Abstract
Perivascular epithelioid cell tumor (PEComa) is a rare entity with unpredictable clinical outcome. It is a group of mesenchymal tumors with presence of perivascular epithelioid cells (PEC), known to stain positively with melanosome markers HMB-45. They may arise from many unusual sites including the kidneys. Renal PEComa was previously groupedas angiomyolipoma and its existence is rare too. Pediatric PEComas are even rarer with less than 40 cases described worldwide and none involving the kidney before. We report a case of a 7 year old boy who presented with incidental findings of an abdominal mass, which confirmed to be a renal PEComa of the atypical epithelioid angiomyolipoma (AAML) type. The clinical management and review of literature for this interesting entity is discussed.
Key words
pediatric, PEComa, atypical angiomyolipoma
Case presentation
A previously well 7 years old boy was noted by his grandmother to have a bulge over the right side of the abdomen for one week duration. She also noticed that the boy had loss of appetite and less active. There was no complaint of abdominal pain, abnormal bowel habit or urinary symptoms such as hematuria. There was neither history of fever to suggest infection nor history of trauma. On examination, the boy appeared healthy and not syndromic. He was not pale or jaundice and his vital signs were stable. There was an obvious bulge on the right lumbar area. The bulge was a large non tender, 9 cm X 9 cm mass. The mass was ballotable suggesting that it was the right kidney. There was no other organomegaly. Blood investigations were done which included full blood count, renal profile, tumor markers for malignant germ cell tumour and teratoma and liver function test. All the blood investigations were normal. Radiological imaging was done concurrently; the abdominal ultrasound showed a mass arising from the right kidney, measuring 11.5 cm x 6.4 cm x 9.4 cm. It was a combined solid cystic mass with no calcifications seen. CT scan of the abdomen confirmed the findings and showed that the mass was exophytic in nature, localised with no invasion to surrounding structure. There was no caval involvement or aortocaval adenopathy. The other solid organs and lung bases were normal (Figure 1). The provisional diagnosis was a renal sarcoma with a differential diagnosis of renal cell carcinoma. An ultrasound guided percutaneous biopsy was performed which was reported as melanocytic angiomyolipoma. We proceeded to do open right nephro-ureterectomy; intraoperatively the tumor was quite vascularized, surgery went well without problems. We bisected the specimen to reveal an unusual looking tumor (Figure 2). Post operatively, the patient was well and discharged home at day 5. The pathological examination of the tumor revealed macroscopically the tumor was irregular, fragile, and blackishin color, located at the lower pole of the right kidney measuring 8.5 cm X 4.5 cm X 6.5 cm. The tumor extended into the renal pelvis and capsule but did not appear to breach the capsule. Microscopically, the tumor was composed of sheets and nests and perivascular cuffing of large polygonal epithelioid cells. These cells had mildly pleomorphic, vesicular nuclei with prominent nucleoli and abundant granular to clear cytoplasm. The cells were densely pigmented by melanin. Mitotic figure were rarely seen. The tumor was seen invading but not breaching the renal capsule. The ureter was free from tumor. The adjacent renal parenchyma was compressed by the tumor bulk. No lymphovascular invasion was seen. Immunohistochemically, the tumor cells were positive for HMB45 and Melan A but negative for CD10. These descriptions were diagnostic for PEComa, non-malignant. The patient came for regular follow up and serial ultrasound imaging; at one year follow up the patient remained well and radiologically there was no recurrence of tumor.
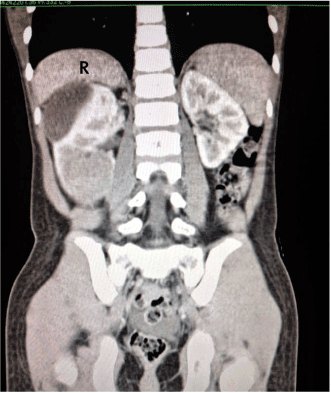
Figure 1. CT scan showing the right renal tumour (R=Right)
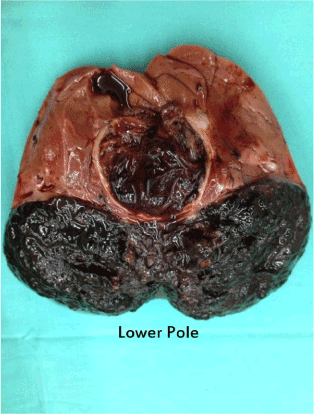
Figure 2. The right renal tumor bisected to reveal a bizarre looking hyperpigmented tumor
Discussion
Renal tumors in a 7 year old child are uncommon and pose a range of unusual diagnosis. Wilms tumor being the commonest renal carcinoma in children is also less common in patients above 5 years of age. The findings of renal tumor arising from perivascular epithelioid cells (PEC) in a child raised concerns on how best to tackle this tumor. The perivascular epithelioid cells (PEC) which characteristically stained the melanosome marker HMB-45 was first described in 1992 by Bonetti et al. [1,2].It was further described as a group of mesenchymal tumors with presence of PEC to be known as PEComa in 1996 by Zamboni et al.[1-3]. Apart from melanosome HMB-45, the PEComas also produce positive staining for Melanin A and melanoma-associated antigen CD63 [1-7].PEComas include a variety of tumors e.g. angiomyolipomas, lymphangioleiomyomatosis, abdominopelvic sarcoma of perivascular epithelioid cells, clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres and clear cell sugar tumor of the lung [8-13]. In 2006, Pan et al. studied 8 cases of PEComas of the genitourinary tract (3 were renal in origin) and found that there was associated deletion of TSC2 locus on 16p [10].The spectrum of renal PEComas include the variants of angiomyolipoma (AML) i.e. the classical angiomyolipoma (CAML) and the epithelioid angiomyolipoma (EAML). The EAML is further divided into atypical epithelioid angiomyolipoma (AAML) and malignant renal epithelioid angiomyolipoma (MEAML). It is difficult to determine the presence of malignancy in renal PEComa, the suggested indicators are high index of mitotic activity, tumor size and presence of vascular invasion [1,8,10,14,15]. In our patient, the mitotic figure was rarely seen and there was no lymphovascular invasion suggesting that is was more likely not to be malignant. However, the presence of abundant granular to clear cytoplasm in the pleomorphic cells raised the suspicion of it being AAML [1,15,16]. It is also known from the literature that the AAML renal PEComa can be difficult to distinguished from Renal cell carcinoma (RCC) [1,10,14,15,16]. We opted to monitor this child by doing 3-monthly ultrasound of the abdomen for the first year post surgery. Many of the literatures on PEComa focus on the histology and identification of the tumor with some assistance on clinico-pathological prognostication but hardly any on standard protocol for treatment and management [1,10,15,16]. It is noted from literature that when malignant change occurs, the tumor tends to have variable response to chemotherapy and in some cases may relapse, requiring multiple surgeries [8,14].
Conclusion
There have been a number of reported cases of pediatric EAML renal PEComa which posed diagnostic and management challenges, but most case reports were the malignant type angiomyolipomas, MEAML [14,17,18]. The AAML variant of the EAML renal PEComa such as in this pediatric patient left us to ponder what to do next in case of recurrence and potential malignant change in the future. For this boy, we plan for lifelong follow-up and monitoring of recurrence. We are hopeful that there will be more studies on AAML variant of the EAML renal PEComa to assist better understanding of tumor behaviour or future development of tumor markers which can help to monitor tumor aggression; these steps are especially more important in any child diagnosed with this unfortunate disease.
2021 Copyright OAT. All rights reserv
Abstract
Perivascular epithelioid cell tumor (PEComa) is a rare entity with unpredictable clinical outcome. It is a group of mesenchymal tumors with presence of perivascular epithelioid cells (PEC), known to stain positively with melanosome markers HMB-45. They may arise from many unusual sites including the kidneys. Renal PEComa was previously groupedas angiomyolipoma and its existence is rare too. Pediatric PEComas are even rarer with less than 40 cases described worldwide and none involving the kidney before. We report a case of a 7 year old boy who presented with incidental findings of an abdominal mass, which confirmed to be a renal PEComa of the atypical epithelioid angiomyolipoma (AAML) type. The clinical management and review of literature for this interesting entity is discussed.
Key words
pediatric, PEComa, atypical angiomyolipoma
Case presentation
A previously well 7 years old boy was noted by his grandmother to have a bulge over the right side of the abdomen for one week duration. She also noticed that the boy had loss of appetite and less active. There was no complaint of abdominal pain, abnormal bowel habit or urinary symptoms such as hematuria. There was neither history of fever to suggest infection nor history of trauma. On examination, the boy appeared healthy and not syndromic. He was not pale or jaundice and his vital signs were stable. There was an obvious bulge on the right lumbar area. The bulge was a large non tender, 9 cm X 9 cm mass. The mass was ballotable suggesting that it was the right kidney. There was no other organomegaly. Blood investigations were done which included full blood count, renal profile, tumor markers for malignant germ cell tumour and teratoma and liver function test. All the blood investigations were normal. Radiological imaging was done concurrently; the abdominal ultrasound showed a mass arising from the right kidney, measuring 11.5 cm x 6.4 cm x 9.4 cm. It was a combined solid cystic mass with no calcifications seen. CT scan of the abdomen confirmed the findings and showed that the mass was exophytic in nature, localised with no invasion to surrounding structure. There was no caval involvement or aortocaval adenopathy. The other solid organs and lung bases were normal (Figure 1). The provisional diagnosis was a renal sarcoma with a differential diagnosis of renal cell carcinoma. An ultrasound guided percutaneous biopsy was performed which was reported as melanocytic angiomyolipoma. We proceeded to do open right nephro-ureterectomy; intraoperatively the tumor was quite vascularized, surgery went well without problems. We bisected the specimen to reveal an unusual looking tumor (Figure 2). Post operatively, the patient was well and discharged home at day 5. The pathological examination of the tumor revealed macroscopically the tumor was irregular, fragile, and blackishin color, located at the lower pole of the right kidney measuring 8.5 cm X 4.5 cm X 6.5 cm. The tumor extended into the renal pelvis and capsule but did not appear to breach the capsule. Microscopically, the tumor was composed of sheets and nests and perivascular cuffing of large polygonal epithelioid cells. These cells had mildly pleomorphic, vesicular nuclei with prominent nucleoli and abundant granular to clear cytoplasm. The cells were densely pigmented by melanin. Mitotic figure were rarely seen. The tumor was seen invading but not breaching the renal capsule. The ureter was free from tumor. The adjacent renal parenchyma was compressed by the tumor bulk. No lymphovascular invasion was seen. Immunohistochemically, the tumor cells were positive for HMB45 and Melan A but negative for CD10. These descriptions were diagnostic for PEComa, non-malignant. The patient came for regular follow up and serial ultrasound imaging; at one year follow up the patient remained well and radiologically there was no recurrence of tumor.
Discussion
Renal tumors in a 7 year old child are uncommon and pose a range of unusual diagnosis. Wilms tumor being the commonest renal carcinoma in children is also less common in patients above 5 years of age. The findings of renal tumor arising from perivascular epithelioid cells (PEC) in a child raised concerns on how best to tackle this tumor. The perivascular epithelioid cells (PEC) which characteristically stained the melanosome marker HMB-45 was first described in 1992 by Bonetti et al. [1,2].It was further described as a group of mesenchymal tumors with presence of PEC to be known as PEComa in 1996 by Zamboni et al.[1-3]. Apart from melanosome HMB-45, the PEComas also produce positive staining for Melanin A and melanoma-associated antigen CD63 [1-7].PEComas include a variety of tumors e.g. angiomyolipomas, lymphangioleiomyomatosis, abdominopelvic sarcoma of perivascular epithelioid cells, clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres and clear cell sugar tumor of the lung [8-13]. In 2006, Pan et al. studied 8 cases of PEComas of the genitourinary tract (3 were renal in origin) and found that there was associated deletion of TSC2 locus on 16p [10].The spectrum of renal PEComas include the variants of angiomyolipoma (AML) i.e. the classical angiomyolipoma (CAML) and the epithelioid angiomyolipoma (EAML). The EAML is further divided into atypical epithelioid angiomyolipoma (AAML) and malignant renal epithelioid angiomyolipoma (MEAML). It is difficult to determine the presence of malignancy in renal PEComa, the suggested indicators are high index of mitotic activity, tumor size and presence of vascular invasion [1,8,10,14,15]. In our patient, the mitotic figure was rarely seen and there was no lymphovascular invasion suggesting that is was more likely not to be malignant. However, the presence of abundant granular to clear cytoplasm in the pleomorphic cells raised the suspicion of it being AAML [1,15,16]. It is also known from the literature that the AAML renal PEComa can be difficult to distinguished from Renal cell carcinoma (RCC) [1,10,14,15,16]. We opted to monitor this child by doing 3-monthly ultrasound of the abdomen for the first year post surgery. Many of the literatures on PEComa focus on the histology and identification of the tumor with some assistance on clinico-pathological prognostication but hardly any on standard protocol for treatment and management [1,10,15,16]. It is noted from literature that when malignant change occurs, the tumor tends to have variable response to chemotherapy and in some cases may relapse, requiring multiple surgeries [8,14].
Conclusion
There have been a number of reported cases of pediatric EAML renal PEComa which posed diagnostic and management challenges, but most case reports were the malignant type angiomyolipomas, MEAML [14,17,18]. The AAML variant of the EAML renal PEComa such as in this pediatric patient left us to ponder what to do next in case of recurrence and potential malignant change in the future. For this boy, we plan for lifelong follow-up and monitoring of recurrence. We are hopeful that there will be more studies on AAML variant of the EAML renal PEComa to assist better understanding of tumor behaviour or future development of tumor markers which can help to monitor tumor aggression; these steps are especially more important in any child diagnosed with this unfortunate disease.
References
- Khan MS, Iram S, O'Brien TS, Dasgupta P (2006) Renal 'perivascular epitheloid cell-omas'. BJU Int 98: 1146-1147. [Crossref]
- Bonetti F, Pea M, Martignoni G, Zamboni G (1992) PEC and sugar. Am J Surg Pathol 16: 307-308. [Crossref]
- Zamboni G, Pea M, Martignoni G, Zancanaro C, Faccioli G, et al. (1996) Clear cell "sugar" tumor of the pancreas. A novel member of the family of lesions characterized by the presence of perivascular epithelioid cells. Am J Surg Pathol 20: 722-730. [Crossref]
- Pea M, Bonetti F, Zamboni G, Martignoni G, Riva M, et al. (1991) Melanocyte-marker-HMB-45 is regularly expressed in angiomyolipoma of the kidney. Pathology 23: 185-188. [Crossref]
- Bacchi CE, BonettiF, Pea M, Martignoni G, Gown AM (1996) HMB-45: a review. Appl Immunohistochem 4: 73–85.
- Eble JN, Amin MB, Young RH (1997) Epithelioid angiomyolipoma of the kidney: a report of five cases with a prominent and diagnostically confusing epithelioid smooth muscle component. Am J Surg Pathol 21: 1123-1130. [Crossref]
- Mai KT, Perkins DG, Collins JP (1996) Epithelioid cell variant of renal angiomyolipoma. Histopathology 28: 277-280. [Crossref]
- Alaggio R, Cecchetto G, Martignoni G, Bisogno G, Cheng L, et al. (2012) Malignant perivascular epithelioid cell tumor in children: description of a case and review of the literature. J Pediatr Surg 47: e31-40. [Crossref]
- Hornick JL, Fletcher CD (2006) PEComa: what do we know so far? Histopathology 48: 75-82.[Crossref]
- Martignoni G, Pea M, Reghellin D, Zamboni G, Bonetti F (2007) Perivascular epithelioid cell tumor (PEComa) in the genitourinary tract. Adv Anat Pathol 14: 36-41. [Crossref]
- Folpe AL, Goodman ZD, Ishak KG, Paulino AF, Taboada EM, et al. (2000) Clear cell myomelanocytic tumor of the falciform ligament/ ligamentumteres: a novel member of the perivascular epithelioid clear cell family of tumors with a predilection for children and young adults. Am J Surg Pathol 24: 1239-1246. [Crossref]
- Folpe AL, McKenney JK, Li Z, Smith SJ, Weiss SW (2002) Clear cell myomelanocytic tumor of the thigh: report of a unique case. Am J Surg Pathol 26: 809-812. [Crossref]
- Bonetti F, Martignoni G, Colato C, Manfrin E, Gambacorta M, et al. (2002) Abdominopelvic sarcoma of perivascular epithelioid cells. Report of four cases in young women, one with tuberous sclerosis. Mod Pathol 14: 563-568. [Crossref]
- Xi S, Chen H, Wu X, Jiang H, Liu J, et al. (2014) Malignant renal angiomyolipoma with metastases in a child. Int J Surg Pathol 22: 160-166. [Crossref]
- Brimo F, Robinson B, Guo C, Zhou M, Latour M, et al. (2010) Renal epithelioid angiomyolipoma with atypia: a series of 40 cases with emphasis on clinicopathologic prognostic indicators of malignancy. Am J Surg Pathol 34: 715-722. [Crossref]
- Gill S, Weber-Shrikant E, Chen F (2011) A review of angiomyolipoma and its morphological variant. N A J Med Sci 4: 84-88.
- Johnson MH, Song JB, Dehner LP, Figenshau RS (2014) Epithelioid angiomyolipoma in a pediatric patient: case report and review of literature. Uro Case Reports 2: 159-161.
- Tchaprassian Z, Mognato G, Paradias G, D'Amore ES, Tregnaghi A, et al. (1998) Renal angiomyolipoma in children: diagnositc difficulty in 3 patients. J Urol 159: 1654-1656. [Crossref]