The “classical” renin-angiotensin system is known to maintain blood pressure and body fluids. However, increased understanding of its individual components and their structures and regulatory mechanisms, and the convergence and crosstalk with related pathways has resulted in the appreciation of a much more complex system. Research has shown that the renin-angiotensin system is implicated in a wide range of biological processes and diseases. This review provides an update on recent research into the structures, functions and metabolism of components of the renin-angiotensin system and its related pathways, and its role in biological processes such as angiogenesis, tumorigenesis, metastasis and cellular proliferation. The observation of the expression of the renin-angiotensin system by cancer stem cells suggests that cancer stem cells may be a novel therapeutic target by modulation of the renin-angiotensin system.
cancer stem cell, renin-angiotensin system, angiotensin converting enzyme, pro-renin receptor, angiotensin receptor
The “classical” renin-angiotensin system (RAS) is a hormone system known to maintain blood pressure and body fluids [1]. Initial studies of the RAS date back to 1898 when renin was first discovered by Tigerstedt and Bergman [2]. Subsequent studies have reshaped the “classical” view of the RAS.
Research has led to the appreciation that the RAS is a much more complex system than first thought (Figure 1), with wide-reaching physiological and pathological implications. One significant area of study is the involvement of the RAS in the biology of cancer. Recent literature has implicated a crucial role of the RAS in the development and maintenance of cancer. Its involvement in carcinogenesis, particularly its effects on cancer stem cells (CSCs), has become an area of focus [3-8]. This review summarises current knowledge of the RAS and its related pathways in the context of stem cells, tumourigenesis and cancer. The structure, metabolism and functions of the major components of the RAS are discussed below.
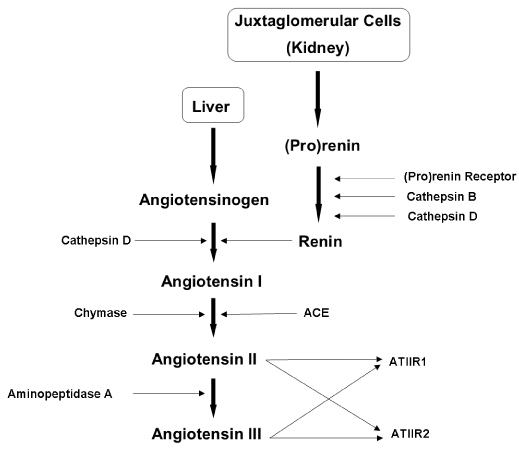
Figure 1. Figure 1: The renin-angiotensin system (RAS). In the RAS, (pro)renin activation is caused by its binding to the (pro)renin receptor. Cathepsins B and D are also renin-activating enzymes. Renin and cathepsin D covert angiotensinogen to angiotensin I (ATI) which is acted upon by angiotensin converting enzyme (ACE) and chymase, to produce angiotensin II (ATII). Aminopeptidase A converts ATII to angiotensin III (ATIII). ATII and ATIII act on angiotensin II receptor 1 (ATIIR1) and angiotensin II receptor 2 (ATIIR2). Redundancy in the pathway has been revealed in the form of cathepsins B and D, aminopeptidase A and chymase.
The RAS was originally associated with the kidneys, which produce renin in response to decreased arterial pressure, reduced sodium in the distal tubule, or sympathetic nervous system activity via the β-adrenergic receptors [9]. Renin is secreted from the juxtaglomerular cells into the bloodstream where it encounters angiotensinogen (AGN), normally produced by the liver [9]. Renin catalyses the conversion of AGN to angiotensin I (ATI), which is quickly cleaved by angiotensin converting enzyme (ACE) to form angiotensin II (ATII). ATII triggers the release of aldosterone from the adrenal glands, which stimulates reabsorption of sodium and water and thereby increases blood volume and blood pressure. ATII also acts on smooth muscle to cause vasoconstriction of the arterioles in different organs of the body [1, 10]. Furthermore, ATII promotes the release of antidiuretic hormone from the posterior pituitary gland, which results in water retention and triggers the thirst reflex [10].
It has been proposed that the development and spread of many cancers is driven by a subpopulation of cancer cells known as CSCs [11]. CSCs have been identified in multiple cancer types including myelogenous leukaemia [12], pancreatic cancer [13], breast cancer [14], oral tongue [15], buccal mucosal [8] and lip [16] squamous cell carcinoma, and glioblastoma multiforme (GBM) [3]. They have been proposed to be generated by mutations in either normal embryonic stem cells (ESCs) or progenitor cells, which may be present at birth or are accumulated over time, resulting in cells possessing the ability for uncontrolled growth and propagation [17-19]. Recent studies have also observed the ability of non-CSCs to ‘de-differentiate’ into CSCs due to epigenetic or environmental factors, which further increases the complexity of tumour biology and treatment [20]. Within a tumour there may only be a small number of CSCs which are highly tumourigenic [21] and have the capacity to divide symmetrically giving rise to additional CSCs that migrate to form new tumours, and to downstream, progenitor cells and differentiated cancer cells that possess no or low tumourigenic potential and form the main bulk of the tumour [22].
Research into the role of the RAS in cancer has gained momentum in recent times. This is underscored by the observation of dysregulation of the RAS in different types of cancers, and the observed efficacy of RAS modulators on cancer in both cancer models and cancer patients [23-29]. Patients administered ACE inhibitors (ACEi) have a reduced risk of developing certain cancers, which has been attributed to specific RAS antagonism rather than their anti-hypertensive action [24,30]. A localised (‘paracrine’) RAS mechanism has been identified in many types of cancers, and interruption of the control of the RAS is thought to be the basis for its role in cancer [23].
Furthermore, CSCs - cells that possess ESC characteristics - have been identified and characterised in many types of cancers [3,8,15,16,31]. Components of the RAS are expressed by these CSCs, supporting the hypothesis of the presence of a ‘paracrine RAS’ in regulating these CSCs [6,7,32] (Figure 2). One theory states that these CSCs develop from normal stem cells following an aberrant process of pluripotency, while an alternative view suggests that differentiated cells become de-differentiated towards a stem-like phenotype caused by one or more genetic mutations [3,33,34].
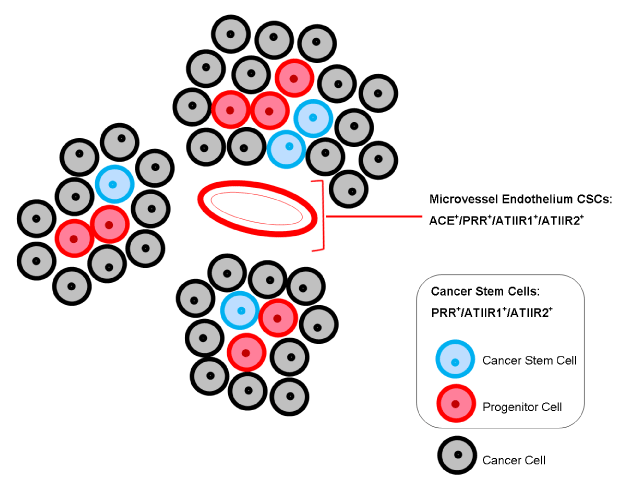
Figure 2. Expression of components of the RAS. Angiotensin converting enzyme (ACE) is localised to the endothelium of the microvessels within the peritumoural stroma, between the tumour nests. (Pro)renin receptor (PRR), angiotensin II receptor 1 (ATIIR1) and angiotensin II receptor 2 (ATIIR2) are expressed on the endothelium of microvessels, as well as the cancer stem cells (CSCs) within the tumour nests and within the peritumoural stroma.
Renin is an enzyme normally released by the kidneys in response to falling arterial pressure [1]. Renin becomes available in two ways: constant release of (pro)renin which becomes activated [35], and controlled β1-adrenoceptor-mediated release of active (mature) renin, especially in response to low levels of renal nerve stimulation [9,36,37]. Interestingly, during acute stimulation renin is exocytosed from secretory granules which only contain mature renin, however, chronic stimulation causes release of both renin and (pro)renin [9]. (Pro)renin is the inactive form of renin, when the active site is blocked by a 43-amino-acid prosegment. It is 7-9 times more abundant in circulation than renin, and both are bound by the same receptor, the (pro)renin receptor (PRR) [38].
PRR is a 35kDa transmembrane protein with a single transmembrane domain and a short cytoplasmic domain, and although it interacts with G-proteins, it has no intrinsic kinase activity [38]. Little is known about its structure because recombinant PRR is difficult to produce in its native form [38]. However, its importance is highlighted by being highly conserved between mammalian species. There is 95% nucleotide sequence homology and 80% amino-acid sequence homology between PRR of humans and those of rats and mice, respectively [38]. PRR is highly abundant in the heart, brain, placenta and visceral adipose tissue, with low abundance in kidney, liver and subcutaneous adipose tissue [10]. Membrane-bound PRR activates intracellular signalling via the MAP-kinase ERK1/2; active renin phosphorylates ERK1/2 which upregulates TGF-β1, collagens, fibronectin and cyclooxygenase-2, and is essential to brain development [10,38].
A soluble form of PRR, 28kDa in size, has been isolated and is found in the blood plasma [39,40]. PRR is cleaved by the protease furin in the trans-Golgi apparatus to produce two fragments. The 28kDa soluble PRR (sPRR) fragment is secreted into the plasma where it binds to renin and (pro)renin, activating the latter [39,41-43]. The 8.9kDa transmembrane segment has been tracked to the cell membrane, constituting the remaining PRR following cleavage of sPRR [38,41]. Like full-length PRR, its 8.9kDa fragment has been demonstrated to co-precipitate with V-ATPase, a hydrogen ion pump essential for pH regulation and vesicle acidification [44]. This association may explain why PRR gene deletion causes the death of embryos before the end of embryogenesis, as seen in zebrafish and flatworm studies [9,38-41].
Physiologically, (pro)renin can be activated in two ways [38]. Proteolytic activation occurs in the juxtaglomerular cells by irreversible removal of the prosegment, thereby exposing renin’s active site [38]. This proteolytic activation is thought to be effected by the enzymes cathepsin B and cathepsin D [45-47]. Alternatively, non-proteolytic activation has been induced in vitro by exposing (pro)renin to low pH (~3) or cold temperatures (~4°C) [38]. This non-proteolytic activation occurs in vivo when (pro)renin binds to the PRR. The prosegment folds away from the active site like a door on a hinge, allowing (pro)renin to be reversibly active while bound to the receptor.
A major focus of PRR research is its relationship with Wnt signalling. The Wnt family of glycoproteins control cell fate and polarity during embryonic development and regulate adult cell homeostasis [48,49]. Alteration of this pathway has been associated with congenital birth defects, neoplastic and degenerative diseases, and cancer [48,49]. Its importance in stem cell biology has also been observed through its role in tissue repair and cancer [50], including a study of GBM demonstrating overexpression of PRR coupled with the observation that inhibition of renin reduces cellular proliferation and promotes apoptosis [51]. Importantly, PRR has been found to be vital for normal Wnt signalling to occur, both canonical (β-catenin) and non-canonical (planar cell polarity) [41]. PRR binds the Wnt receptor complex proteins Fz and LRP6, and is responsible for linking this complex with V-ATPase. The PRR gene (ATP6AP2/PRR) received its name because PRR co-precipitates with V-ATPase by this functional link [41, 44]. Once activated, LRP6 appears to require an acidic environment to become phosphorylated and causes downstream signalling, with PRR linking V-ATPase to LRP6 to enable this to occur [41,44].
A study by Shibayama et al. (2015) [40] investigated PRR being key to the regulatory mechanism responsible for increased Wnt/β-catenin signalling in pancreatic ductal adenocarcinoma (PDAC) and its premalignant lesions known as pancreatic intraepithelial neoplasias (PanINs). Cells of later stage PanINs (stages 2 and 3) and PDAC have significantly increased β-catenin in the cytoplasm and nuclei. PDAC patients have increased sPRR and PRR expression from stage 2 PanIN onwards relative to normal pancreatic ducts, implying that abnormal PRR expression begins in the early stages of tumourigenesis [40]. Wnt/β-catenin dependence on PRR has been confirmed in a study of embryonic development [41], and when PRR is overexpressed such as in PDAC there is a loss of control of cell proliferation [40].
The presence of PRR has also been documented in the CSCs in oral cavity cancer including oral tongue [32], buccal mucosal [6,8] and lip [16] SCC, and also in GBM [7].
This would suggest a crucial role for PRR activation on the proliferation of CSCs, possibly via Wnt/β-catenin signalling, leading to carcinogenesis.
Angiotensin converting enzyme (ACE), also known as CD143, is the endothelial-bound peptidase which physiologically converts ATI to ATII (Figure 1). It is found in many endothelial cell types, especially in lung capillaries and epithelial cells of the kidney [52]. ACE is crucial in the regulation of blood pressure, angiogenesis and inflammation [53]. It is a common target for diseases of the cardiovascular system via ACEi which prevent ACE acting on ATI and hence inhibits its conversion to ATII [10,24,45]. It is also a cellular marker for primitive haematopoietic cells called haemangioblasts [5, 6,53-55].
Interestingly, recent reports have demonstrated expression of ACE on the endothelium of the microvessels within the stroma surrounding tumour nests, inferring a possible putative tumour vasculature reminiscent of tumour-associated ‘vascular mimicry’ or tumour angiogenesis [6,7,32].
Studies investigating the role of ACE in cancer have been carried out across a range of different tumours. ACE is expressed in infantile haemangioma (IH), the most common tumour of infancy. ATI production increases in response to high renin levels, and is subsequently converted by ACE into ATII which drives proliferation and differentiation [54,55]. ACE is expressed on the endothelium of the microvessels within the peritumoural stroma surrounding tumour nests in oral cavity SCC [6,15,16] and GBM [7].
Studies on the role of ACE in cancer have shown intriguing findings. Different ACE alleles have been correlated with prognosis: II and AA alleles produce a low-activity form of ACE and humans homozygous for these alleles have a lower incidence of cancer, while DD and TT homozygotes have more highly active ACE and higher risks of cancer [25,56]. Furthermore, ACEis have a well-documented cancer-protective effect [23-25,53,56]. A review by Ager et al. (2008) [24] cites studies showing the effect of Perindopril, an ACEi, in reducing tumour growth in head and neck SCCs. Captopril, another ACEi, inhibits tumour growth, metastasis and angiogenesis in orthopotic models of metastatic colorectal cancer to the lung [24].
Taken together, these results suggest that an overactive ACE promotes cancer growth and progression, and an inhibited or low-activity ACE may have cancer-protective effects.
Angiotensin II receptor 1 (ATIIR1) binds both ATII and angiotensin III (ATIII) [10] (Figure 1). ATIII, which consists of seven amino acids, is the product of aminopeptidase A removing the N-terminal residue from ATII [10]. It has been suggested that ATIII is more important than ATII in terms of the ATIIR1-mediated actions [10]. ATIIR1 is primarily involved in blood pressure regulation. When bound to ATII or ATIII it causes vasoconstriction by stimulating the release of vasopressin, reabsorption of water and sodium by promoting secretion of aldosterone and insulin, fibrosis, cellular growth and migration, pro-inflammation, glucose release from the liver, increased plasma triglyceride concentration, and reduced gluconeogenesis [10]. As these actions are associated with cardiovascular diseases, ATIIR1 is seen to produce downstream adverse effects if unregulated, upregulated, or unopposed [10, 24]. Furthermore, ATIIR1 is a G-protein-coupled receptor, with downstream signalling involved in vasodilation, hypertrophy and NF-κB activation leading to TNF-α and PAI-1 expression [10].
ATIIR1 has well-documented links with cancer, with one study demonstrating its overexpression in ~20% of breast cancer patients, but only in oestrogen receptor-positive tumours and not in those with Erbb2 overexpression. This links the RAS to oestrogen signalling and suggests that it functions redundantly to signalling of the NUE oncogene Erbb2 [23]. Furthermore, increased ATIIR1 expression correlates with a transition from benign to malignant tumours in the development of breast, ovarian and gastric cancers [23,24]. In addition, the effect of RAS dysregulation has been associated with increased VEGF expression and angiogenesis in cancers [23].
ATIIR1 is overexpressed in SCC, with the expression level correlated with worsening differentiation status of these tumours [25]. In ovarian and cervical cancer, ATIIR1 overexpression has been shown to be an indicator of tumour invasiveness [25]. Furthermore, administration of ATIIR1 blockers (ARBs) have been associated with reduced tumour size, reduction in tumour vascularisation, lower occurrence of metastases, and lower VEGF levels [25]. The connective tissue around the tumours in this study [25] has high expression levels of ATIIR1, increasing VEGF levels and therefore VEGF signalling. This is thought to have caused the angiogenesis seen around and within tumours [25,27]. Surprisingly, the tissue-associated macrophages around the tumour also display increased ATIIR1 levels and release more VEGF, suggesting that ATIIR1 can act on the tumour microenvironment by preventing immune cell infiltration [25]. This observation may partly explain the finding that ATIIR1 expression correlates with poor overall survival and a greater likelihood of progression of the cancer to a higher grade, compared to those not expressing ATIIR1 [27].
ATIIR1 appears to play a crucial role in the production of macrophages with tumour-promoting functions; ATII has been found to facilitate the production of haematopoietic stem and progenitor cells (HSPCs), which generate a tumour-associated macrophage (TAM) [57]. TAMs are the most abundant cell type in the tumour stroma, and are known to promote inflammation, angiogenesis, metastasis, migration and suppression of anti-tumour immune responses [57]. Reducing ATII production leads to lower numbers of HSPCs and TAMs in the spleen, suggesting that the interaction between ATII and the ATIIR1 drives the presence of HSPCs and TAMs within the tumour stroma [57].
ATII interaction with ATIIR1 promotes epithelial-to-mesenchymal transition (EMT), a process essential to cancer evasiveness and metastasis [58]. Furthermore, ATII causes upregulation of CSC markers and aggressiveness of cancer, highlighting a possible link between the RAS, EMT and CSCs in terms of metastasis and cancer progression [58].
Expression of ATIIR1 by the endothelium of the microvessels, as well as nuclear and cytoplasmic expression both within tumour nests and the peritumoural stroma of oral cavity SCC [6,15], and GFAP+ GBM CSCs [7], has been observed.
As with ATIIR1, ATIIR2 binds both ATII and ATIII [10]. The overall action of ATIIR2 is thought to oppose that of ATIIR1 [10,24], and is associated with vasodilation, growth inhibition, increased excretion of sodium in the urine and growth of nervous tissue [10]. ATIIR2 also plays an essential role in normal foetal development [59, 60]. A study into ATIIR2 mRNA expression in rats shows that its expression is transient in all cardiac and pulmonary structures throughout the perinatal period, and reaches its lowest basal level of expression around 21 days post-partum [59]. This suggests that the gene for ATIIR2 is not highly expressed in adult rats. This contrasts with the mRNA levels of ATIIR1, which is expressed throughout cardiac muscle and large blood vessels and persist into adulthood [59]. Another study using radioligand binding demonstrates transient expression of ATIIR2 in the foetal mesenchyme of rats [60]. This mesenchyme has the potential to differentiate into tissues such as smooth muscle and striated muscle, connective tissue sheaths, osteoblasts and fibroblasts, hence, transient ATIIR2 expression in this tissue during development suggests it is involved in regulating differentiation and development [60]. The presence of all components of the RAS in the developing rat foetus has been confirmed, and ATII/ATIIR2 binding does occur at this stage of maturation [60].
ATIIR2 plays a role in counteracting the effects of ATIIR1 in cancer and cardiovascular health. Studies in ATIIR2-deficient mice demonstrate an increase in ATIIR1-mediated VEGF upregulation in response to ATII, while another investigation saw inhibition of VEGFR/Flk-1 signals due to ATIIR2 action, indicating inhibitory cross-talk between ATIIR2 and the VEGF pathways [24]. ATIIR2 plays another role in this pathway by blocking VEGFR2 to reduce cell migration and metastasis [25].
A study using ATIIR2-knockout mice reports significantly faster growth of subcutaneously transplanted mouse pancreatic ductal carcinoma and a much higher degree of tumour vasculature when compared to mice with functional ATIIR2 [28]. This indicates further protective roles of ATIIR2 against cancer.
ATIIR2 induces apoptosis of cancer cells in a manner independent of ATII, via an extrinsic pathway which utilises caspase 3 [29]. Interestingly, ATIIR1 acts in the opposite way, preventing apoptosis by suppressing caspase 3 as well as upregulating the apoptosis-inhibitor survivin [25].
Further evidence of cancer-protective activity of ATIIR2 is the discovery of a gene coding for ATIIR2-interacting proteins (ATIPs) which inhibit EGFR-mediated proliferation. The ATIP gene has been identified as a putative tumour suppressor gene [25].
Treatment with Candesartan, an ATIIR1 blocker, and Irbesartan, another ARB, stopped tumour growth, metastasis and angiogenesis in a mouse model of metastases of colorectal cancer to the lung [24].
Previous reports have highlighted a role for activation of ATIIR2 by ATII on proliferation of stem cells derived from brain [61] and IH [55]. As with ATIIR1, nuclear and cytoplasmic expression of ATIIR2 has also been demonstrated in CSCs within the tumour nests and the peritumoural stroma of oral cavity SCCs [6,15,16] and the SOX2+ but not GFAP+ CSCs in GBM [7], as well as on the microvessel endothelium. It is clear from the literature that ATIIR2 plays a role in stem cell development; whether this is through its effects on apoptosis of CSCs or proliferation remains to be determined [6,7,15].
It has become increasingly recognised that the RAS is involved in a much wider range of biological processes than blood pressure and body fluid regulation. There has been increased understanding of the activities and signalling of individual components of the RAS, and the appreciation of the complex interplay between the RAS and other related pathways including the IGF/IGFR1 pathway, VEGF pathway and the existence of putative bypass loops consisting of cathepsins B, D and G. The interactions within this complex system individually and collectively via crosstalks with the related pathways are implicated in numerous biological processes such as angiogenesis, cell migration and proliferation, tumourigenesis, metastasis, and haematopoiesis. The sum of the activities within this integrated and complex system may create a conducive microenvironment for carcinogenesis, by promoting the proliferation and differentiation of CSCs.
The RAS is far more biologically important and relevant than first imagined. This is suggested by additional levels of redundancy in the RAS pathway in the form of cathepsins and chymase, as well as the roles of ATII receptors in tumourigenesis, expression of ACE as a novel marker of haemangioblasts. Crosstalks between the RAS and significant processes such as cancer progression, stem cell biology, inflammation and embryonic development highlight the extent of its influence. With the presence of components of the RAS confirmed in CSCs and their implications in processes such as angiogenesis, cell migration and proliferation, metastasis, haematopoiesis and tumourigenesis, targeting the RAS may potentially be a novel way to a prevent self-renewal of CSCs and restrict their ability to drive cancer progression and spread.
The role of the local (‘paracrine’) RAS is unique to each tissue or organ. While ATIIR1 is generally associated with harmful cancer-inducing activity, ATIIR2 appears to have a beneficial anti-cancer activity in most situations, the opposite has been seen to occur in cancers of other tissues and organs [24]. Whether RAS dysfunction is causative or a consequence of carcinogenesis remains uncertain.
2021 Copyright OAT. All rights reserv
This review shows that the RAS plays a role in regulating some interdependent functional capabilities required for malignant transformation – angiogenesis, cellular proliferation and growth signal sensitivity among them – largely via ATII and the ATIIR1 [23, 24], and indicates that restriction of CSC functions by manipulation of the RAS pathways may have an effect on the growth and development of cancer.
The expression of components of the RAS by CSCs in numerous types of cancer suggests that CSCs many be a novel therapeutic target by manipulation of the RAS and related pathways.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. TI, PDF and STT are inventors of the PCT patent applications Cancer Diagnosis and Therapy (No. PCT/NZ2015/050108), and Cancer Therapeutic (62/452479).
- Peach MJ (1977) Renin-angiotensin system: biochemistry and mechanisms of action. Physiol Rev 57: 313-370. [Crossref]
- Basso N, Terragno NA (2001) History about the discovery of the renin-angiotensin system. Hypertension 38: 1246-1249.
- Bradshaw A, Wickremesekera A, Brasch HD, Chibnall AM, Davis PF, et al. (2016) Cancer Stem Cells in Glioblastoma Multiforme. Front Surg 3: 48. [Crossref]
- Jokubaitis VJ, Sinka L, Driessen R, Whitty G, Haylock DN, et al. (2008) Angiotensin-converting enzyme (CD143) marks hematopoietic stem cells in human embryonic, fetal, and adult hematopoietic tissues. Blood 111: 4055-4063. [Crossref]
- Sinka L, Biasch K, Khazaal I, Péault B, Tavian M (2012) Angiotensin-converting enzyme (CD143) specifies emerging lympho-hematopoietic progenitors in the human embryo. Blood 119: 3712-23. [Crossref]
- Featherston T, Yu HH, Dunne JC, Chibnall AM, Brasch HD, et al. (2016) Cancer stem cells in moderately differentiated buccal mucosal squamous cell carcinoma express components of the renin-angiotensin system. Front Surg 3: 52. [Crossref]
- Bradshaw AR, Wickremesekera AC, Brasch HD, Chibnall AM, Davis PF, et al. (2016) Glioblastoma multiforme cancer stem cells express components of the renin-angiotensin system. Front Surg 3: 51. [Crossref]
- Yu HH, Featherston T, Tan ST, Chibnall AM, Brasch HD, et al., (2016) Characterization of cancer stem cells in moderately differentiated buccal mucosal squamous cell carcinoma. Front Surg 3: 46. [Crossref]
- Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM (2014) Classical Renin-Angiotensin system in kidney physiology. Compr Physiol 4: 1201-1228. [Crossref]
- Fyhrquist F, Saijonmaa O (2008) Renin-angiotensin system revisited. J Intern Med 264: 224-236. [Crossref]
- Kreso A, Dick JE (2014) Evolution of the cancer stem cell model. Cell Stem Cell 14: 275-291. [Crossref]
- Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, et al. (2009) Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458: 776-779. [Crossref]
- Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, et al. (2007) Identification of pancreatic cancer stem cells. Cancer Res 67: 1030-1037. [Crossref]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100: 3983-3988. [Crossref]
- Baillie R, Itinteang T, Yu HH, Brasch HD, Davis PF, et al. (2016) Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma. J Clin Pathol 69: 742-744. [Crossref]
- Ram R, Brasch HD, Dunne JC, Davis PF, Tan ST, et al. (2017) The Identification of Three Cancer Stem Cell Subpopulations within Moderately Differentiated Lip Squamous Cell Carcinoma. Front Surg 4: 12. [Crossref]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, et al. (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367: 645-648. [Crossref]
- Shipitsin M, Polyak K (2008) The cancer stem cell hypothesis: in search of definitions, markers, and relevance. Lab Invest 88: 459-463. [Crossref]
- Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, et al. (2016) Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front Surg 3: 21. [Crossref]
- Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K (2015) Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis 2: 152-163. [Crossref]
- Adams JM, Strasser A (2008) Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res 68: 4018-4021. [Crossref]
- Tamura K, Aoyagi M, Ando N, Ogishima T, Wakimoto H, et al. (2013) Expansion of CD133-positive glioma cells in recurrent de novo glioblastomas after radiotherapy and chemotherapy. J Neurosurg 119: 1145-1155. [Crossref]
- George AJ, Thomas WG, Hannan RD (2010) The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer 10: 745-759. [Crossref]
- Ager EI, Neo J, Christophi C (2008) The renin-angiotensin system and malignancy. Carcinogenesis 29: 1675-1684. [Crossref]
- Deshayes F, Nahmias C (2005) Angiotensin receptors: a new role in cancer? Trends Endocrinol Metab 16: 293-299. [Crossref]
- Feng Y, Wan H, Liu J, Zhang R, Ma Q, et al. (2010) The angiotensin-converting enzyme 2 in tumor growth and tumor-associated angiogenesis in non-small cell lung cancer. Oncol Reports 23: 941-948. [Crossref]
- Ino K, Shibata K, Kajiyama H, Yamamoto E, Nagasaka T, et al. (2006) Angiotensin II type 1 receptor expression in ovarian cancer and its correlation with tumour angiogenesis and patient survival. Br J Cancer 94: 552-560. [Crossref]
- Doi C, Egashira N, Kawabata A, Maurya DK, Ohta N, et al. (2010) Angiotensin II type 2 receptor signaling significantly attenuates growth of murine pancreatic carcinoma grafts in syngeneic mice. BMC Cancer 10: 67. [Crossref]
- Li H1, Qi Y, Li C, Braseth LN, Gao Y, et al. (2009) Angiotensin type 2 receptor-mediated apoptosis of human prostate cancer cells. Mol Cancer Ther 8: 3255-3265. [Crossref]
- Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, et al. (1998) Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet 352: 179-184. [Crossref]
- Jordan CT, Guzman ML, Noble M (2006) Cancer stem cells. N Engl J Med 355: 1253-1261. [Crossref]
- Itinteang T, Dunne JC, Chibnall AM, Brasch HD, Davis PF, et al. (2016) Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma express components of the renin-angiotensin system. J Clin Pathol 69: 942-945. [Crossref]
- Chen GY, Zhong,JF (2012) Pluripotent Human Stem Cells: An Overview, in Stem Cells and Cancer Stem Cells, Volume 1: Stem Cells and Cancer Stem Cells, Therapeutic Applications in Disease and Injury: Volume 1, Hayat MA, Edt, Springer Netherlands: Dordrecht: 3-12.
- Kasai T, Chen L, Mizutani A, Kudoh T, Murakami H, et al. (2014) Cancer stem cells converted from pluripotent stem cells and the cancerous niche. J Stem Cells Regen Med 10: 2-7. [Crossref]
- Pratt RE, Flynn JA, Hobart PM, Paul M, Dzau VJ (1988) Different secretory pathways of renin from mouse cells transfected with the human renin gene. J Biol Chem 263: 3137-41.
- Kopp U, Aurell M, Nilsson IM, Ablad B (1980) The role of beta-1-adrenoceptors in the renin release response to graded renal sympathetic nerve stimulation. Pflugers Arch 387: 107-113. [Crossref]
- Weber F, Brodde OE, Anlauf M, Bock KD (1983) Subclassification of human beta-adrenergic receptors mediating renin release. Clin Exp Hypertens A 5: 225-238. [Crossref]
- Nguyen G, Muller DN (2010) The biology of the (pro)renin receptor. J Am Soc Nephrol 21: 18-23. [Crossref]
- Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, et al. (2009) Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension 53: 1077-1082. [Crossref]
- Shibayama Y, Fujimori T, Nguyen G, Hirose T, Totsune K, et al. (2015) (Pro)renin receptor is crucial for Wnt/β-catenin-dependent genesis of pancreatic ductal adenocarcinoma. Sci Rep 5: 8854. [Crossref]
- Nguyen G (2011) Renin, (pro)renin and receptor: an update. Clin Sci (Lond) 120: 169-178. [Crossref]
- Nguyen G, Blanchard A, Curis E, Bergerot D, Chambon Y, et al. (2014) Plasma soluble (pro)renin receptor is independent of plasma renin, prorenin, and aldosterone concentrations but is affected by ethnicity. Hypertension 63: 297-302. [Crossref]
- Gonzalez AA, Lara LS, Luffman C, Seth DM, Prieto MC (2011) Soluble form of the (pro)renin receptor is augmented in the collecting duct and urine of chronic angiotensin II-dependent hypertensive rats. Hypertension 57: 859-864. [Crossref]
- Ray LB (2010) Of Wnt, Prorenin Receptor, and V-ATPase. Sci Signaling 3: ec29-ec29.
- Itinteang T, Chudakova DA, Dunne JC, Davis PF, Tan ST (2015) Expression of Cathepsins B, D, and G in Infantile Hemangioma. Front Surg 2: 26. [Crossref]
- Neves FA, Duncan KG, Baxter JD (1996) Cathepsin B is a prorenin processing enzyme. Hypertension 27: 514-517. [Crossref]
- Morris BJ (1978) Activation of human inactive ("pro-") renin by cathepsin D and pepsin. J Clin Endocrinol Metab 46: 153-157. [Crossref]
- MacDonald BT, Tamai K, He X (2009) Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9-26. [Crossref]
- Komiya Y, Habas R (2008) Wnt signal transduction pathways. Organogenesis 4: 68-75. [Crossref]
- Lucero OM, Dawson DW, Moon RT, Chien AJ (2010) A re-evaluation of the "oncogenic" nature of Wnt/beta-catenin signaling in melanoma and other cancers. Curr Oncol Rep 12: 314-318. [Crossref]
- Juillerat-Jeanneret L1, Celerier J, Chapuis Bernasconi C, Nguyen G, Wostl W, et al. (2004) Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br J Cancer 90: 1059-1068. [Crossref]
- Riordan JF (2003) Angiotensin-I-converting enzyme and its relatives. Genome Biol 4: 225. [Crossref]
- Zambidis ET, Park TS, Yu W, Tam A, Levine M, et al. (2008) Expression of angiotensin-converting enzyme (CD143) identifies and regulates primitive hemangioblasts derived from human pluripotent stem cells. Blood 112: 3601-3614. [Crossref]
- Itinteang T, Brasch HD, Tan ST, Day DJ (2011) Expression of components of the renin-angiotensin system in proliferating infantile haemangioma may account for the propranolol-induced accelerated involution. J Plast Reconstr Aesthet Surg 64: 759-765. [Crossref]
- Itinteang T, Marsh R, Davis PF, Tan ST (2015) Angiotensin II causes cellular proliferation in infantile haemangioma via angiotensin II receptor 2 activation. J Clin Pathol 68: 346-350. [Crossref]
- Rosenthal T, Gavras I (2009) Angiotensin inhibition and malignancies: a review. J Hum Hypertens 23: 623-635. [Crossref]
- Cortez-Retamozo V, Etzrodt M, Newton A, Ryan R, Pucci F, et al. (2013) Angiotensin II drives the production of tumor-promoting macrophages. Immunity 38: 296-308. [Crossref]
- Tawinwung S, Ninsontia C, Chanvorachote P (2015) Angiotensin II Increases Cancer Stem Cell-like Phenotype in Lung Cancer Cells. Anticancer Res 35: 4789-4797. [Crossref]
- Shanmugam S, Corvol P, Gasc JM (1996) Angiotensin II type 2 receptor mRNA expression in the developing cardiopulmonary system of the rat. Hypertension 28: 91-97. [Crossref]
- Grady EF, Sechi LA, Griffin CA, Schambelan M, Kalinyak JE (1991) Expression of AT2 receptors in the developing rat fetus. J Clin Invest 88: 921-933. [Crossref]
- Chao J, Yang L, Buch S, Gao L (2013) Angiotensin II increased neuronal stem cell proliferation: role of AT2R. PLoS One 8: e63488. [Crossref]