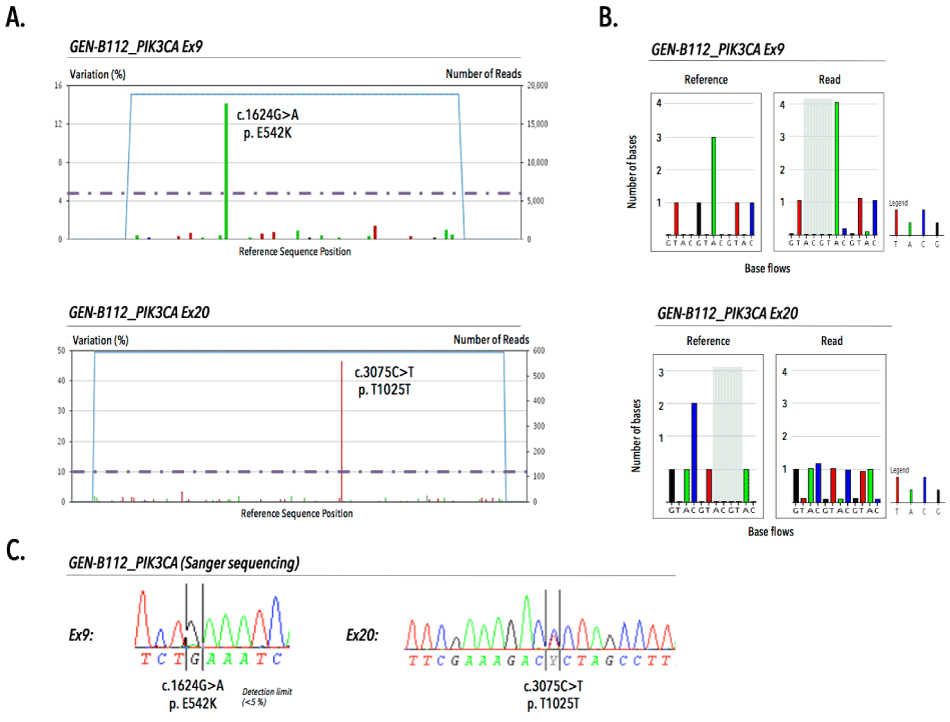
Figure 3. Validation of the 454 pyrosequencing assay to determine PIK3CA mutations in genomic DNA from FFPE tissue sections. A) Targeted amplicon sequencing of GEN-B112 reveals presence of the missense E542K in exon 9 (upper) and synonymous T1025T mutation in exon 20 (lower) above a variant read frequency of 5 and 10%, respectively. B) 454 pyrograms demonstrate validity of picked-up mutations via comparative analysis of number of bases between reference and read base flows for exon 9 (upper) and exon 20 (lower). C) External quality assurance of sample B112 displaying both E542K (at detection limit of less than 5%) and T1025T mutations by sanger sequencing.
Single-cell PIK3CA mutational analysis in breast cancer cell lines
We assessed the performance of our workflow via independent sorting experiments with spiked tumor cells (TCs) in donor blood. After CellSearch enumeration we isolated single (n=64) and groups (n=9) of MCF-7, BT-20 and SK-BR-3 TCs and WBCs (i.e., internal negative control) by DEPArray. All group TC samples (n=9) obtained after spiking and DEPArray purification consisted of 20 cells per group. Upon WGA the QC PCR revealed a genomic integrity index (GII, scale 0-4) ≥ 1 and ≥3 in 92,2% and 60% of the single TCs, respectively. A GII=4 was obtained in 66,67% and 83,33% of the recovered pools of CTCs and WBCs, respectively. GII≥1 cells were selected for downstream analysis. We tested the ability of our assay to discriminate mutant cells from those with a WT PIK3CA genotype at various VAF cut-off levels (Figure 4). Filtering variant reads of Ex9 at ≥5% delivered a sensitivity and specificity of 85.3% and 90%, whereas Ex20 reached highest discriminating power at ≥10% with a sensitivity of 71% and specificity of 100%. With the stringency filter set, 18 single and 2 pools of TCs and a pool of WBCs were analyzed and compared to genomic DNA (gDNA) from the parental cell line. Heterozygous WT/MT allelic frequencies were preserved in the DEPArray-recovered pooled TCs (Figure 5). Single cell samples (n=54) could demonstrate an imbalance in the allelic frequency. A dropout of the expected mutant allele was observed in 7 and 5 cells for Ex9 and 20, respectively. Loss of the WT allele was observed in 6 and 1 cell for Ex9 and 20, respectively. Two false-positive E545A (VAF: 100%) missense mutations were detected, indicating pseudogene amplification. Qualitatively the single cell assay identified 29/36 (80,5%) and 13/18 (72,2%) of the expected mutations in Ex9 and 20, respectively. All SK-BR-3 and WBC samples revealed a WT PIK3CA genotype.
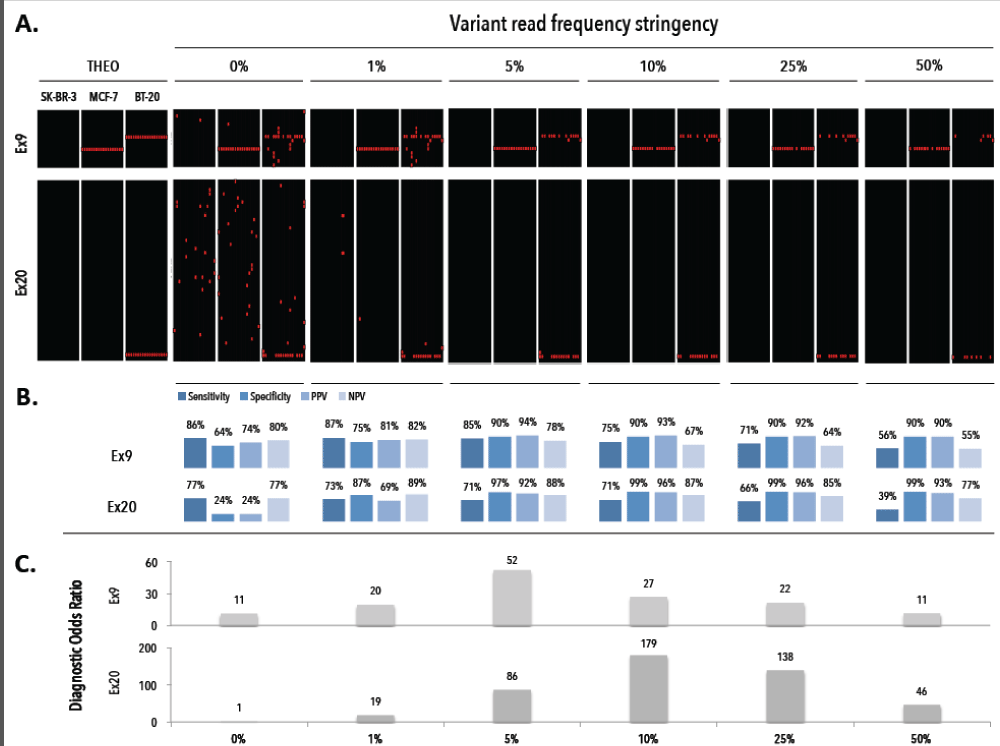
Figure 4. Assessment of the diagnostic threshold of the variant read frequency for a positive test result. A) Obtained mutational profiles of single cell (n=54) from breast cancer cell lines SK-BR-3, MCF-7 and BT-20 at alternating variant read frequency stringency levels (0%, 1%, 5%, 10%, 25%, 50%). B) Assessment of sensitivity, specificity, PPV and NPV for different VR% stringency levels. C) The diagnostic odds ratio for different VR% stringency levels. THEO denotes the theoretical expected profiles, PPV denotes positive predictive value, NPV denotes negative predictive value.
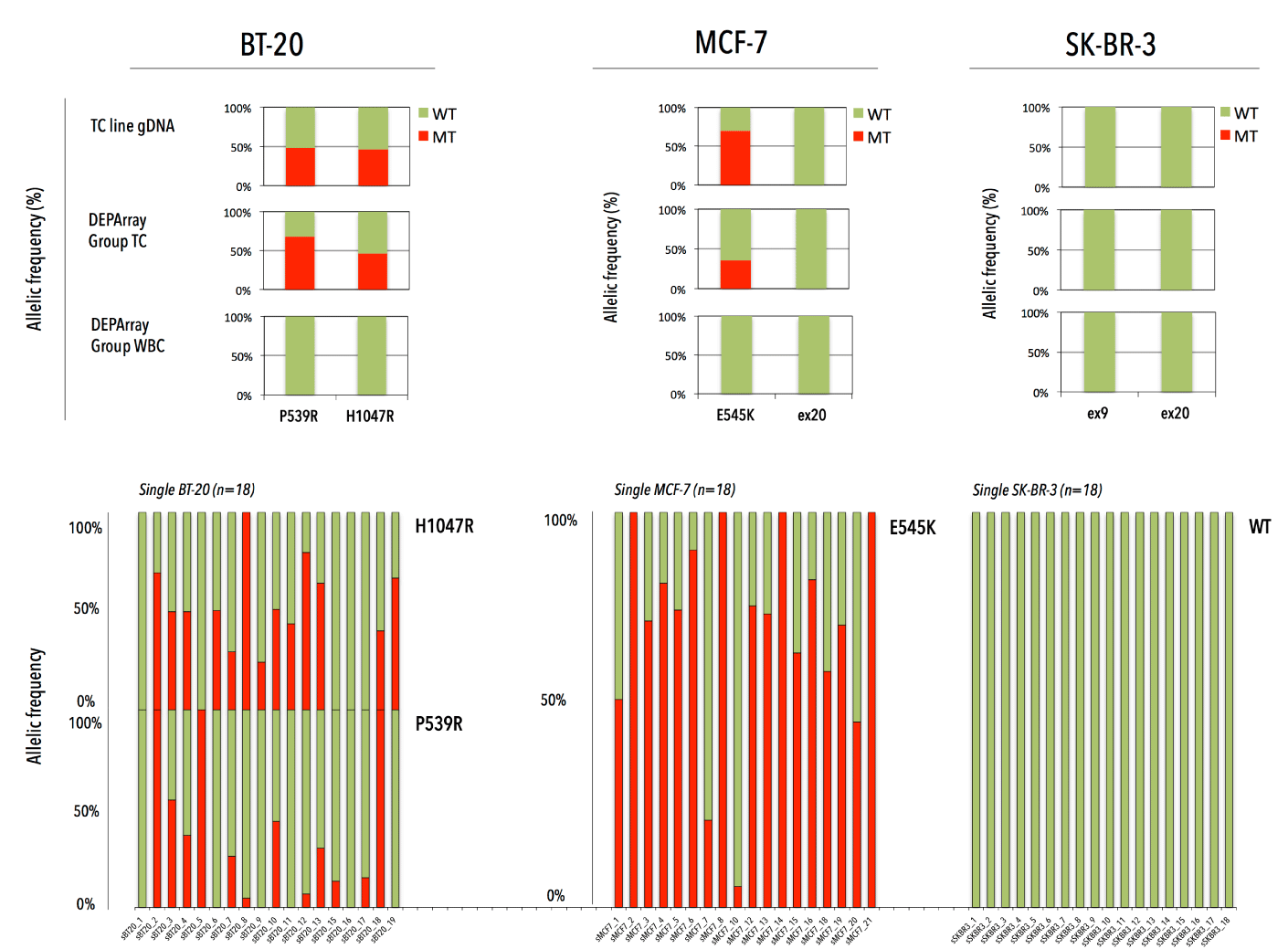
Figure 5. PIK3CA WT/MT allelic ratio in bulk DNA and DEPArray-recovered tumor cells in three breast cancer cell lines (BT-20, MCF-7 and SK-BR-3). The upper panel represents the WT/MT allelic frequency in bulk DNA and groups of DEPArray-purified TCs and WBCs. The lower panel presents the WT/MT allelic balance for single TCs after whole genome amplification.
Patient population and sample collection
A blood collection for CTC enumeration was performed in patients with HR+ MBC (n=60), before the start of a new line of systemic therapy. We defined a cohort of 38 patients (38/60, 63.3%) with five or more CTCs (Table 2). Median CTC count was 29 CTCs/7.5 mL, with 25th and 75th percentile CTC values corresponding to 13 and 182 CTCs, respectively. High CTC counts correlated with the presence of more bone metastases (p=0.024484, c2-test) or with the presence of combined visceral and bone metastases (p=0.017163, c2-test). Apart from CTC biopsy samples, FFPE PT samples (n=27), metastases (n=11) and plasma samples (n=31, CTC-matched) were collected from patients enrolled in this study (Figure 6). The metastases encompassed nine FFPE tissues, consisting of an abdominal debulking sample (n=1), BM biopsy (n=1), pleural lesion (n=1), skin metastasis (n=2), bone metastasis (n=1) and lymph nodes (n=3). All, except the abdominal and one skin lesion, were temporally matched with the CTC blood draw. Additionally, we analyzed DEPArray-purified DTCs from ascites, pleura and/or bone marrow from two patients (15 single and 11 group samples, covering 139 DTCs in total).
Table 2. Clinical, pathological and biological characteristics of study cohort (n=60). Clinicopathological data are presented for the total cohort and stratified by a CTC cut-off of 5 CTCs/7.5mL. In treatment characteristics the absolute numbers (or percentages) of total population can exceed the total number of patients due to combined therapy regimes.
|
|
|
# CTCs per 7,5 mL of PB
|
|
|
Total No.
of patients
|
<5
|
≥5
|
P
|
|
All patients
|
60
|
100.0%
|
22
|
36.7%
|
38
|
63.3%
|
|
|
Time between primary surgery and CTC sampling
|
|
|
|
|
|
|
|
|
≤ 5 y
|
32
|
53.3%
|
13
|
21.7%
|
19
|
31.7%
|
|
|
> 5 y
|
28
|
46.7%
|
9
|
15.0%
|
19
|
31.7%
|
|
|
Age at advanced disease
|
|
|
|
|
|
|
|
|
mean ± SD (y)
|
65 ± 13.8
|
|
|
|
|
|
|
|
≤ 50 y
|
9
|
15.0%
|
4
|
6.7%
|
5
|
8.3%
|
|
|
> 50 y
|
51
|
85.0%
|
18
|
30.0%
|
33
|
55.0%
|
|
|
Grade (Nottingham system)
|
|
|
|
|
|
|
|
|
I, well differentiated
|
9
|
15.0%
|
4
|
6.7%
|
5
|
8.3%
|
|
|
II, moderately differentiated
|
18
|
30.0%
|
7
|
11.7%
|
11
|
18.3%
|
|
|
III, poorly differentiated
|
22
|
36.7%
|
9
|
15.0%
|
13
|
21.7%
|
|
|
Unknown
|
11
|
18.3%
|
2
|
3.3%
|
9
|
15.0%
|
|
|
Histological type
|
|
|
|
|
|
|
|
|
Invasive ductal carcinoma (IDC)
|
44
|
73.3%
|
18
|
30.0%
|
26
|
43.3%
|
|
|
Invasive lobular carcinoma (ILC)
|
12
|
20.0%
|
2
|
3.3%
|
10
|
16.7%
|
|
|
Both
|
2
|
3.3%
|
1
|
1.7%
|
1
|
1.7%
|
|
|
Unknown
|
2
|
3.3%
|
1
|
1.7%
|
1
|
1.7%
|
|
|
Hormone receptor status
|
|
|
|
|
|
|
|
|
ER+/PR+/HER2+
|
7
|
11.7%
|
3
|
5.0%
|
4
|
6.7%
|
|
|
ER+/PR+/HER2-
|
45
|
75.0%
|
16
|
26.7%
|
29
|
48.3%
|
|
|
ER+/PR-/HER2+
|
1
|
1.7%
|
1
|
1.7%
|
0
|
0.0%
|
|
|
ER+/PR-/HER2-
|
6
|
10.0%
|
2
|
3.3%
|
4
|
6.7%
|
|
|
ER-/PR+/HER2-
|
1
|
1.7%
|
0
|
0.0%
|
1
|
1.7%
|
|
|
Therapy at CTC sampling
|
|
|
|
|
|
|
|
|
Chemotherapy
|
27
|
45.0%
|
6
|
10.0%
|
21
|
35.0%
|
|
|
Hormonal therapy
|
26
|
43.3%
|
12
|
20.0%
|
14
|
23.3%
|
|
|
Targeted therapy
|
16
|
26.7%
|
6
|
10.0%
|
10
|
16.7%
|
|
|
Combined palliative therapy
|
|
|
|
|
|
|
|
|
Hormonal + Targeted
|
4
|
6.7%
|
1
|
1.7%
|
3
|
5.0%
|
|
|
Chemotherapy + Targeted
|
8
|
13.3%
|
3
|
5.0%
|
5
|
8.3%
|
|
|
Start # Line of therapy at CTC sampling
|
|
|
|
|
|
|
|
|
1
|
38
|
63.3%
|
18
|
30.0%
|
20
|
33.3%
|
|
|
2
|
8
|
13.3%
|
2
|
3.3%
|
6
|
10.0%
|
|
|
3
|
2
|
3.3%
|
0
|
0.0%
|
2
|
3.3%
|
|
|
≥4
|
12
|
20.0%
|
2
|
3.3%
|
10
|
16.7%
|
|
|
Site of primary metastasis
|
|
|
|
|
|
|
|
|
Visceral
|
15
|
25.0%
|
10
|
16.7%
|
5
|
8.3%
|
|
|
Non-viscerral
|
27
|
45.0%
|
7
|
11.7%
|
20
|
33.3%
|
*
|
|
Both
|
16
|
26.7%
|
4
|
6.7%
|
12
|
20.0%
|
**
|
|
Unknown
|
2
|
1.7%
|
1
|
1.7%
|
1
|
1.7%
|
|
* P for 2-tailed Chi-square test = 0.024484
** P for 2-tailed Chi-square test = 0.017163
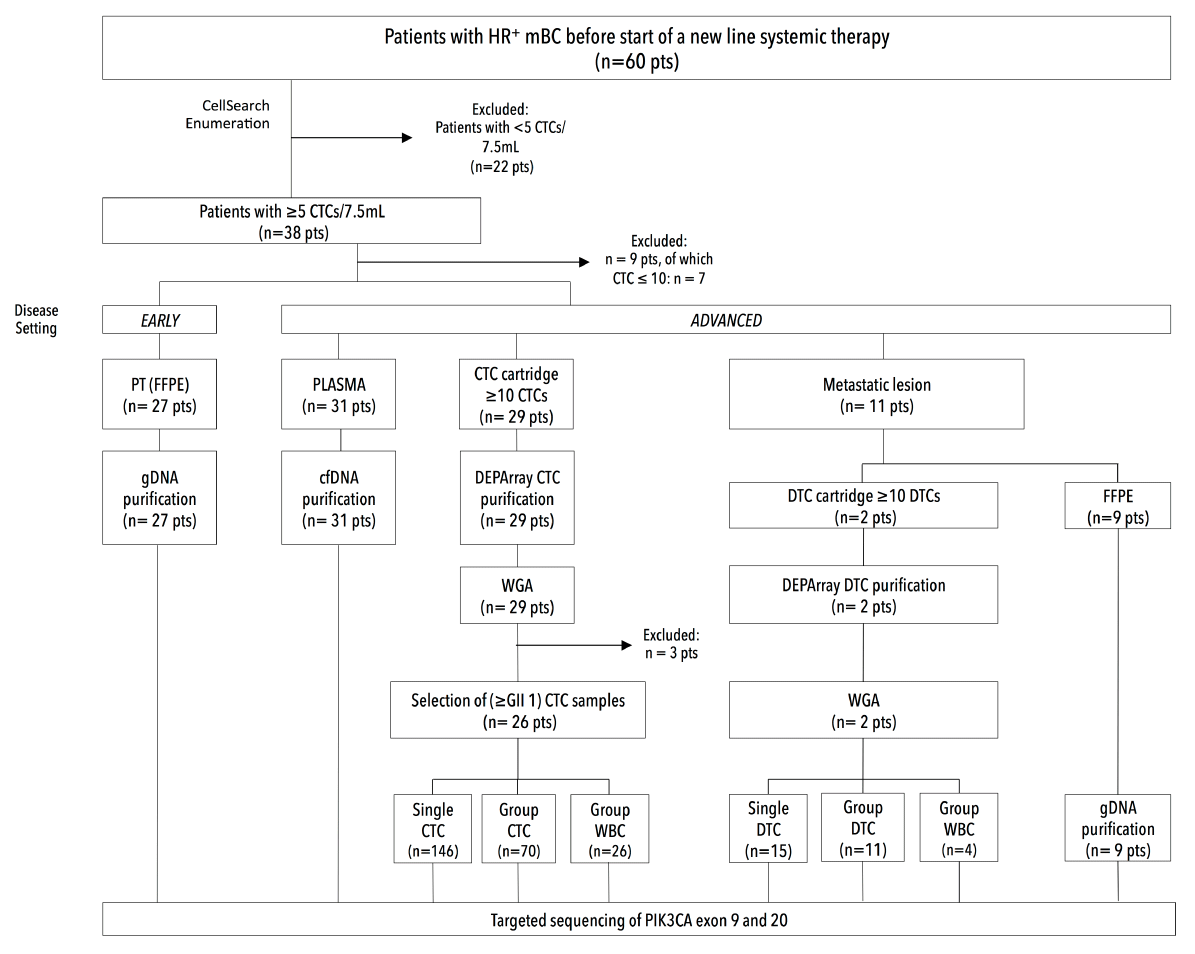
Figure 6. Study diagram: Enrolment of patients and collection of clinical samples. Sixty patients (n=60) with hormone receptor-positive breast cancer were enrolled in the study, of whom 38 had over 5 CTCs/7.5mL. From the latter we collected clinical samples from both an early (i.e. primary tumor) disease and advanced (i.e. metastatic) disease setting.
Enumerated CTC cartridges with ≥10 CTCs/7.5mL were selected for DEPArray sorting. Mean sample age (i.e., calculated as the time between CellSearch enumeration and DEPArray isolation) was 1.29 ± 0.61 years and did not correlate to overall WGA success rate (Figure 7B). In total, we isolated 249 single CTCs (mean 8 and median 10 single CTC recoveries per patient), 134 CTC pools covering 2709 CTCs in total (mean 4 and median 3 CTC pool recoveries per patient, ranging from 2-120 CTCs/pool) and 69 WBC recoveries (5 single and 64 pools, n=20 WBCs/pool). CellSearch CTC counts correlated well with DEPArray detection (Figure 8) in the lower (<250 CTCs/7.5 mL) CTC range (Spearman's rho correlation, n=20, r=0.96, p<0.01). A mean recovery rate of 47% ± 22%, ranging from 5.3% to 80% (median 52.2%) was achieved. If calculated towards the theoretical expected value (accounting for the dead volume during cell suspension transfer from CellSearch to DEPArray) a mean recovery rate of 70% ± 35%, ranging from 7.9% to 114% (median 80%) was obtained.
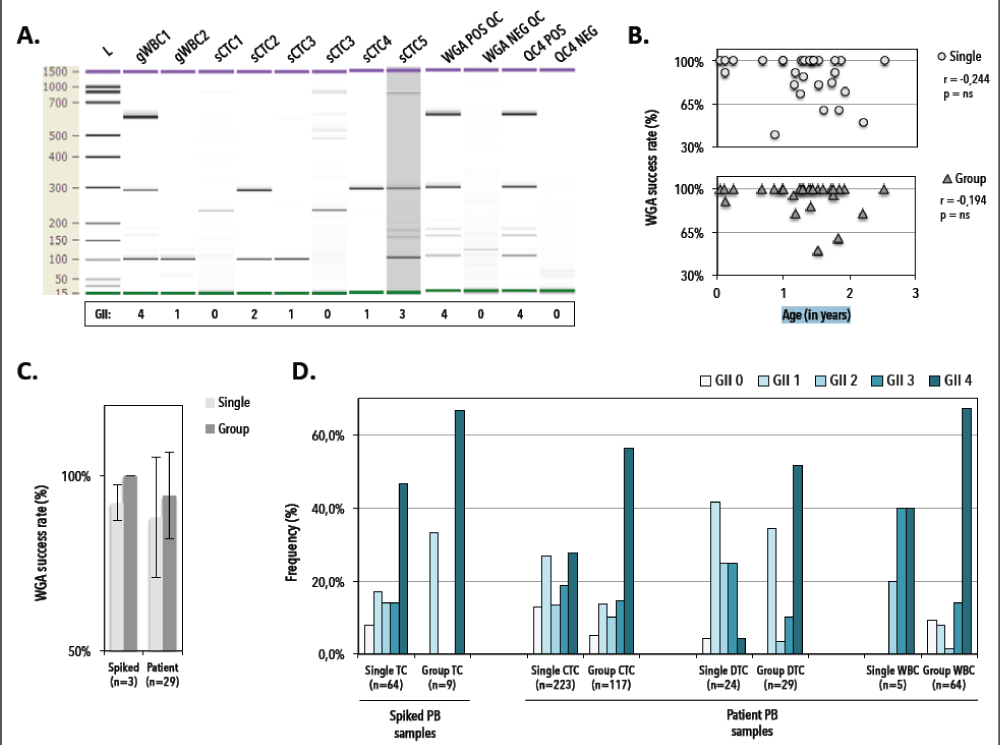
Figure 7. Quality control to assess genomic integrity index (GII) of Ampli1-amplified spiked TC and patient circulating (CTC) and disseminated (DTC) tumor cells and white blood cells (WBC). A) Representative DNA 1000 (Agilent) gel electropherogram of QC4 fragment analysis of DEPArray-recovered BT20. QC4 POS and QC4 NEG denote positive and negative controls of the QC4 PCR, 20ng of fresh genomic DNA and molecular grade water (which was used in the PCR mastermix), respectively. WGA POS QC and NEG QC denote positive and negative control samples for the Ampli1 WGA procedure, i.e. 0.5ng of fresh genomic DNA and molecular grade water (which was used in the WGA mastermix), respectively. sCTC denotes single DEPArray-recovered tumor cell. gWBC and gCTC denote group recoveries of white blood cells and tumor cells, respectively. L denotes DNA size ruler. B) WGA success rate (defined as GII ≥1) in function of sample age (n=29). C) WGA success rates in spiked breast cancer cells (n=3) and patient CTC samples (n=29). D) Distribution of Genomic integrity indices (GII 0 (light) till GII 4 (dark)) across single and group spiked tumor cells (TC), circulating tumor cells (CTC), disseminated tumor cells (DTC) and white blood cells (WBC). PB denotes peripheral blood. GII denotes genomic integrity index.
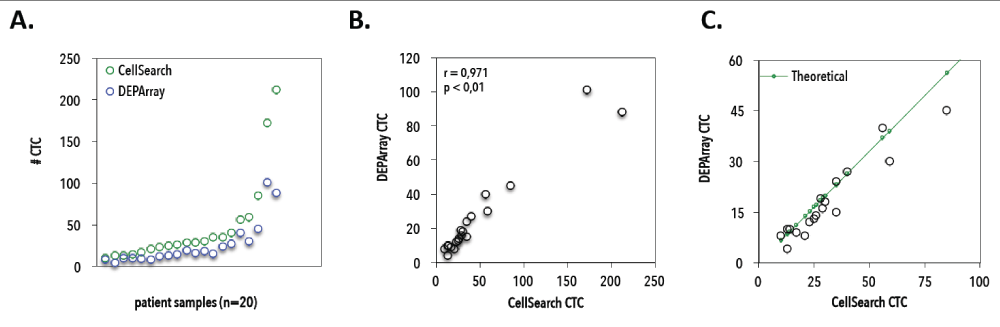
Figure 8. Correlation between CellSearch and DEPArray counting experiments. A) Number of CTCs as detected by CellSearch (green) and DEPArray (blue) for 20 patients with <250 CTCs/7.5mL on CellSearch. B) Correlation between observed CellSearch and DEPArray CTC counts from 20 patients with hormone receptor-positive breast cancer (Spearman’s rho correlation coefficient r = 0.97, P < 0.01). C) CellSearch and DEPArray counts in <100 CTC range, compared to theoretical expected CTC numbers on DEPArray (green line).
We subjected 340 of 383 (88.7%) CTC recoveries (i.e., 223 singles, 117 pools), 54/54 (100%) DTC recoveries (24 singles, 29 pools) and 69/69 (100%) WBC recoveries (5 single and 64 pools) to WGA. We interrogated the genomic integrity index (GII) of DEPArray-recovered cells by Ampli1 QC4 analysis to identify single and group cell samples suitable for downstream analysis. A representative electropherogram of a WGA quality control experiment is presented in Figure 7, in which the number of detected amplicons (from 0 to 4) determines the GII value. Overall, whole genome amplification succeeded (defined as generating at least one PCR fragment upon Ampli1 QC4 analysis) in 87% and 95% of single and group CTC samples, respectively, and did not correlate to the age of a given sample. Genomic integrity indices revealed a good distribution with approximately 50% (n=104 CTCs) of the single recovered patient CTC having a GII ³3, i.e., high quality DNA (27). Absence of QC4 PCR fragments was noted in 7.8% (5/64); 13% (29/223) and 4.2% (1/24) of the single spiked, CTCs and DTCs, respectively. From 26 patients we selected 216 DEPArray-purified CTC samples (146 single and 70 group samples, covering 1036 CTCs in total) for downstream analysis.
Assessment of the PIK3CA status across different compartments
PIK3CA mutation screening was performed by massive parallel sequencing of specimens (n=447, which include patient samples, spiked tumor cell samples and control samples) at sufficient coverage (Figure 9). A mean of 243.809 ± 50.517 read signals were generated, resulting in 133.270 ± 32.118 filtered reads. On average unfiltered sequenced libraries contained 12% ± 5,6% dot and mixed reads. In the analyzed patient samples (n=347) a dropout of 2.6% (n=9) and 4% (n=14) was noted for PIK3CA exon 9 and 20, respectively. On average (excluding outliers with extreme sequencing depths, see coverages >5000) we obtained a mean fold coverage of 1384 ± 788 reads (median 1274, range 4-5029) and 1567 ± 811 reads (median 1530, range 5-5822) for PIK3CA exon 9 and 20, respectively.
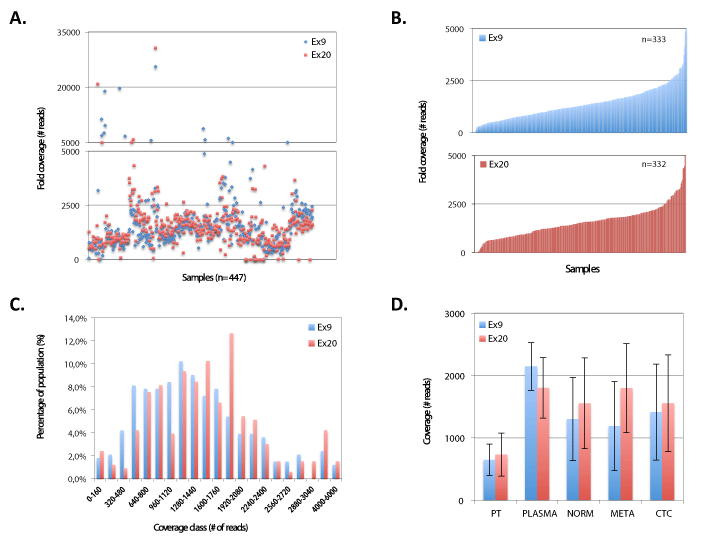
Figure 9. Sequencing depth during 454 pyrosequencing runs. A) Scatterplot of coverage depth for PIK3CA exon 9 and 20 for all sequenced samples (n=447). B) Patient sample coverage depth range for exon 9 (n=333, upper) and 20 (n=332, lower). C) Coverage distribution for PIK3CA Ex9 (n=333) & Ex20 (n=332). D) Coverage depth of PIK3CA exon 9 and 20 for different sample types. PT denotes primary tumor, NORM denotes normal (i.e. white blood cells). META denotes metastases. CTC denotes circulating tumor cells
Primary tumor mutational analysis
Analysis of hematoxylin/eosin-stained PT tissue sections (n=27, with nIDA=20 and nILA=7) showed a mean tumor cell content (TCC) of 58% ± 21% (median 50%, range 10%-90%) (Figure 10 and 11). We empirically set a VAF cut-off of 5% ± 2.5%, assuming the heterozygous nature of eventual mutations and the assumption that the lowest TCC=10%, resulting in 20-30 variant reads for an eventual mutation at the used sequencing depth. All PT samples (23 technical replicates) were sequenced in two independent runs. We characterized one synonymous Q546Q (1638G>A) substitution, which was detected in 2/27 patients (7.4%, see samples 1529 and 1839) and five missense substitutions: P539R (3/27; 11.1%), E542K (3/27; 11.1%), E545K (18/27; 66,7%), a previously unknown L997F (2989C>T; 2/27; 7.4%) and the H1047R (7/27; 26%). Additionally, double hotspot-mutated genotypes were detected in 8/27 (29.6%) patients. We observed how the TCC of a mutant PT correlated (n=16, r=0.5393, p=0.0311) with the VAF of the hotspot mutation (Figure 12).
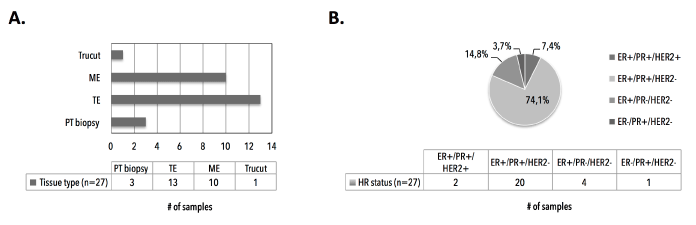
Figure 10. Primary tumor FFPE tissue sections characteristics. A) Type of surgery or intervention to obtain primary tumor tissue. B) Distribution of hormone receptor statuses in primary tumor tissues, as assessed by immunohistochemistry. ME denotes mastectomy. TE denotes tumorectomy. PT denotes primary tumor. HR denotes hormone receptor. ER denotes estrogen receptor. PR denotes progesterone receptor. HER2 denotes Human Epidermal growth factor Receptor 2
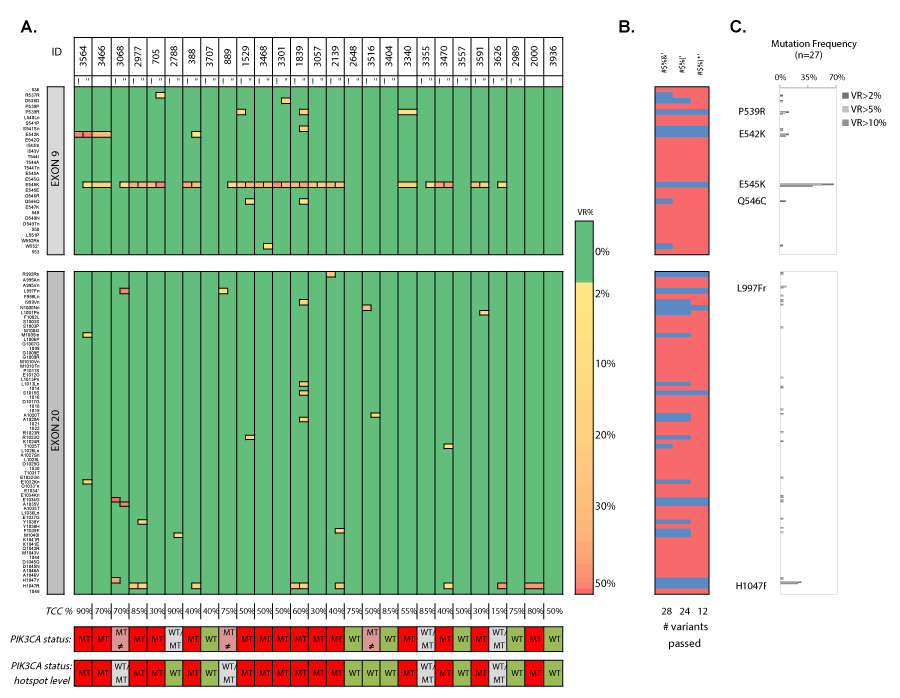
Figure 11. Heatmap en frequency plots of missense and synonymous mutations in PT samples in two independent sequencing runs. A) Heatmap of detected variants, presented with the detected variant read frequency (see color scale), ranging from >2% to 50%. For unique variants, see supplementary table 14. Lower: PIK3CA status exon wide (upper) and hotspot-focused (bottom). B) Assessment of the number of passed (blue) variants for different variant read frequency cutoffs (2%, 5% and 10%) based on the maximal variant read frequency that was picked up for a given mutation. C) Mutation frequencies (%) in FFPE primary tumor samples (n=27) with P539R, E542K, E545K, Q546Q, L997Fn (new) and H1047R as most frequently detected substitutions. VR% denotes variant read frequency. MT denotes MT. WT denotes WT. MT ≠ denotes inconsistent variants between technical replicates. WT/MT denotes WT or MT genotypes between technical replicates. a/b denote independent sequencing runs I and “ denote replicate 1 and 2, respectively.
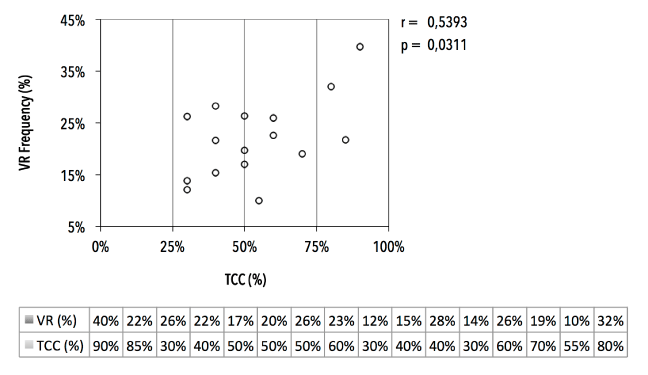
Figure 12. Correlation of tumor cellularity in FFPE tissue sections and mutation frequencies. Variant read frequency (VR%, in %) of major PIK3CA hotspot mutations in function of tumor cell content (TCC, in %) of FFPE primary tumor tissue section (n=16).
Fifteen patients (15/27, 55.5%) exhibited clear hotspot mutations, which were reproducible in thirteen selected patient samples. The majority of mutations occurred in Ex9 (12/15 patients (80%)) or in Ex9 and 20 combined (2/15 patients (13.3%)). One patient sample (1/15; 6.7%) showed an exclusive Ex20 mutation (sample 2000). The double-mutated E545K/H1047R genotype was reproducible in patients 2977 and 1839. Coefficient of variation (CV) for the detected VAFs ranged from 2.98% to 54.74%. E.g. sample 3564 (TCC=90%) displays an E542K missense mutation with a VAFMEAN=39.7% and CVVAF=2.98%, which could indicate a homogenous and clonal TC population. However, one replicate additionally presented a low-frequent E545K mutation. Alternatively, sample 2977 (TCC=85%) harbors next to an E545K mutation (CVVAF=3.48%) a reproducible Ex20 H1047R mutation with lower and more variable VAFs (CVVAF=44.03%), which may indicate the existence of a H1047R-harboring subpopulation. Six of the 27 tumors (22.2%) exhibited WT genotypes (patients 3707, 2648, 3404, 3557, 2989 and 3936) and this result was reproduced in five selected samples. MPS of technical replicates delivered also inconsistent results. Three samples (11%) gave either a WT or MT genotype (patients 2788, 3355 and 3626) and irreproducible variants (with VAF <5%) were detected in 3/27 samples (patients 3068, 889 and 3516).
Analysis of cfDNA at disease progression
We extracted cfDNA from patient (n=31) and donor (n=5) plasma samples. CfDNA yield was significantly higher (p=0.012) in patients with MBC (mean 0.665 ± 0.42 ng/μL plasma), but did not correlate with CTC numbers (Figure 13). Applying a VAF cut-off of 2% at 1600x coverage resulted in the characterization of 28 substitutions (Figure 14). Six out of 31 cfDNAs (19.3%) exhibited WT genotypes (patients 889, 3301, 3495, 3713, 3754 and 2749), which was reproduced in one selected sample (i.e., 889). Twenty-five patients (25/31, 80%) harbored a MT PIK3CA genotype. Most prevalent variants were P539R (n=5/31, 16.1%), E542K (n=4/31, 12.9%), E545K (n=17/31, 54.8%), L1036L (n=5/31, 16.1%) and H1047R (n=6/31, 19.4%). Four intra-run cfDNA replicate samples (1 healthy and 3 patient samples) reproduced the double MT (E542K/E545K, mean CVVAF=14.49%) and WT genotype in patients 3564 and 889, respectively. Discordant results were obtained for healthy control (H3) and patient (3470) samples. H3 delivered both a WT and MT genotype (i.e., synonymous S1015S mutation (VAF=2.09%)). Sample 3470 generated a reproducible WT Ex9, but inconsistent Ex20 substitutions. Double hotspot-mutated genotypes were detected in 9/31 (29%) patients.
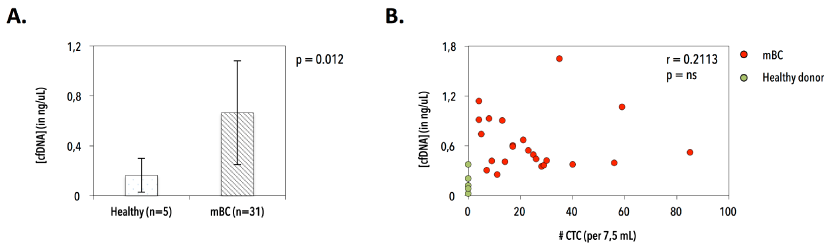
Figure 13. Purification of circulating cell free DNA (cfDNA) in patients with metastatic breast cancer and healthy control individuals. A) Significant difference in cfDNA yield in healthy individuals (n=5, mean 0.164 ± 0.14 ng/μL plasma) and patients with metastatic breast cancer (n=31, mean 0.665 ± 0.42 ng/μL plasma). B) Correlation between cfDNA yield (in ng/μL plasma) and CTC number (N per 7.5 mL blood), excluding outlier CTC numbers.
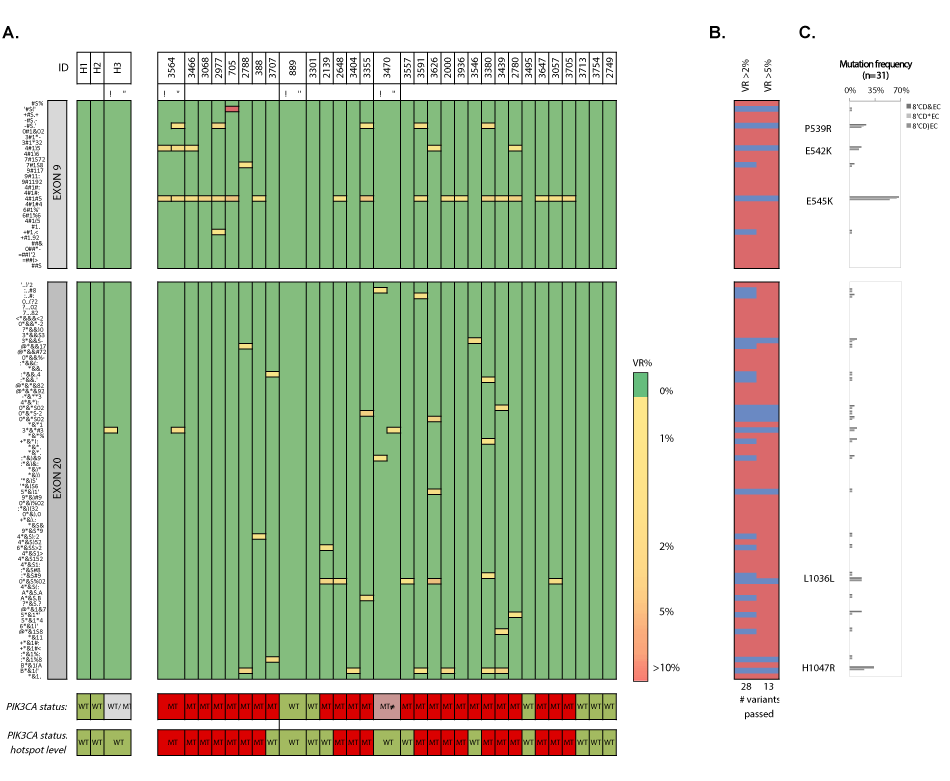
Figure 14. Heatmap en frequency plots of missense and synonymous mutations within exon 9 and 20 in cfDNA samples. A) Heatmap of detected variants, presented with the detected variant read frequency (see color scale), ranging from >2% to >10%. Lower: PIK3CA status exon wide (upper) and hotspot-focused (bottom). For unique variants, see supplementary table 15. B) Assessment of the number of passed (blue) variants for different variant read frequency cutoffs (2% and 5%) based on the maximal variant read frequency that was picked up for a given mutation. C) Mutation frequencies (%) in cfDNA samples (n=31) with P539R, E542K, E545K, Q546Q, L1036L (new) and H1047R as most frequently detected substitutions. VR% denotes variant read frequency. MT denotes mutant. WT denotes wild-type. MT ≠ denotes inconsistent variants between technical replicates. WT/MT denotes WT or MT genotypes between technical replicates.
Analysis of CTCs at disease progression
Mutational analysis was performed on purified CTCs from 26 patients with poor prognosis and >10CTCs/7.5mL. Pooled WBCs (recovered during CTC purification) showed WT sequences, indicating a high specificity. We selected a pool of 10 CTCs and bulk DNA from an enriched CTC fraction; together with three pools of CTCs and the PT from patient 3466 and 2000, respectively, for cross-validation by whole exome sequencing (WES) on the Illumina HiSeq platform. Concordant E542K and H1047R missense substitutions were detected in all samples from patients 3466 and 2000, respectively (Figure 15).
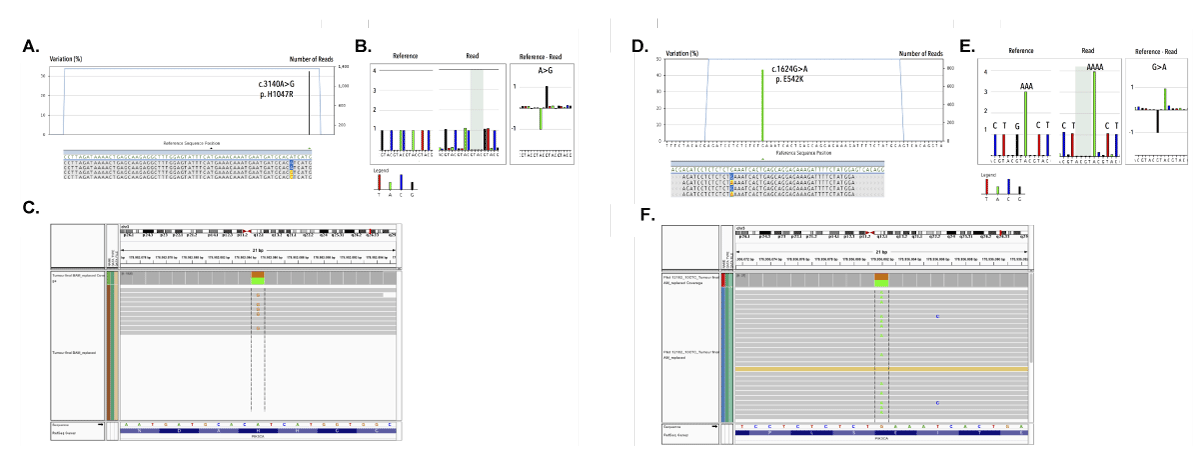
Figure 15. Cross-validation by comparing the mutational status of a pooled CTC sample by 454 targeted amplicon sequencing and whole exome sequencing on the Illumina HiSeq. For patient 2000: A) Targeted amplicon sequencing reveals presence of the missense H1047R in exon 20. B) 454 pyrograms demonstrate validity of picked-up mutations via comparative analysis of number of bases between reference and read base ows. C) Cross validation of pool CTC sample on Illumina HiSeq reproducing the H1047R missense substitution. For patient 3466: D) Targeted amplicon sequencing reveals presence of the missense E542K in exon 9. E) 454 pyrograms demonstrate validity of picked-up mutations via comparative analysis of number of bases between reference and read base ows. F) Cross validation of pool CTC sample on Illumina HiSeq reproducing the E542K missense substitution
A WT status was observed in all CTCs from 4/26 (15.4%) patients (3707, 1529, 3403 and 3557). The remaining 22/26 patients (84.6%) demonstrated a variety of both driver and low-frequent mutations (n=34), occurring in both a homogeneous and heterogeneous manner (Figure 16). When grouping all 216 CTC samples, we observed how the hotspot substitutions P539R (4.2%), E542K (12.5%), E545K (19.9%) and H1047R (1.6%) were most frequently detected. The less frequent and rare PIK3CA variants (n=23/34, 67.6%) were unique events occurring in single CTC samples with an overall lower GII. Vice versa we observed three Ex20 substitutions, which were uniquely detected in pooled CTC samples. In the selected patient cohort we confirmed the overall highest mutation frequency for the P539R (6/26, 23.1%), E542K (6/26, 23.1%), E545K (13/26, 50%) and H1047R (10/26, 38.5%) missense substitutions.
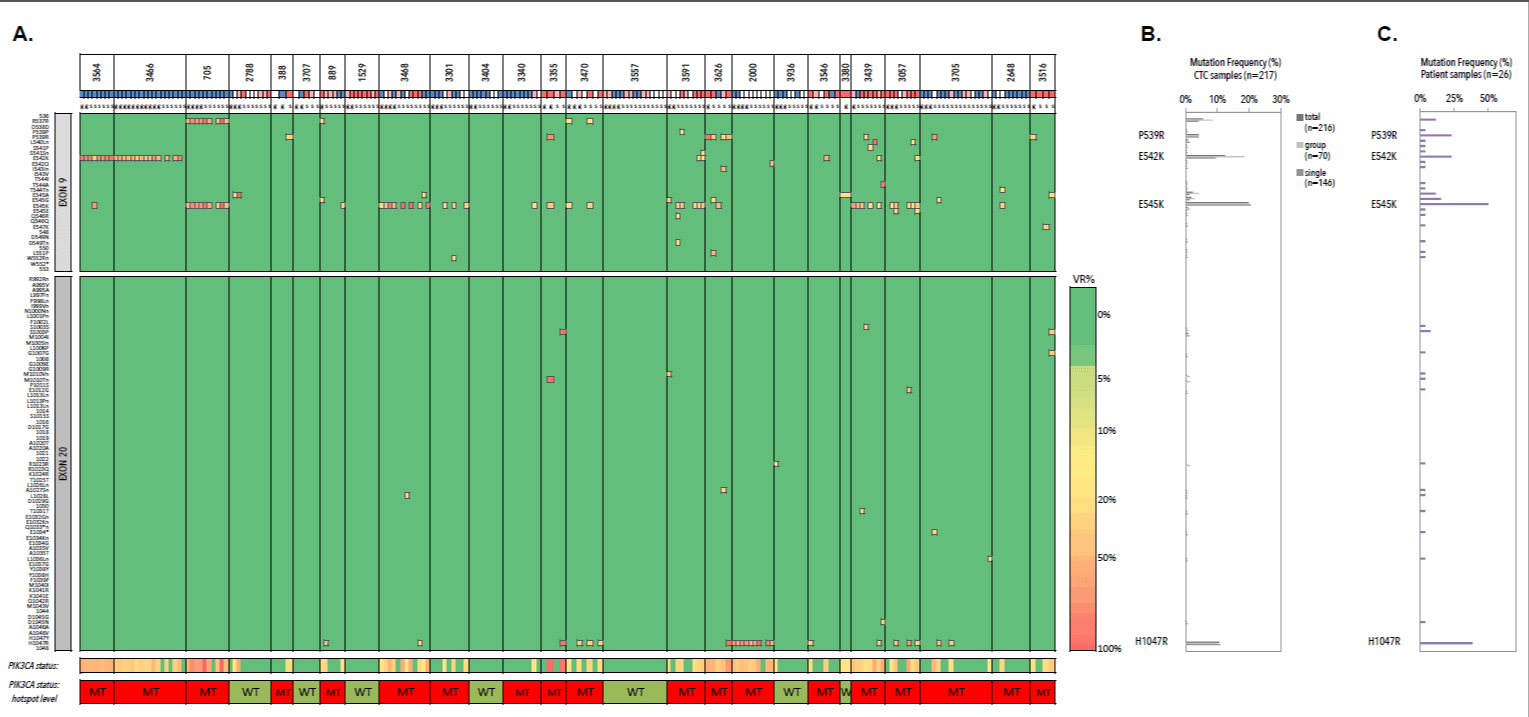
Figure 16. Heatmap andfrequency plots of missense and synonymous mutations within exon 9 and 20 of CTC samples. A) Heatmap of detected variants, presented with the detected variant read frequency (see color scale), ranging from >5% to >50%. Lower: PIK3CA status exon wide (upper) and hotspot-focused (bottom). For unique variants, see supplementary table 17. CTC samples are additionally presented with their genomic integrity score (see color scheme below patient ID). Blue denotes a GII of 4, white denotes a GII of 3, pink denotes a GII of 2 and red denotes a GII of 1. B) Mutation frequency of PIK3CA mutation across all CTC samples (n=216). C) Mutation frequencies (%) in CTC compartment of patients with metastatic hormone receptor-positive breast cancer (n=26) with P539R, E542K, E545K and H1047R as most frequently detected substitutions
Intra-patient mutational analysis revealed the coexistence of both WT and MT CTCs, with the occasional occurrence of CTCs having a double mutated genotype. To explore the degree of this intra-patient mutational heterogeneity we subjected the hotspot loci to an in-depth analysis (Figure 17). We defined pooled CTC samples, harboring a PIK3CA mutation, as homogeneous since preclinical performance studies demonstrated an imbalance in MT and WT allele amplification, making the assessment of the percentage MT cells within group samples fraught with some uncertainty. The majority of the mutational heterogeneity was detected in single cells. The hotspot genotypes allowed the classification of patients in 4 categories: WT, MT clonal (i.e., >75% of the CTCs having one hotspot mutation), MT subclonal (i.e., >25% of MT CTCs) and mix populations (i.e., displaying a mixture of both WT and MT genotypes). Hotspot analysis revealed how all CTCs from 7/26 patients (26.9%) harbored a WT genotype (patients 2788, 3707, 1529, 3404, 3557, 3936 and 3380), resulting in a mutation frequency of 73%. CTCs from 6 patients (6/26, 23.1%) demonstrated an overall clonal hotspot-mutated CTC population (patients 3564, 3466, 705, 3468, 3626 and 2000). The minor subpopulations in these ‘clonal’ samples consisted of CTCs harboring a WT (in 4/6 patients), a different hotspot (in 1/6 patients) or a double mutated (in 3/6 patients) genotype. Seven out of 26 patients (26.9%) had minor MT subpopulation(s) (patients 388, 889, 3301, 3340, 3591, 3705 and 2648). Finally, we defined 6 patients (23.1%) having mix populations of both WT, hotspot- and double mutated CTCs and thus exhibiting mutational heterogeneity in the highest degree (patients 3355, 3470, 3546, 3439, 3057 and 3516). Double mutated CTCs were detected in 8/26 patients (30.7%). Of note, patient follow-up data indicated how presence of PIK3CA mutations in CTCs is associated with survival (log rank, p=0.002) in patients with metastatic breast cancer, when stratifying patients according to their level of PIK3CA mutational heterogeneity (Figure 18).
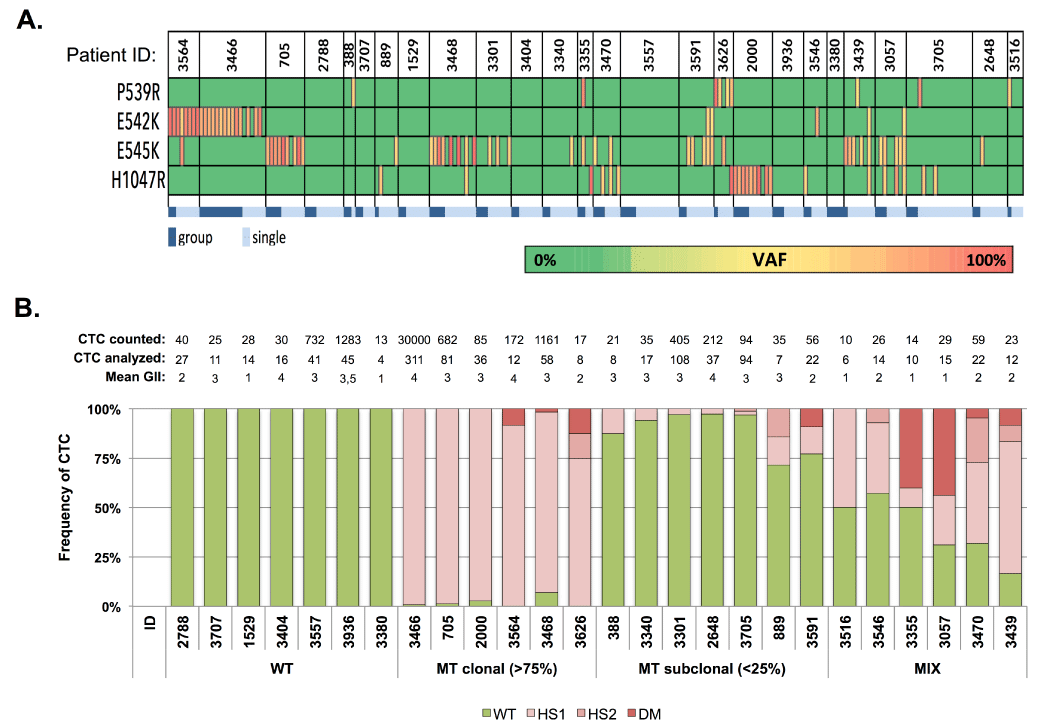
Figure 17.
PIK3CA hotspot mutational analysis in CTC compartment of patients with HR+ MBC. A) Hotspot mutational spectrum of CTC. Dark blue denotes group CTC samples. Light blue denotes single cell samples. B) PIK3CA mutational heterogeneity in CTC from patients with HR+ MBC (n=26). Above the bar chart the number of counted (CellSearch) and analyzed CTCs are presented, together with the mean genomic integrity index (GII). Patients were classified as having a WT, MT clonal, MT subclonal or MIX PIK3CA genotype. Clonal CTC samples indicate the presence of a hotspot mutation (E542K, E545K or H1047R) in >75% of the analyzed cells.
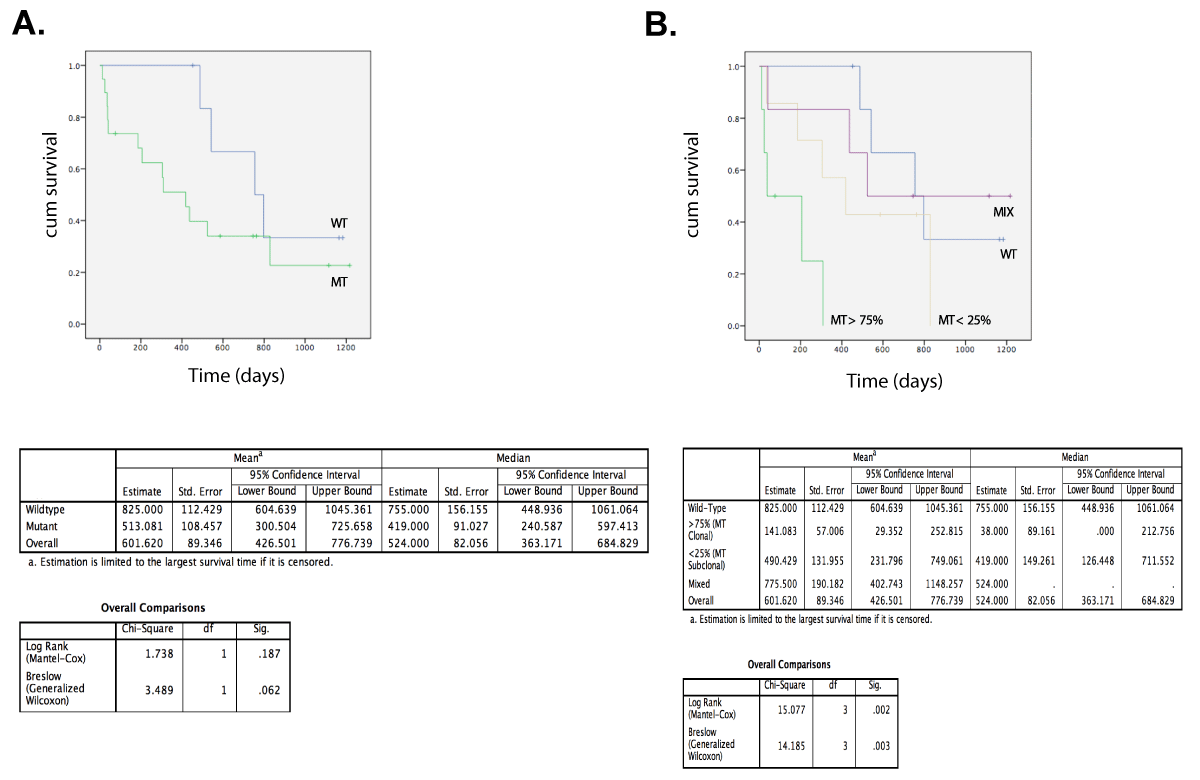
Figure 18. Overall survival from patients with HR+ MBC. Stratification according to MT or WT PIK3CA genotype in (A) and level of heterogeneity (B) in their CTC compartment: WT (n=7) and MT (n=19) with MIX (n=6), MT < 25% (n=7) and MT > 75% (n=6).
2021 Copyright OAT. All rights reserv
Analysis of metastases at disease progression:
We collected metastases from 11 patients, of which seven FFPE samples were analysed as intra-run replicates (Figure 19). Three patients (3/11; 27.3%) exhibited clear WT genotypes, which were reproducible in selected samples 3707 and 3705. All BM and pleural DTCs from patient 3936 exhibited a WT status. Mutant genotypes were observed in 7/11 (63.6%) patients (samples 705, 3546, 3380, 3439, 2780, 2486 and 3647) and were reproducible at the level of mutational hotspots. Missense substitutions E542K (3/11, 27.3%) and E545K (6/11, 54.5%) were the most commonly detected variants. Intra-run CVs of VAFs ranged from 0.1% to 59.2%. Replicate samples of the pleural lesion from patient 2486 (TCC>90%) carry an E542K missense mutation with CVVAF=0,1%. Alternatively, duplicate analysis of the LN sample from patient sample 2780 (TCC=85-90%) delivers E545K in 46.37% and 19.01% of the total reads (CV=59.2%).
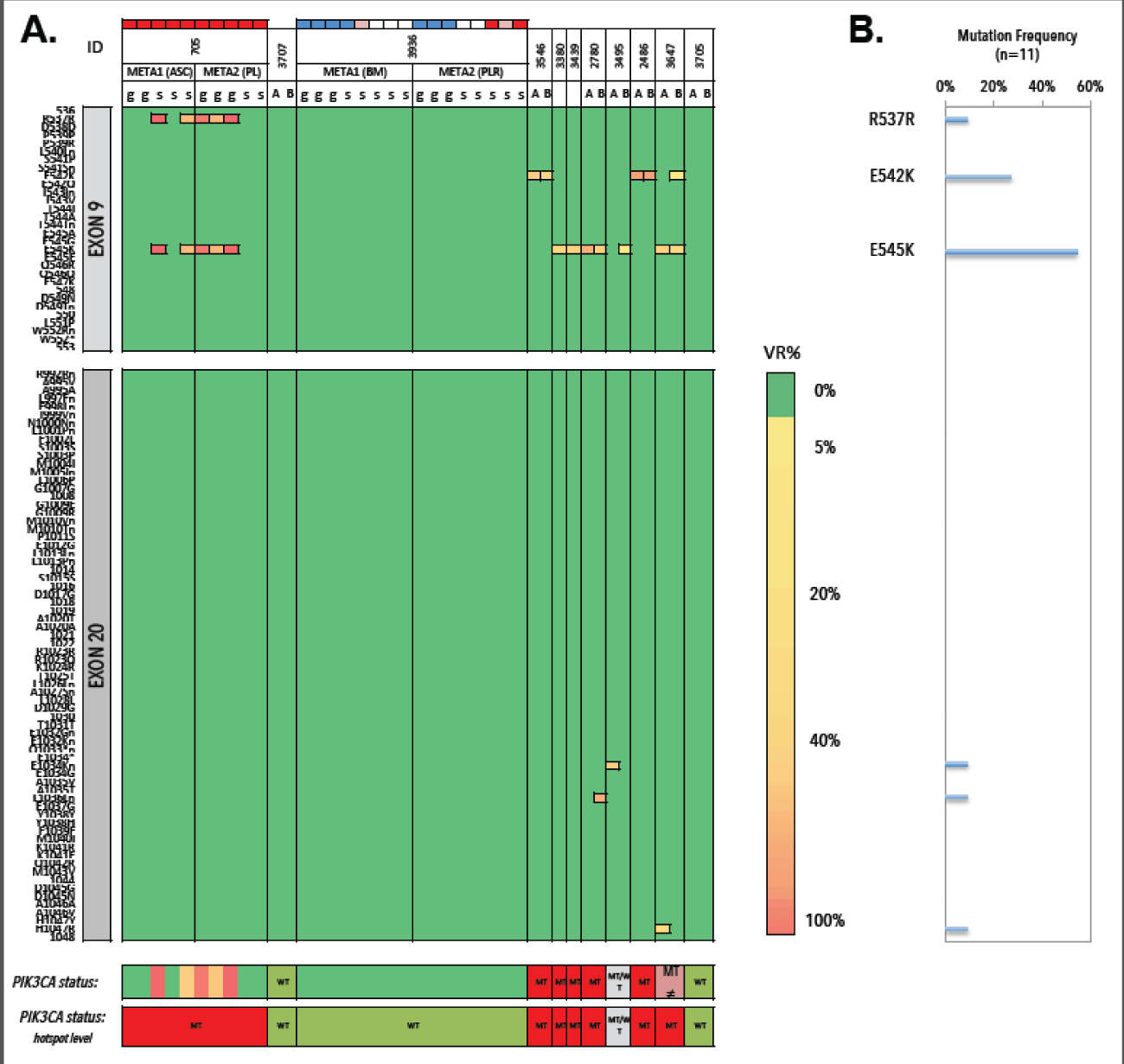
Figure 19. Heatmap of detected variants, presented with the detected variant read frequency. Range: >2% to >50%. Lower: PIK3CA status exon wide (upper) and hotspot-focused (bottom). DTC samples are additionally presented with their genomic integrity score (see color scheme above patient ID). Blue denotes a GII of 4, white denotes a GII of 3, pink denotes a GII of 2 and red denotes a GII of 1. B) Mutation frequencies (%) in metastases (n=11) with E542K and E545K as most frequently detected substitutions. VR% denotes variant read frequency. MT denotes MT. WT denotes WT. MT ≠ denotes inconsistent variants between technical replicates. WT/MT denotes WT or MT genotypes between technical replicates.
Comparative analysis of PIK3CA mutational status
Assessment of the PIK3CA mutational status was performed in different compartments, allowing a comparative analysis (Figure 20). Focusing on the validated mutational hotspots (P539R, E542K, E54K and H1047R) PIK3CA status in PT and cfDNA or CTC was available in 21 and 22 cases, respectively. Comparison of a metastatic lesion and temporally matched CTCs and cfDNA was possible in 7 and 10 patients, respectively. The level of agreement between the compartments was determined by focusing on the mutational hotspots (Figure 21). MT or WT statuses from PT tissues showed slight and substantial agreement with cfDNA (n=21; 33% disparity; k=0.14) and CTCs (n=22; 14% disparity; k=0.6733), respectively. Comparison of CTCs and temporally matched cfDNA samples revealed slight agreement (n=22; 36% disparity; k=0.0833). When comparing CTCs and metastases, a proper agreement was observed (n=7; 29% disparity, k=0.4167).
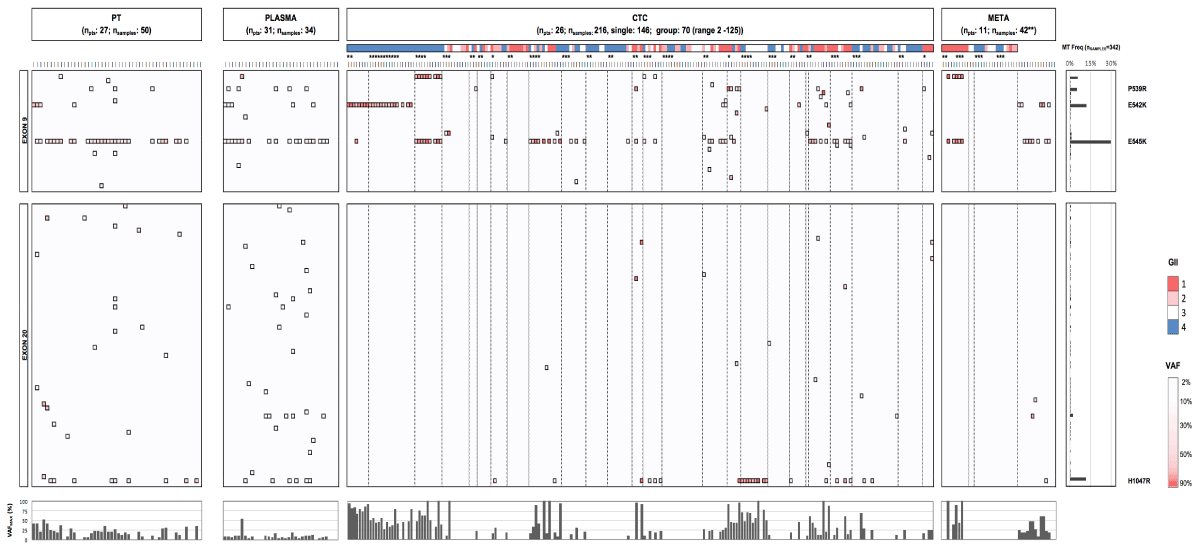
Figure 20. Comparative analysis of PIK3CA genotypes and VAFs in primary tumor, plasma, CTCs and metastases. PIK3CA ex9 and 20 mutational heatmaps across different compartments in patients with HR+ MBC. Color gradient from light to dark red in ex9 and 20 boxes indicates low (2) and high (>80%) VAFs for a particular substitution, respectively. The color scale above CTC and DTC samples (which are grouped per patient by boxes with dotted borders) denotes the genomic integrity index (GII), as determined by Ampli1 QC4 analysis (red: GII1; pink: GII2; white: GII3 and blue: GII4). Bottom bar chart represents the maximum VAF detected in analyzed patient samples and reveals (in comparison to PT and plasma) the enhancement in the VAF of a given mutation when analyzing purified CTC. PT denotes primary tumor, CTC denotes circulating tumor cell, META denotes metastatic lesion, VAF denotes variant allele frequency. (*) denotes group CTC/DTC samples consisting of more than one cell. (**) The 42 metastases samples encompassed FFPE tissues (n=9 patients) and DEPArray-purified disseminated tumor cells (DTCs) from two patients (15 single and 11 group samples, covering 139 DTCs in total).
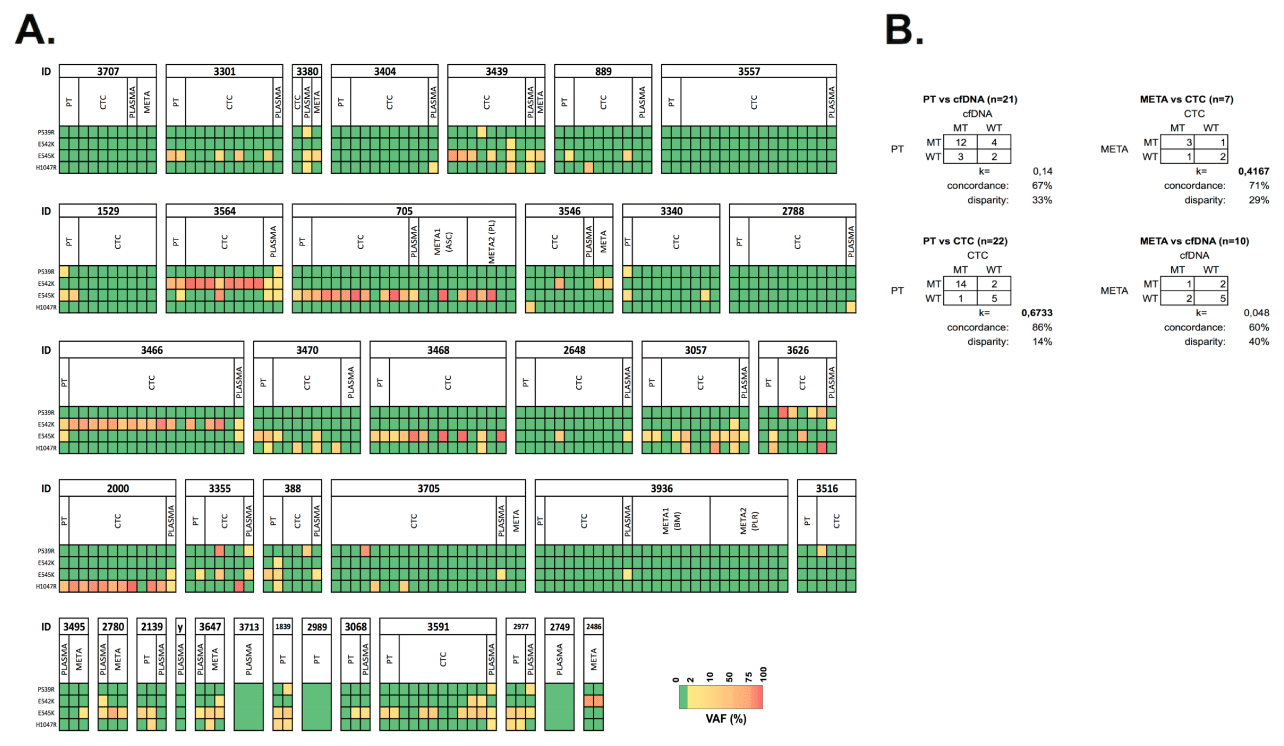
Figure 21. Comparative analysis of PIK3CA genotypes in PT, plasma, CTCs and metastases. A) PIK3CA mutational profiles of hotspot loci across different compartments in patients with HR+ MBC (n=38). Color gradient from yellow to red indicates low (2-10%) and high (>85%) VAFs, respectively. (y) denotes patient id 3754 B) Level of agreement of the PIK3CA mutational status in CTC or cfDNA and corresponding PTs and metastatic lesions. k denotes Cohen Kappa statistics
PIK3CA data across PT, cfDNA and CTCs was available in 18 cases. In this group 12 patients harbored PIK3CA aberrations in the PT, which were undetected in plasma-derived cfDNA in three cases (3/12, 25%), whereas the corresponding CTCs demonstrated the expected mutation (patients 3301, 889 and 3470). It is worth mentioning that cfDNA samples from patients 3301 and 3470 did carry the expected mutations, however below the selected VAF stringency level. In 3/18 evaluable patients (16.7%) a mutation was detected in cfDNA whereas we did not detect any aberration in the PT and matched CTCs (patient 3404, 2788 and 3936). Additional bone marrow and pleural DTCs from patient 3936 also revealed a WT genotype. A MT PIK3CA status across all compartments (PT, cfDNA and CTCs) was observed in 11/18 (61.1%) samples. In the six cases where a metastatic lesion was analyzed next to the cfDNA and CTC samples, a concordant PIK3CA status was observed in three cases. In cases 3380 and 3546, CTCs and cfDNA samples missed the MT genotype, respectively, which was present in the respective metastasis. In patient 3705 the WT status from the metastasis was not reproduced in the mutation-harboring CTCs and cfDNA. In general, mutational discordance between early and advanced disease was observed in four cases (patients 1529, 2139, 2648 and 3516). Gain of mutation was observed in two patients, which was confirmed by cfDNA in patient 2648.
In two cases, CTCs were capable to dissect the mutational heterogeneity of the hotspot mutations observed in PT or cfDNA. In patient 3564 we noticed how duplicate analysis of the PT demonstrated an E542K (VAF=38.85%) or E542K (VAF=40.52%) with low frequent E545K (VAF=2.94%) mutation. Duplicate analysis of cfDNA reproduced the double mutated genotype (VAFs>5%). Analysis of CTC samples revealed an overall homogenously E542K-mutated population, with one CTC harboring the double mutated genotype. The same feature was seen in patient 3470, who’s PT revealed a E545K (VAF=38.85%) or E545K (VAF=29.45%) with a low frequent H1047R (VAF=3.36%) mutation. These aberrations were present in one duplicate cfDNA sample. A single and two pools of CTCs revealed the existence of a mutually exclusive E545K and H1047R-positive subpopulations. Additionally, we identified a single CTC harboring the double-mutated E545K/H1047R genotype.
We present an in-depth validation, feasibility assessment and clinical application of PIK3CA mutation detection in primary tumors, CTCs, cfDNA and metastases from patients with HR+ MBC. All analyzed metastases samples (both tissue and disseminated tumor cell effusions) were temporally matched with the CTC blood draw, except the abdominal and one skin lesion. We previously reported the use of CellSearch and DEPArray to obtain pure CTCs (with a CellSearch to DEPArray recover efficiency of 62±12%) for molecular characterization down to the single cell level, and has demonstrated its applicability in different cancers [13,25,27,31-34]. Using the FDA-cleared CellSearch CTC counts has proven its applicability as a prognostic biomarker in clinic. However, the technique is based on positive selection of EpCAM+ cells, which may have missed tumor cells that underwent epithelial-to-mesenchymal transition (EMT). Recently, Sawada et al. have demonstrated the enhanced capture and enumeration efficiency of a novel marker-independent fluidic devise, when comparing to CellSearch. In patient samples, their platform detected significantly more CTCs than CellSearch, and detected CTCs in 9 cases, who were negative according to CellSearch [49]. Exploring PIK3CA mutational heterogeneity in CTCs obtained by marker-independent techniques would be a logical extension of this study.
We theoretically calculated that a CellSearch cartridge with 5 CTCs would allow us to minimally recover 2 and maximally 4 CTCs on DEPArray. However, due to variability in sample age and nature, keeping in mind the WGA unsuccessfulness in 7.8% of the spiked single cells, we empirically selected a subset of patients having more than 10 CTCs, to have at least 1-2 cells analyzed. In our cohort of 60 patients with HR+ MBC, 29 patients (ca. 50% of the total population) had equal to or more than 10 CTCs/7.5mL. We selected 26 patients for PIK3CA mutational analysis in whom it was feasible to determine the PIK3CA status in at least one CTC. CTCs were subjected to MPS of PIK3CA Ex9 and 20 after WGA, using a specific Ex9 reverse primer, which avoids pseudogene amplification [29]. Within this study we determined a rationale for a VAF stringency filter for the analysis of NGS data from single CTCs. By spiking breast cancer cells, carrying heterozygous PIK3CA mutations, and subjecting these cells to the same workflow of CTC enrichment, purification and WGA, we determined a VAF cut-off value for a reliable qualitative assessment of hotspot variants by calculation of the sensitivity and specificity for various VAF cutoff levels. Single cell data from spiked TCs revealed an WT/MT allelic imbalance compared to fresh gDNA, which was also noted, albeit to a lesser extent, in pools of CTCs. The goal of our workflow was to assess whether a sample harbors a PIK3CA mutation or not. We report an imbalance in the detected variant allele frequencies when performing the mutational analysis on small pools of cells or on a single cell level. The origin of this imbalance is uncertain but might be traced back to manipulation steps, encompassing a cell fixation, permeabilisation and staining procedure. Furthermore, on average CellSearch cartridges were stored for 1.5 years at 4°C, upon which cells were subjected to dielectrophoresis and downstream WGA, which for all reasons described above could have resulted in bias. This observation has also been described in the recent report by Gash and colleagues. Regardless of which WGA method was used, a preferential amplification of one allele was noted in a subset of the analyzed CTCs [48]. Due to the variability and sometimes low TCC of PT, metastases and cfDNA samples, compared to purified CTCs, we empirically defined a VAF cut-off for these matrices, based on the coverage depth of the performed sequencing experiments. Additionally, we need to emphasize that when analyzing cfDNA samples we did not correct for WT cfDNA originating from lysed white blood cells between samples, which could have resulted in the poor concordance between PT/CTC and cfDNA.
Besides hotspot we detected other PIK3CA mutations within the coding sequences. These low-frequent and rare substitutions were detected in unique samples, and may in some occasions have gain-of-function activity [35,36]. We emphasize the nature of the PFA-fixed cells and tissues, which may have affected the DNA quality, resulting in fixation bias and subsequent sequencing errors [37]. Hence, this raises the question of false discovery due to technical errors. Therefore, lack of validation or reproducibility renders uncertainty regarding the validity of these rare variants. In this way it is unclear why there was variability in some duplicate primary tumor samples. During the sequencing experiments we occasionally performed a no template control (H2O used to generate PIK3CA sequencing libraries), which did not deliver any DNA sequences. Additionally, internal negative control samples (i.e., patient WBCs co-purified with CTCs) revealed a wild-type PIK3CA genotype on all occasions. However, we noticed how these inconsistent results were achieved during sequencing of primary tumor samples, which were performed in two independent sequence runs, of which one sequence run only delivered unidirectional reads, instead of bi-directional coverage of the targeted exons, which may form the basis for the observed inconsistent results. On the contrary, we validated our methodology to detect and reproduce hotspot P539R, E542K, E545K and H1047R mutations by different approaches. We analysed several breast cancer cell lines harboring a known PIK3CA WT or MT status, both on bulk and single cell level. We reproduced the mutational status of a FFPE tumor tissue section that was reported by an external laboratory to carry two distinct PIK3CA mutations, and finally we performed cross validation of obtained results for selected patient CTC samples by whole exome sequencing (WES). Also, to validate the somatic nature of the non-hotspot mutations we sequenced germline DNA from pools of white blood cells, both in the assay validation and patient analysis phase. Here, all WBC samples generated wild-type PIK3CA genotypes, which could indicate the true somatic nature of the non-hotspot picked-up variants.
We selected PIK3CA as it is the most prevalent mutated oncogene in HR+ breast cancer with reported mutation frequencies in up to 40% of patients [38]. Over 80% of these alterations occur in the helical (Ex9) and kinase (Ex20) domains [39]. We chose to study the mutational heterogeneity in these hotspot loci by analysis of single CTCs as real-time liquid biopsy in patients with metastatic breast cancer. CTC analysis showed mutant PIK3CA genotypes in 21/26 (80%) patients, whereas previous studies have reported mutation frequencies ranging from 15% to 37% [27,33,40-42]. This higher rate could be in part explained by selecting a pure HR+ MBC cohort with a CTC count >10 CTCs/7.5mL. This resulted in the interrogation of 147 single and several pools of CTCs, covering total of 1036 CTCs from 26 patients. The increased CTC sampling size may have enhanced our sensitivity to detect low frequent MT cells.
The concept of PIK3CA mutational heterogeneity in metastatic breast cancer has been described by other research groups, where different levels of intra-patient heterogeneity were observed [27,33,40-42]. Here we performed mutational analysis on patients, all having a HR+ subtype at diagnosis and having advanced disease at the moment of blood draw, making it a more homogenous target population. Additionally, we want to remark that here we are among the first to demonstrate the dissection capabilities of intra-patient heterogeneity in mBC by comparative analysis of single circulating tumor cells to the primary tumor, circulating cell-free DNA and tissue and DTC metastases. Most recently, Gash et al. observed similar mutational patterns as the presented study with 4 cases having both PIK3CA wild-type and mutant subpopulations. Overall, the researchers concluded that mutational heterogeneity was frequent, as is the case in our presented study [48]. Additionally, different PIK3CA mutations were detected in asynchronously collected blood samples from the same patient. In our study, temporal heterogeneity was observed in four patients, with gain of mutation detected in two patients. This has also been observed in studies where comparison of asynchronously collected patient samples revealed mutational discordance in 7% up to 40% of the cases [43-45]. Besides temporal, mutational heterogeneity was detected between individual, synchronous collected CTCs, harboring different mutations or even double mutated and WT genotypes. Interestingly, it was reported how mutational discordance may occur in CTC samples from temporally-matched blood samples within a particular patient, indicating how one needs to approach PIK3CA mutational analysis by a single blood draw with caution [40,41]. We report how PIK3CA mutations are frequent and may occur in a homo- or heterogeneous manner in synchronously-collected CTCs. However, the relatively low number of patients analyzed limits the possibility to draw strong conclusions with regards to mutation incidence and frequency of mutational discordance between primary tumor, CTC, ctDNA and metastatic lesions. Therefore, the observed degree of heterogeneity emphasizes the need for an in depth single and pooled CTC analysis in the metastatic setting, via larger prospective clinical studies.
Single cell analysis allows the identification of minor CTC subpopulations. Besides the hotspot and rare PIK3CA alterations, we also observed patient samples harbouring a double mutated genotype within the hotspot loci. These events are rare, but indicate the heterogeneous nature of the PIK3CA oncogene [46,47]. Here, we observed two cases where the molecular heterogeneity of the PT and plasma samples were dissected by single and pooled CTC analysis. We identified how a CTC population with a double mutated PIK3CA or heterogeneous CTC subpopulations each containing one mutation may co-exist next to each other. Therefore, the exploration of the PIK3CA mutational heterogeneity demonstrates how within a given PT minor PIK3CA MT subclones can exist, which can be detected in circulation at an advanced stage of the disease. Performing the analysis on small pools of CTCs or down to the single cell level has allowed us to demonstrate the existence of these independent subpopulations. Our study provides an insight into the complexity of the mutational heterogeneity of PIK3CA in breast cancer at an advanced disease stage. We observed how the presence of PIK3CA mutations in CTCs is not associated with survival in patients with HR+ MBC, which is in contrast with previous reported results [42]. Interestingly we did observe how the presence of clonal PIK3CA mutations in CTCs is associated with worse survival.
We provide a validated and sensitive MPS method to determine PIK3CA mutations in CTCs, allowing the study of mutational heterogeneity. The presented liquid biopsy can be performed in high throughput on several single and pools of CTCs with 100% purity from one or several blood samples, making it suitable for use in a diagnostic setting. We provide supportive evidence that PIK3CA hotspot mutations are frequently present in EpCAM–positive CTCs from patients with HR+ MBC and may occur in a homo- or heterogeneous manner, which needs to validated via larger prospective clinical studies.
Besides mutational heterogeneity we observed temporal discordance when comparing early to advanced disease. Using our methodology, sequencing depth and VAF stringency levels, mutational analysis of CTCs may resemble better primary and metastasis tumor tissue sections, compared to cfDNA samples from plasma. However, we do emphasize the sequencing depth of our cfDNA samples and high VAF cutoff level. Nonetheless, using a mean sequencing depth of 1600x and a VAF cutoff of 2% allowed us to detect PIK3CA hotspot mutations in plasma from patients with advanced MBC which in 67% of the cases showed concordant results with the PT. The low sample input, sequencing depth and stringent VAF filtering may lay at the basis of the observed discordance. However, we want to emphasize that we targeted a patient population with advanced metastatic disease, having a high circulating tumor burden, in whom perhaps a lower sequencing depth is sufficient to detect clonal driver mutations, as the PIK3CA oncogene. In conclusion, the study presents the utilization of a liquid biopsy on a single cell level, which dissected the PIK3CA mutational heterogeneity within the intravascular compartment of individual patients, thereby paving the way towards the the successful implementation of the liquid biopsy model as part of the personalized medicine in the management of patients with metastatic cancer.
This work was supported by the Belgian Foundation Against Cancer. We acknowledge Dr. Op de Beeck for providing the Sanger sequencing service during the assay development phase and Dr. Denkert Carsten for providing reference FFPE tumor tissue sections. We acknowledge Diana Cunati and Elena Peruzzi (both from Silicon Biosystems, Bologna, Italy) for their help with protocols and DEPArray system operation.
We acknowledge the financial support from the TRANSLATIONAL & CLINICAL RESEARCH GRANTS 2012 (LD, SVL, BDL) by the Belgian Foundation Against Cancer (Leuvensesteenweg 479, B-1030 Brussels, Belgium).
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, et al. (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486: 400–404. [Crossref]
- Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, et al. (2012) The life history of 21 breast cancers. Cell 149: 994–1007. [Crossref]
- Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, et al. (2012) Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 366: 520-529. [Crossref]
- Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, et al. (2014) Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep 6: 377-387. [Crossref]
- Cancer Genome Atlas Network, Koboldt DC, Fulton RS, McLellan MD, Schmidt H, et al. (2012) Comprehensive molecular portraits of human breast tumors. Nature 490: 61–70. [Crossref]
- Lee JW, Soung YH, Kim SY, Lee HW, Park WS, et al. (2005) PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene 24: 1477-1480. [Crossref]
- Meyer DS, Koren S, Leroy C, Brinkhaus H, Müller U, et al. (2013) Expression of PIK3CA mutant E545K in the mammary gland induces heterogeneous tumors but is less potent than mutant H1047R. Oncogenesis 2: e74. [Crossref]
- Bader AG, Kang S, Vogt PK (2006) Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A 103: 1475–1479. [Crossref]
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, et al. (2005) Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res 65: 10992-11000. [Crossref]
- Alix-Panabières C, Pantel K (2013) Circulating tumor cells: liquid biopsy of cancer. Clin Chem 59: 110-118. [Crossref]
- Ignatiadis M, Piccart M (2012) Liquid biopsy to test new treatment strategies in breast cancer: are we there yet? Ann Oncol 23: 1653-1655. [Crossref]
- Rack B, Schindlbeck C, Juckstock J, Andergassen U, Hepp P, et al. (2014) Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J Natl Cancer Inst 2014: 106. [Crossref]
- Hodgkinson CL1, Morrow CJ1, Li Y2, Metcalf RL3, Rothwell DG3, et al. (2014) Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med 20: 897-903. [Crossref]
- Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, et al. (2014) Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol 32: 479–484. [Crossref]
- Maheswaran S1, Sequist LV, Nagrath S, Ulkus L, Brannigan B, et al. (2008) Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med 359: 366-377. [Crossref]
- Poplawski AB, Jankowski M, Erickson SW, Díaz de Ståhl T, Partridge EC, et al. (2010) Frequent genetic differences between matched primary and metastatic breast cancer provide an approach to identification of biomarkers for disease progression. Eur J Hum Genet 18: 560–568. [Crossref]
- Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68-73. [Crossref]
- Marusyk A, Almendro V, Polyak K (2012) Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 12: 323-334. [Crossref]
- Klein CA, Blankenstein TJF, Schmidt-Kittler O, Petronio M, Polzer B, et al. (2002) Genetic heterogeneity of single disseminated tumor cells in minimal residual cancer. Lancet 360: 683–689. [Crossref]
- Magbanua MJM, Sosa EV, Roy R, Eisenbud LE, Scott JH, et al. (2013) Genomic profiling of isolated circulating tumor cells from metastatic breast cancer patients. Cancer Res 73: 30–40. [Crossref]
- Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, et al. (2013) Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res 73: 2965–2975. [Crossref]
- Wang Y, Waters J, Leung ML, Unruh A, Roh W, et al. (2014) Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512: 155–160. [Crossref]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, et al. (2011) Tumour evolution inferred by single-cell sequencing. Nature 472: 90-94. [Crossref]
- Lee M-CW, Lopez-Diaz FJ, Khan SY, Tariq MA, Dayn Y, et al. (2014) Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc Natl Acad Sci USA 111: E4726–E4735. [Crossref]
- Peeters DJ, De Laere B, Van den Eynden GG, Van Laere SJ, Rothé F, et al. (2013) Semiautomated isolation and molecular characterisation of single or highly purified tumour cells from CellSearch enriched blood samples using dielectrophoretic cell sorting. Br J Cancer 108: 1358-1367. [Crossref]
- Hollestelle A1, Nagel JH, Smid M, Lam S, Elstrodt F, et al. (2010) Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res Treat 121: 53-64. [Crossref]
- Polzer B, Medoro G, Pasch S, Fontana F, Zorzino L, et al. (2014) Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol Med 6: 1371-1386. [Crossref]
- Nosho K, Kawasaki T, Ohnishi M, Suemoto Y, Kirkner GJ, et al. (2008) PIK3CA mutation in colorectal cancer: relationship with genetic and epigenetic alterations. Neoplasia 10: 534-541. [Crossref]
- Baker CL, Vaughn CP, Samowitz WS (2012) A PIK3CA pyrosequencing-based assay that excludes pseudogene interference. J Mol Diagn 14: 56-60. [Crossref]
- Glas AS, Lijmer JG, Prins MH, Bonsel GJ, Bossuyt PM (2003) The diagnostic odds ratio: a single indicator of test performance. J Clin Epidemiol 56: 1129-1135. [Crossref]
- Carpenter EL, Rader J, Ruden J, Rappaport EF, Hunter KN, et al. (2014) Dielectrophoretic capture and genetic analysis of single neuroblastoma tumor cells. Front Oncol 4: 201. [Crossref]
- Fabbri F, Carloni S, Zoli W, Ulivi P, Gallerani G, et al. (2013) Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett 335: 225-231. [Crossref]
- Pestrin M, Salvianti F, Galardi F, De Luca F, Turner N, et al. (2015) Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Molecular Oncology 9: 749–757. [Crossref]
- Fernandez SV, Bingham C, Fittipaldi P, Austin L, Palazzo J, et al. (2014) TP53 mutations detected in circulating tumor cells present in the blood of metastatic triple negative breast cancer patients. Breast Cancer Res 16: 445. [Crossref]
- Müller CI, Miller CW, Hofmann W-K, Gross ME, Walsh CS, et al. (2007) Rare mutations of the PIK3CA gene in malignancies of the hematopoietic system as well as endometrium, ovary, prostate and osteosarcomas, and discovery of a PIK3CA pseudogene. Leuk Res 31: 27–32. [Crossref]
- Gymnopoulos M, Elsliger MA, Vogt PK (2007) Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A 104: 5569-5574. [Crossref]
- Swennenhuis JF, Reumers J, Thys K, Aerssens J, Terstappen LW (2013) Efficiency of whole genome amplification of single circulating tumor cells enriched by CellSearch and sorted by FACS. Genome Med 5: 106. [Crossref]
- Sabine VS, Crozier C, Brookes CL, Drake C, Piper T, et al. (2014) Mutational analysis of PI3K/AKT signaling pathway in tamoxifen exemestane adjuvant multinational pathology study. J Clin Oncol 32: 2951–2958. [Crossref]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, et al. (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304: 554. [Crossref]
- Schneck H, Blassl C, Meier-Stiegen F, Neves RP, Janni W, et al. (2013) Analysing the mutational status of PIK3CA in circulating tumor cells from metastatic breast cancer patients. Mol Oncol 7: 976–86. [Crossref]
- Deng G, Krishnakumar S, Powell AA, Zhang H, Mindrinos MN, et al. (2014) Single cell mutational analysis of PIK3CA in circulating tumor cells and metastases in breast cancer reveals heterogeneity, discordance, and mutation persistence in cultured disseminated tumor cells from bone marrow. BMC Cancer 14: 456. [Crossref]
- Markou A, Farkona S, Schiza C, Efstathiou T, Kounelis S, et al. (2014) PIK3CA mutational status in circulating tumor cells can change during disease recurrence or progression in patients with breast cancer. Clin. Cancer Res 20: 5823–5834. [Crossref]
- Dupont Jensen J, Laenkholm A-V, Knoop A, Ewertz M, Bandaru R, et al. (2011) PIK3CA mutations may be discordant between primary and corresponding metastatic disease in breast cancer. Clin Cancer Res 17: 667–677. [Crossref]
- Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, Sahin A, Liu S, et al. (2011) PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther 10: 1093–1101. [Crossref]
- Arthur LM, Turnbull AK, Renshaw L, Keys J, Thomas JS, et al. (2014) Changes in PIK3CA mutation status are not associated with recurrence, metastatic disease or progression in endocrine-treated breast cancer. Breast Cancer Res Treat 147: 211-219. [Crossref]
- Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, et al. (2008) An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 68: 6084-6091. [Crossref]
- Cizkova M, Susini A, Vacher S, Cizeron-Clairac G, Andrieu C, et al. (2012) PIK3CA mutation impact on survival in breast cancer patients and in ERα, PR and ERBB2-based subgroups. Breast Cancer Res 14: R28. [Crossref]
- Gasch C, Oldopp T, Mauermann O, Gorges TM, Andreas A, et al. (2016) Frequent detection of PIK3CA mutations in single circulating tumor cells of patients suffering from HER2-negative metastatic breast cancer. Mol Onc 2016.
- Sawada T, Araki J, Yamashita T, Masubuchi M, Chiyoda T, et al. (2016) Prognostic Impact of Circulating Tumor Cell Detected Using a Novel Fluidic Cell Microarray Chip System in Patients with Breast Cancer. EBIOM Elsevier 2016.