Abstract
Repetitive mild traumatic brain injury (r-mTBI) has been gaining increasing attention from the researchers since several studies have reported that the cognitive dysfunctions after single mTBI become measurably long-term deficits, such as delayed speed of processing and memory dysfunction, after r-mTBI, contributing to the emerging hypothesis that r-mTBI may cause cumulative damage to the brain, and in the absence of cell death, could result in cognitive deficits which may ultimately progress to memory and learning dysfunction. Studies also associated r-mTBI with “second-impact syndrome” and chronic traumatic encephalopathy (CTE) as possible consequences of r-mTBI. However, the potential injury mechanisms involved in r-mTBI still remain unclear and research on r- mTBI is still in early stages. Thereby, in this study an in vitro uniaxial stretching model was used to investigate the r-mTBI related cell damage for clarifying the pathology and post-injury sequelae of r- m TBI in comparison with single mTBI. 3 types of stretching experiments were conducted including the single loading groups that were subjected once to stretching with a strain of 0.1 at a strain rate of 5 s-1, the repetitive loading groups that were subjected again to the same stretching 1 h after the initial stretching, and the sham control groups that were set in and removed from the uniaxial stretching device without receiving mechanical loading. Results shows that even though initial insult induced some level of swelling formation, swelling formation increased by second stretching confirming that r-mTBI causes increased amounts of cellular damage when compared with single insults of the same magnitude. Moreover, the absence of progression to cell death at 24 h post injury is detected after swelling formation by repetitive stretching. These data indicate that if injured neurons are subsequently subjected to low strain at the same level, the rate of injury significantly increases confirming the emerging hypothesis on repetitive mTBI exacerbating traumatic axonal injury.
Key words
repetitive mild traumatic brain injury, concussion, impulsive strain, axonal stretching, axonal swellings, cell death
Background
To this day, traumatic brain injury (TBI) continues to be a leading cause of mortality and morbidity worldwide, making TBI a significant public health problem [1]. In the United States alone, the estimated number for annual occurrence of TBI is 1,700,000 [2-3] whereas in Asia 300-400, and in European countries (average of the 23 countries) 200-300 people per 100 000 are hospitalized annually due to TBI [4-6]. Moreover, majority of these cases categorized as mild TBI (mTBI) or concussion [2, 7] while, in spite of the name “mild”, approximately 15% of mTBI patients suffering persistent cognitive dysfunction in the United States alone [8]. Furthermore, several studies on professional athletes and military personnel have reported that these cognitive dysfunctions become measurably long-term deficits, such as delayed speed of processing and memory dysfunction, after “repetitive” mTBI (r-mTBI), contributing to the emerging hypothesis that r-mTBI may cause cumulative damage to the brain, and in the absence of cell death, could result in cognitive deficits which may ultimately progress to memory and learning dysfunction [9-17]. Aiding to the human cases, studies on animal models have also demonstrated a worsened outcome with repetitive TBI, where cell death and tissue damage are common consequences of moderate and severe TBI, not mTBI, yet cellular dysfunction and memory deficits are overt after mTBI [18-22].
Additionally, increasing evidence suggests that mTBI patients suffer from diffuse axonal injury (DAI), which is one of the most common pathology of TBI, associated with rapid brain deformation, stretching, inertial forces occurring as a result of traumatic incidents such as accidents, falls and assaults resulting in the stretching of neuronal axons [23-27] where primary damage to axons progressively develops into secondary cascades such as neuronal degeneration and axonal cytoskeletal disconnection [28-31]. Considering that DAI is involved in the immediate loss of conscioussness after TBI and several studies have been shown white matter abnormalities consistent with DAI in mTBI patients [32-34], there is a strong possibility of a potential mechanism which leads to vulnerability with a repeat injury since phenotypic or physiologic changes of the injured axons would likely influence outcome following r-mTBI and that DAI has an important role in r-mTBI. Studies also associated r-mTBI with “second-impact syndrome” where individuals displaying post-concussion symptoms after initial TBI, which can include visual, sensory or motor dysfunctions or mental difficulty such as cognitive and memory problems, and devastating brain swelling resulting in semi- comatose situation with dilating pupils, loss of eye movement and respiratory failure after a second TBI occurring days or weeks after the first injury [35]. More recently, similar pathology, referred to as chronic traumatic encephalopathy (CTE), a neurodegenerative brain disorder, has been gaining growing awareness as another consequence of r-mTBI [36-40].
However, the potential injury mechanisms involved in r-mTBI still remain unclear and research on r-mTBI is still in early stages. Although in vivo models contribute substantially in understanding pathological and physiological sequelae on macroscopic and microscopic levels, complementing these with in vitro studies that simulate specific aspects of r-mTBI is crucial to address questions concerning post-injury sequelae and r-mTBI-related sub-lethal cellular dysfunction at the cellular and subcellular levels [41].
This study introduces an in vitro model in which subsequent injuries were induced by using a uniaxial stretching device for realization of the post-injury sequelae of r-mTBI. Proposed in vitro uniaxial stretching model provides a reliable environment to study the mechanisms underlying cellular dysfunction and for a better characterization of the cellular degradation and dysfunction following both single and repeated injuries. Results showed that swellings along neurites stretched twice at an hour interval increased immediately after stretching and were sustained for 24 h whereas swellings along neurites stretched once transiently increased within several hours following stretching. However, the neurites of neurons in cultures that received repeated insults showed signs of damage that were not evident after a single mild injury. Furthermore, no significant cell death was detected after r-mTBI.
These data indicate that if injured neurons are subsequently subjected to low strain at the same level, the rate of injury significantly increases confirming the emerging hypothesis on repetitive mTBI exacerbating traumatic axonal injury.
Methods
Uniaxial stretching device
The uniaxial stretching device mainly consists of a servo actuator, a linear sensor for measuring tensile displacement, a load cell for measuring tensile loading and a polydimethylsiloxane (PDMS) chamber, and is shown in Figure 1. A full description of the device configuration and loading mechanism has been published [42]. The Green-Lagrange strains of the culture substrate in the PDMS chamber were calculated by microscope images before and after the stretching. The strains on the central point of the culturing substrate in the stretching direction were obtained for every 0.5 mm displacement to 4 mm in total and plotted in Figure 2A. In this study, the history of the displacement shown in Figure 2B was applied to the chamber with a strain of 0.10 at a strain rate of 5 s-1. The strain rate was obtained by dividing the maximum strain by the time to maximum strain. The curves stood up in the first 10 msec by stretching, and converged in the last 10 msec by releasing with the same ratio as the standing up. It is expected that the neuron on the substrate is stretched/compressed with a similar deformation history through the substrate.
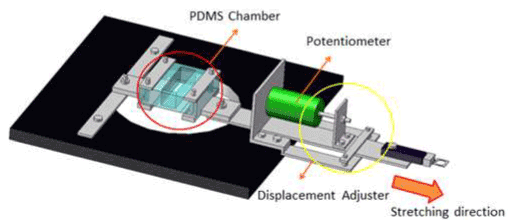
Figure 1. Uniaxial stretching device. The device consists of a PDMS chamber on a microscope stage, a linear sensor, a load cell, a servo actuator. The PDMS chamber is clamped to the microscope stage at one edge, and the other edge of the chamber is connected to the actuator.
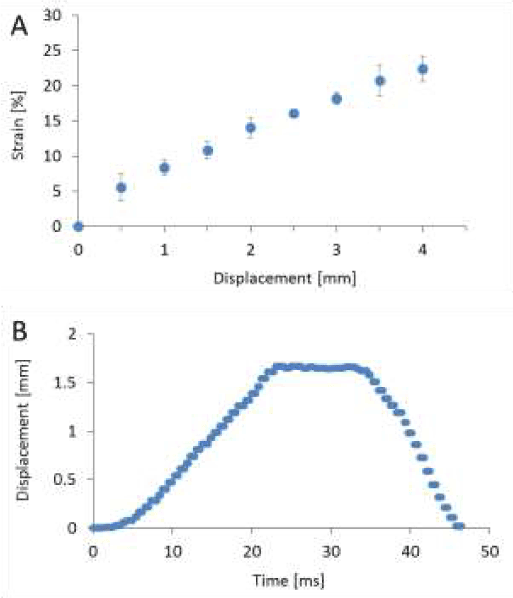
Figure 2. Displacement measurement of PDMS chamber. Relation between the forced displacement and the longitudinal strain around the center where cells are cultured was measured (A). The displacement corresponds to the impact (strain 0.10, strain rate 5 s-1) is expressed as the function of time (B).
Cell culture
PC12 cells—tumor cells derived from adrenal chromaffin cells of a rat [43] —were provided by the RIKEN Cell Bank (Tsukuba, Japan). After exposure to nerve growth factor (NGF), the cells extend neurites, which are structurally axon-like cylindrical protrusions, and differentiate into cholinergic sympathetic neurons [44]. The cells were cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Gibco), 10% horse serum (horse serum), 100 U/mL penicillin (Sigma-Aldrich), and 0.1 mg/mL streptomycin (Sigma-Aldrich) under conditions of 5% CO2 and 100% humidity at 37°C. The cells were seeded into the PDMS chamber at 1500–3000 cells/cm2 in the presence of 100 ng/mL NGF (Invitrogen) for 5 days. As a sham control, neurons matched in age to those subjected to strains were cultured in the PDMS chamber, after which they were placed in and removed from the uniaxial stretching device without mechanical loading. The medium in the PDMS chamber was not removed during the experiment, which was completed within 5 min. The surrounding temperature of the device was held at 37°C. The PDMS chamber was returned to the CO2 incubator after the experiment.
Repetitive stretching experiment
3types of stretching experiments were conducted. The single loading groups were subjected to initial stretching with a strain of 0.1 at a strain rate of 5 s-1, the repetitive loading groups were subjected again to the same stretching 1 h after first stretching with a strain of 0.1 at a strain rate of 5 s-1. The sham control groups were set in and removed from the uniaxial stretching device without receiving mechanical loading. The medium in the PDMS chamber was not removed during the experiment which was completed within 5 min. The temperature of the device and surroundings were kept at 37°C. The PDMS chamber was returned to the CO2 incubator after the experiment.
Morphological observation
Cells were observed using an inverted fluorescence microscope (FSX100, Olympus, Tokyo,Japan). Phase-contrast images were obtained immediately before stretching and at 5 min, 1 h, 3 h, 6 h and 24 h after first stretching. The number of cells and length of neurites were manually measured with ImageJ (National Institutes of Health, Bethesda, MD, USA). A neurite swelling was defined as a thick part in a neurite. Injured neuron was defined as the rate of neurons that have neurite swellings.
Cell viability
Cell viability was assessed using fluorescent probes (LIVE/DEAD Viability/Cytotoxicity Assay Kit; Lonza, Walkersville, MD, USA) for distinguishing live and dead cells. Cells were rinsed with Dulbecco’s phosphate buffered saline (D-PBS) and were incubated with 4 μM ethidium homodimer-1 (EthD-1; excitation and emission wavelengths of 528 and 617 nm, respectively) and 2 μM calcein-AM (excitation and emission wavelengths of 494 and 517 nm, respectively) at 37°C for 30 min. After rinsing with D-PBS, fluorescence images were obtained at 24 h after injury using the FSX-100. Live and dead cells were manually quantified using 5 randomly selected regions per experiment.
Statistical analysis
Results were expressed as the mean ± standard deviation (SD) of 4 independent experiments. 100–200 neurons per a PDMS substrate were analysed totally. Means were compared by Steel’s multiple comparison test. A p value of less than 0.05 was considered significant.
Results
Neurite swellings were observed in PC12 cells exposed to stretching (Figure 3); however, they were present in low amounts in the sham control groups and at pre-stretching time points (Figure 4). The formation of neurite swellings in the single loading groups increased significantly immediately after stretching and reached a peak at 3 h, but there was no significant difference at 24 h compared to the sham control group. However, swelling formation in the repetitive loading groups increased two times after second stretching more than after first stretching and was significantly higher at all time points than that in the sham control group.

Figure 3. Morphological changes of neurites after stretching. Phase-contrast images are PC12 cells in sham control (A, B, C and D), exposed to a single stretching (E, F, G and H) and exposed to a repetitive stretching (I, J, K and L) at pre-loading (A, E and I), at 1 h post-loading (B, F and J), at 3 h post-loading (C, G and K), 24 h post-loading (D, H and L). Scale bar is 50 μm. Arrows indicate neurite swellings.
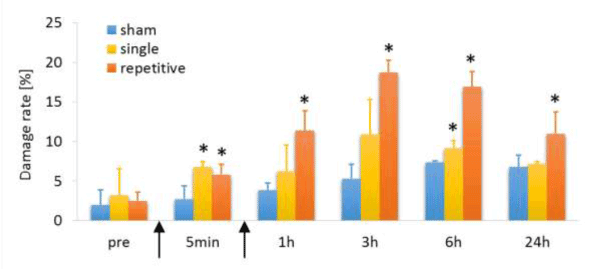
Figure 4. Time course of damage rate after stretching. The * symbol represents a statistically significant difference (p < 0.05) versus sham controls at each time point. Arrows indicates loading points.
The length of neurites, the number of cells and the percentage of cell viability at 24 h after loading are shown in Table 1. There was no significant difference in the neurite length and the cell number among all groups. Although cell viabilities of all groups were significantly unchanged, dead cells were observed only in the repetitive loading groups shown in Figure 5. Additionally, neurites of the repetitive loading groups were not stained by calcein-AM even without swelling formation.
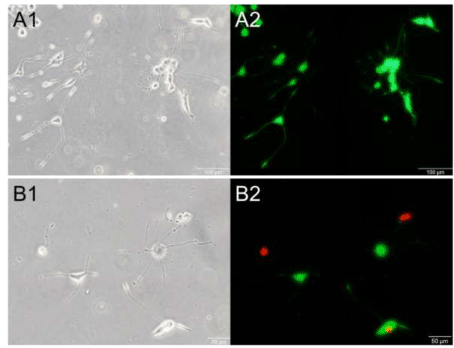
Figure 5. Alive cells and dead cells after stretching. Phase-contrast images (left) and fluorescence images (right) of PC12 cells of the sham control groups (A1 and A2), the single loading groups (B1 and B2) and the repetitive loading groups (C1 and C2). Live cells are shown in green and the nuclei of dead cells are shown in red.
Table 1. Neurite length, cell number and cell viability at 24 h after stretching
|
Neurite length [μm] |
Cell number [/mm2] |
Cell viability [%] |
Sham control |
86 ± 10 |
37 ± 7 |
100 ± 0 |
Single loading |
86 ± 4 |
33 ± 1 |
100 ± 0 |
Repetitive loading |
91 ± 14 |
35 ± 7 |
97.6 ± 3.4 |
|
Discussion
Repetitive mild traumatic brain injury (r-mTBI) recently gaining more interest from the researchers yet the pathology or the underlying mechanisms of detrimental effects of multiple mTBI remains unclear. To address this gap in the literature, several studies have proposed animal models in which studies indicated more persistent behavioral impairments and axonal injury compared to single injury without gross pathological injury or neuronal cell loss, behavioral deficits without apparent neuronal loss, neuronal cytoskeletal abnormalities, accelerated β-amyloid deposition and a potential “vulnerable state” lasting approximately between 3 and 5 days following the first TBI [18-19,21,45-48]. Although these in vivo studies provided valuable information for understanding that the changes in behavior post-injury is possible to be measured and correlated to cell damage, the extend of these studies can only reach to theorizing the underlying mechanisms of cognitive impairment at the cellular level under r-mTBI [41]. Therefore, it is crucial to complement in vivo studies with thoroughly devised in vitro studies to address the post-injury sequelae at the cellular level.
Moreover, despite several in vitro models on axonal injury induced by severe, moderate and mild TBI have been proposed ranging from direct axonal transection, transient axonal stretch injuries using pressurized fluid deflection, uniaxial in vitro models controlled by air pulse, focusing on the electrophysiogical responses of neural cells, axonal swelling formation, neurofilament structure, changes in ionic homeostasis, and axolemma permeability [29,49-62], even fewer in vitro studies exist for investigating r-mTBI mainly focusing on the inter-injury interval, pathophysiologic response, dependence on the severity of insult and characterization of the cellular degradation and dysfunction [41,48,63-64]. Thereby, in this study an in vitro uniaxial stretching model was used to investigate the r-mTBI related cell damage for clarifying the pathology and post-injury sequelae of repetitive mild TBI in comparison with single mTBI. The uniaxial stretching model is chosen since uniaxial stretching of the axon is clinically more relevant to axonal injury than biaxial stretching [65-67]. Furthermore, the widely employed models involving fluid or air pressure induced deformation is nonuniform where the strain at the center of the membrane is being assumed as the representative value which can lead to inconsistencies when relating cell injury to the amount of applied strain [56]. 3 types of stretching experiments were conducted including the single loading groups that were subjected once to stretching with a strain of 0.1 at a strain rate of 5 s-1, the repetitive loading groups that were subjected again to the same stretching 1 h after the initial stretching, and the sham control groups that were set in and removed from the uniaxial stretching device without receiving mechanical loading. Results shows that even though initial insult induced some level of swelling formation, swelling formation increased by second stretching confirming that r-mTBI causes increased amounts of cellular damage when compared with single insults of the same magnitude. Moreover, the absence of progression to cell death at 24 h post injury is detected after swelling formation by repetitive stretching.
In conclusion, the proposed model in this study is a promising method for investigating 8 secondary pathways of damage, combination with the directional control of axonal elongation therefore accurate control and observation of axonal injury, the effects of injury on cultures from various cell types, the long term consequences of r-mTBI, and the influence of different numbers of injuries and different inter-injury intervals which also can be listed as the future work of this study.
Repetitive mild TBI covers a substantial portion of all TBI cases although the attention given to experimental repetitive TBI studies has remained scarce until recent years. However, recent research involving both in vivo and in vitro studies seems promising for interpreting the pathology of r-mTBI. The identification of molecular targets specific to mTBI and ultimately the development of novel, effective therapeutics will be enabled after a thorough recognition of mTBI.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
HN designed the study. EK and SS carried out the stretching test and observed the cell morphology and viability. SA conceived the study, participated in its design and coordination, and helped to draft the manuscript. All authors have read and approved the final manuscript.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (B) (25289064) and a Grant-in- Aid for Scientific Research (C) (26350509) from the Japan Society for the Promotion of Science.
Reference
- Jennett B (1996) Epidemiology of head injury. J Neurol Neurosurg Psychiatry 60: 362-369. [Crossref]
- Faul M, Xu L, Wald MM, Coronado VG ( 2010) Traumatic brain injury in the United States: emergency department visits, hospitalizations and deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control.
- Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, et al. (2012) Repetitive Mild Traumatic Brain Injury in a Mouse Model Produces Learning and Memory Deficits Accompanied by Histological Changes. J Neurotrauma 29: 1–13. [Crossref]
- McGregor K, Pentland B (1997) Head injury rehabilitation in the UK: An economic perspective. Soc Sci Med 45: 295-303. [Crossref]
- Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J (2006) A systematic review of brain injury epidemiology in Europe. Acta Neurochir (Wien) 148: 255-268. [Crossref]
- Roozenbeek B, Maas AI, Menon DK (2013) Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol 9: 231-236. [Crossref]
- Laker SR (2011) Epidemiology of concussion and mild traumatic brain injury. PM R 3: S354-358. [Crossref]
- National Center for Injury Prevention Control. Report to Congress on Mild Traumatic Brain Injury in the United States: Steps to Prevent a Serious Public Health Problem. 2003. Atlanta: Centers for Disease Control and Prevention.
- Corsellis JA, Bruton CJ, Freeman-Browne D (1973) The aftermath of boxing. Psychol Med 3: 270-303. [Crossref]
- Collins MW, Grindel SH, Lovell MR, Dede DE, Moser DJ, et al. (1999) Relationship between concussion and neuropsychological performance in college football players. JAMA 282: 964–970. [Crossref]
- Guskiewicz KM, McCrea M, Marshall SW, Cantu RC, Randolph C, et al. (2003) Cumulative effects associated with recurrent concussion in collegiate football players: the NCAA concussion study. JAMA 290: 2549–2555. [Crossref]
- Iverson GL, Gaetz M, Lovell MR, Collins MW (2004) Cumulative effects of concussion in amateur athletes. Neurosurgery 69: 173–183. [Crossref]
- Guskiewicz KM, Marshall SW, Bailes J, McCrea M, Harding HP Jr, et al. (2007) Recurrent concussion and risk of depression in retired professional football players. Med Sci Sports Exerc 39: 903-909. [Crossref]
- Covassin T, Stearne D, Elbin R (2008) Concussion history and postconcussion neurocognitive performance and symptoms in collegiate athletes. J Athl Train 43: 119-124. [Crossref]
- Omalu BI, Hamilton RL, Kamboh MI, DeKosky ST, Bailes J (2010) Chronic traumatic encephalopathy in a national football league player: case report and emerging medico-legal practice questions. J Forensic Nurs 6: 40–46.
- Rosenfeld JV, Ford NL (2010) Bomb blast, mild traumatic brain injury and psychiatric morbidity: a review. Injury 41: 437-443. [Crossref]
- Kamnaksh A, Kovesdi E, Kwon SK, Wingo D, Ahmed F, et al. (2011) Factors affecting blast traumatic brain injury. J Neurotrauma 28: 2145-2153. [Crossref]
- Uryu K, Laurer H, McIntosh T, Praticò D, Martinez D, et al. (2002) Repetitive mild brain trauma accelerates Aß deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci 22, 446–454. [Crossref]
- Laurer HL, Bareyre FM, Lee VM, Trojanowski JQ, Longhi L, et al. (2001) Mild head injury increasing the brain's vulnerability to a second concussive impact. J Neurosurg 95: 859-870. [Crossref]
- Yoshiyama Y, Uryu K, Higuchi M, Longhi L, Hoover R, et al. (2005) Enhanced Neurofibrillary Tangle Formation, Cerebral Atrophy, and Cognitive Deficits Induced by Repetitive Mild Brain Injury in a Transgenic Tauopathy Mouse Model. J Neurotrauma 22: 1134–1141. [Crossref]
- Shitaka Y, Tran HT, Bennett RE, Sanchez L, Levy MA, et al. (2011) Repetitive Closed-Skull Traumatic Brain Injury in Mice Causes Persistent Multifocal Axonal Injury and Microglial Reactivity. J Neuropathol Exp Neurol 70: 551–567. [Crossref]
- Fujita M, Wei EP, Povlishock JT (2012) Intensity- and interval-specific repetitive traumatic brain injury can evoke both axonal and microvascular damage. J Neurotrauma 29: 2172-2180. [Crossref]
- Smith DH, Meaney DF (2000) Axonal Damage in Traumatic Brain Injury. Neuroscientist 6: 483.
- Meythaler JM, Peduzzi JD, Eleftheriou E, Novack TA (2001) Current concepts: Diffuse axonal injury associated traumatic brain injury. Archives of physical medicine and rehabilitation 82: 1461-1471.
- Browne KD, Chen XH, Meaney DF, Smith DH (2011) Mild traumatic brain injury and diffuse axonal injury in swine. J Neurotrauma 28: 1747-1755. [Crossref]
- Johnson VE, Stewart W, Smith DH (2012) Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol 22: 142-149. [Crossref]
- Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246: 35-43. [Crossref]
- Stone JR, Okonkwo DO, Dialo AO, Rubin DG, Mutlu LK et al. (2004) Impaired axonal transport and altered axolemmal permeability occur in distinct populations of damaged axons following traumatic brain injury. Exp Neurol 190: 59–69. [Crossref]
- Chung RS, Staal JA, Mccormack GH, Dickson TC, Cozens MA, et al. (2005) Mild axonal stretch injury in vitro induces a progressive series of neurofilament alterations ultimately leading to delayed axotomy. J Neurotrauma 22: 1081–1091. [Crossref]
- Kelley BJ, Farkas O, Lifshitz J, Povlishock JT (2006) Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Exp Neurol 198: 350–360. [Crossref]
- Staal JA, Dickson TC, Chung RS, Vickers JC (2007) Cyclosporin-A Treatment Attenuates Delayed Cytoskeletal Alterations and Secondary Axotomy Following Mild Axonal Stretch Injury. Dev Neurobiol 67: 1831. [Crossref]
- Inglese M, Makani S, Johnson G, Cohen BA, Silver JA, et al. (2005) Diffuse axonal injury in mild traumatic brain injury: a diffusion tensor imaging study. J Neurosurg 103: 298-303. [Crossref]
- Bazarian JJ, Zhong J, Blyth B, Zhu T, Kavcic V (2007) Diffusion tensor imaging detects clinically important axonal damage after mild traumatic brain injury: a pilot study. J Neurotrauma 24: 1447–1459. [Crossref]
- Wilde EA, McCauley SR, Hunter JV, Bigler ED, Chu Z, et al. (2008) Diffusion tensor imaging of acute mild traumatic brain injury in adolescents. Neurology 70: 948-955. [Crossref]
- Cantu RC (1996) Head injuries in sport. Br J Sports Med 30: 289-296. [Crossref]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, et al. (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neuro 68: 709– 735. [Crossref]
- Gavett BE, Stern RA, McKee AC (2011) Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med 30: 179-188. [Crossref]
- Stern RA, Riley DO, Daneshvar DH, Nowinski CJ, Cantu RC, et al. (2011) Long-term consequences of repetitive brain trauma: chronic traumatic encephalopathy. PM R 3: S460-467. [Crossref]
- Costanza A, Weber K, Gandy S, Bouras C, Hof PR (2011) Review: Contact sport-related chronic traumatic encephalopathy in the elderly: clinical expression and structural substrates. Neuropathol Appl Neurobiol 37: 570–584. [Crossref]
- Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9: 211-221. [Crossref]
- Slemmer JE, Matser EJ, De Zeeuw CI, Weber JT (2002) Repeated mild injury causes cumulative damage to hippocampal cells. Brain 125: 2699- 2709. [Crossref]
- Aomura S, Nakadate H, Kaneko Y, Nishimura A, Willinger R (2016) Stretch-Induced functional disorder of axonal transport in the cultured rat cortex neuron. Inte Mol Med 3: 654–660.
- Greene LA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor, Proceedings of the National Academy of Sciences of the United States of America 73: 2424– 2428.
- Dichter, M.A., Tischler A.S., Greene L.A. Nerve growth factor-induced change in electrical excitability and acetylcholine sensitivity of a rat pheochromocytoma cell line. 1977. Nature 268: 501–504.
- Kanayama G, Takeda M, Niigawa H, Ikura Y, Tamii H, et al. (1996) The effects of repetitive mild brain injury on cytoskeletal protein and behavior. Methods Find Exp Clin Pharmacol 18: 105-115. [Crossref]
- DeFord SM, Wilson MS, Rice AC, Clausen T, Rice LK, et al. (2002) Repeated mild brain injuries result in cognitive impairment in B6C3F1 mice. J Neurotrauma 19: 427-438. [Crossref]
- Longhi L, Saatman KE, Fujimoto S, Raghupathi R, Meaney DF (2005) Temporal window of vulnerability to repetitive experimental concussive brain injury. Neurosurgery 56: 364–374. [Crossref]
- Yuen TJ, Browne KD, Iwata A, Smith DH (2009) Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. J Neurosci Res 87: 3620-3625. [Crossref]
- Galbraith JA, Thibault LE, Matteson DR (1993) Mechanical and electrical responses of the squid giant axon to simple elongation. J Biomech Eng 115: 13-22. [Crossref]
- Ellis EF, McKinney JS, Willoughby KA, Liang S, Povlishock JT (1995) A new model for rapid stretch-induced injury of cells in culture: characterization of the model using astrocytes. J Neurotrauma 12: 325-39. [Crossref]
- Zhang L, Rzigalinski BA, Ellis EF, Satin LS (1996) Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science 274: 1921-1923. [Crossref]
- Tavalin SJ, Ellis EF, Satin LS (1997) Inhibition of the electrogenic Na pump underlies delayed depolarization of cortical neurons after mechanical injury or glutamate. J Neurophysiol 77: 632-638. [Crossref]
- Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF (1999) High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci 19: 4263-4269. [Crossref]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001) Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci 21: 1923-1930. [Crossref]
- Chuckowree JA, Vickers JC (2003) Cytoskeletal and morphological alterations underlying axonal sprouting after localized transection of cortical neuron axons in vitro. J Neurosci 23: 3715. [Crossref]
- Pfister BJ, Weihs TP, Betenbaugh M, Bao G (2003) An in vitro uniaxial stretch model for axonal injury. Ann Biomed Eng 31: 589-598. [Crossref]
- Geddes-Klein DM, Schiffman KB, Meaney DF (2006) Mechanisms and consequences of neuronal stretch injury in vitro differ with the model of trauma. J Neurotrauma 23: 193-204. [Crossref]
- Chen YC, Smith DH, Meaney DF (2009) In-vitro approaches for studying blast-induced traumatic brain injury. J Neurotrauma 26: 861-876. [Crossref]
- Monnerie H, Tang-Schomer MD, Iwata A, Smith DH, Kim HA, et al. (2010) Dendritic alterations after dynamic axonal stretch injury in vitro. Exp Neurol 224: 415-423. [Crossref]
- Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH (2012) Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol 233: 364. [Crossref]
- Higgins S, Lee JS, Ha L, Lim JY (2013) Inducing neurite outgrowth by mechanical cell stretch. Biores Open Access 2: 212-216. [Crossref]
- Yap YC, Dickson TC, King AE2, Breadmore MC3, Guijt RM4 (2014) Microfluidic culture platform for studying neuronal response to mild to very mild axonal stretch injury. Biomicrofluidics 8: 044110. [Crossref]
- Slemmer JE, Weber JT (2005) The extent of damage following repeated injury to cultured hippocampal cells is dependent on the severity of insult and inter-injury interval. Neurobiol Dis 18, 421–431. [Crossref]
- Weber J T( 2007) Experimental models of repetitive brain injuries. Progress in Brain Research, vol. 16, Neurotrauma: New Insights into Pathology and Treatment, pp. 252-261. Elsevier, ISBN 978-0-444-53017-2.
- Holbourn AHS (1943) Mechanics of head injury. Lancet 2: 438–441.
- Holbourn AHS (1945) The mechanics of brain injuries. Br Med Bull 3: 147–149.
- Strich SJ (1961) Shearing of nerve fibres as a cause of brain damage due to head injury. Langmuir, 443–448.