Retinopathy is a one of the major clinical manifestation of Hemoglobinopathy. It is acquired secondary to another retinal disorder. Retinopathy especially retinal occlusions are painless loss of monocular vision it's from a vascular disorder. Ocular stroke caused by embolism in a retinal artery, that may emboli travel to distal branches of the retinal artery, causing loss of other section in the visual field. The manifestations of Sickle cell disease ocular manifestations came due to vascular occlusion, which may exist in the conjunctiva, iris, retina, and choroid. Because the ocular changes produced by SCD could be shown in other diseases, it's important to except other occlusions' causes, which have included central retinal vein occlusion, Eales disease, and retinopathy secondary to other chronic disorders. Other ocular changes cause that includes polycythemia vera, familial exudative vitreoretinopathy, talc and cornstarch emboli, and uveitis. Diagnosis of hemoglobenopathies is performed exclusively through Hb electrophoresis. The treatments and their results vary from one condition to the other.
retinal diseases, hemoglobin, sickle cell trait, hemoglobin SC disease, vitreoretinal surgery, retinopathy
Sickle-cell anaemia is a hereditary condition (SS or SC haemoglobin) common in African people. Owing to occlusion of small vessels at the retinal periphery and ischemia, fibro-vascular proliferation occurs. Localized chorio-retinal scars are also characteristic of the condition. May be it causes loss of vision from traction retinal detachment or vitreous haemorrhage but it could be prevented with photocoagulation. Para-orbital ptosis it's showing as a facial infarction. A proliferative retinitis is common in Hb SC disease and may.
Normal adult hemoglobin (HbA) comprises two A- and two B-globin chains associated with a heme ring. In sickle hemoglobinopathies, there is a mutant hemoglobin, such as HbS (B-chain residue 6 Glu l Val), which behaves abnormally in response to hypoxia or acidosis. This causes “sickling” and hemolysis of red blood cells. Many other mutant hemoglobins have been described, the most common one being HbC. In thalassemias the problem is one of inadequate production of one or more of the A- or B-chains. Although systemic disease is most severe in sickle-cell disease (HbSS), ocular disease is most severe in HbSC and HbS-Thal disease. Sickle hemoglobinopathies are seen in Africans and their descendents (Table 1) [1,2].
Table 1. Sickle Hemoglobenopathies.
Sickle Hemoglobenopathies |
Disease |
Hb |
Prevalence in African-American Population |
Sickle trait |
HbAS |
5-10% |
Sickle-Cell disease |
HbSS |
0.4% |
Hemoglobin SC disease |
HbSC |
0.2% |
Sickle-Cell Thalassemia |
HbS-Thal |
0.5-1.0% ; 0.03% Severe |
Epidemiology and pathogenesis
The pathophysiology of sickle cell disease is not limited to abnormalities of erythrocytes; its clinical manifestations are related to processes and complex metabolic pathways that include endothelial activation, inflammation, nitric oxide bioavailability, oxidative stress and regulation of the adhesiveness of several types of blood cells [3].
The Hb molecule is composed of two alpha and two beta polypeptide chains. In hemoglobins S and C, an amino acid substitution occurs at position 6 of the beta chain. In HbS, glutamic acid is replaced by valine, while in HbC it is replaced by lysine. The diagnosis of hemoglobinopathies is performed exclusively through hemoglobin electrophoresis [4]. The sickling test cannot be used for this purpose because it is nonspecific [5].
The frequency of retinopathy is greatest in adulthood, but retinopathy has also been described in children [6]. Individuals with sickle-cell trait comprise 8% of the African-Caribbean [7]. Although these individuals often show no relevant clinical manifestations, they may present various retinal changes such as hemorrhages, exudates, angioid streaks, acute chorioretinal infarction, chorioretinitis, and vitreous hemorrhage, retinal vascular abnormalities such as tortuous / dilated retinal veins, microaneurysms, central retinal artery occlusion and retinal proliferation [7].
Clinical feature (Sings and symptoms)
Vaso-occlusion of conjunctival vessels leads to the development of “comma” shaped vessels due to the accumulation of sickled RBCs at the distal end of the capillaries [8]. Vascular changes in the optic disc are transient, but present as dark red lesions also visible as dilated dark capillary vessels on fluorescein angiography [9]. Patients may see flashes, floaters or dark shadows, which may be indicative of vitreoretinal traction or detachment.
Retinal vein occlusion [2]
Retinal vein occlusions are common, can occur at almost any age, and range in severity from asymptomatic to the painful, blind eye. They are divided into branch, hemi- or central retinal vein occlusions (equating to occlusion anterior or posterior to the cribriform plate), and ischemic or nonischemic types. Most occlusions occur in those over age 65, but up to 15% may affect patients under 45. BRVO is three times more common than CRVO.
Central retinal vein occlusion (CRVO)
- Nonischemic
- Painless dVA (mild to moderate), metamorphopsia.
- Dilated, tortuous retinal veins with retinal hemorrhages in all four quadrants; occasional cotton-wool spots (CWS); mild optic disc edema.
Branch retinal vein occlusion (BRVO)
- May be asymptomatic; dVA, metamorphopsia, visual fi eld defect (usually altitudinal).
- Acute: retinal hemorrhages (dot, blot, fl ame), CWS, edema in the distribution of a dilated, tortuous vein; superotemporal arcade most commonly affected; usually arise from an arteriovenous (AV) crossing.
- Chronic: venous sheathing, exudates, pigment disturbance, collateral vessels.
Retinal artery occlusion[2]
Retinal artery occlusion is an ocular emergency in which rapid treatment may prevent irreversible loss of vision. CRAO has an estimated incidence 0.85/100,000/year and causes almost complete hypoxia of the inner retina. Experimental evidence shows that this causes lethal damage to the primate retina after 100 min. acute coagulative necrosis is followed by complete loss of the nerve fiber layer, ganglion cell layer, and inner plexiform layer.
Central retinal artery occlusion (CRAO)
- Sudden painless, unilateral dVA (usually CF or worse).
- White swollen retina with a cherry-red spot at the macula (Fig.1); arteriolar attenuation + box-carring; RAPD; visible emboli in up to 25%.
- Variants: a cilioretinal artery (present in 30%) may protect part of the papillomacular bundle, allowing relatively good vision; ophthalmic artery occlusion causes choroidal ischemia with retinochoroidal whitening (no cherry-red spot) and complete loss of vision (usually NLP).
-
Branch retinal artery occlusion (BRAO)
- Sudden painless unilateral altitudinal field defect.
- White swollen retina along a branch retinal arteriole; branch arteriolar attenuation + box-carring; visible emboli common in over 60% (Figure 1).
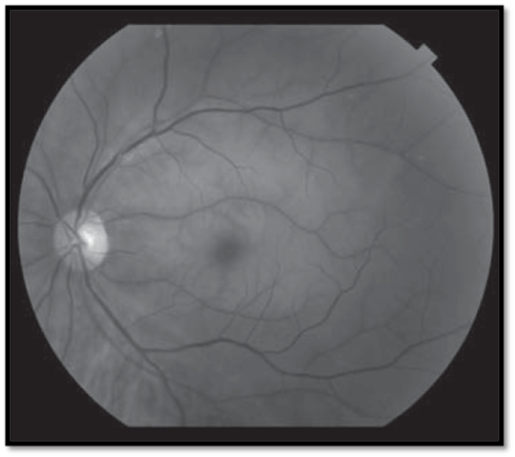
Figure 1. White swollen retina with a cherry-red spot at the macula (Central retinal artery occlusion with extensive retinal edema, whitening, and a cherry-red spot in the fovea. See insert for color version [2]).
Sickle cell retinopathy [10]
Given the patient’s age, race, medical history, and FA findings, the patient most likely has early sickle cell retinopathy. Although the clinical appearance does not completely resemble the typical sickle cell retinopathy picture, the left eye findings likely represent an early glimpse at the evolution of the disease. The lesions identified in his left eye likely are early changes consistent with a salmon patch hemorrhage, a well circumscribed pre-retinal hemorrhage between the retina and internal limiting membrane.
Central retinal vein occlusion has not been reported previously in patients with sickle cell anemia. We describe the case of a 31-year-old man with sickle cell anemia who developed this complication. The search for risk factors for central retinal vein occlusion in this young patient revealed protein S deficiency and a history of iron deficiency. He was treated with anticoagulation therapy, and his vision improved gradually [11].
Central retinal artery occlusion rare causes: Migraine, Behçet disease, syphilis, sickle cell disease. Venous tortuosity, mid-peripheral fundus pigmented lesions with speculated borders (black sunbursts), superficial intraretinal hemorrhages (salmon patch), refractile (iridescent) intraretinal deposits following hemorrhage resorption, angioid streaks, comma-shaped capillaries of the conjunctiva (especially along the inferior fornix). VH and traction bands, RD, CRAO, macular arteriolar occlusions, and enlargement of the foveal avascular zone occasionally develop (Table 2) [12].
Table 2. Goldberg staging of proliferative Sickle Cell Retinopathy.
Goldberg staging of proliferative Sickle Cell Retinopathy |
Stage 1 |
Peripheral arteriolar occlusions |
Stage 2 |
Peripheral arteriovenous anastomoses |
Stage 3 |
Neovascular proliferation "sea-fans" |
Stage 4 |
Vitreous hemorrhage |
Stage 5 |
Retinal detachment |
Retinal alterations
The retinal manifestations of sickle-cell disease can be non-proliferative (yielding diagnostic value) or proliferative; in the latter case, there is a real risk of ocular morbidity. Both non-proliferative and proliferative manifestations are caused by erythrostasis secondary to sickling.
Non-proliferative sickle retinopathy (NPSR):
The retinal changes in NPSR occur secondary to vaso-occlusion and local ischemia. Occlusion of retinal vasculature first appears in the peripheral retina as “salmon patches”, which represent retinal hemorrhage from superficial blood vessels. These lesions are round or oval in shape and can be elevated or flattened [13]. Although they are initially red, they become salmon-colored due to the hemolysis of red blood cells. Over time, the hemorrhage is resorbed and the area appears normal with refractile deposits, which are iridescent spots of hemosiderin and macrophage deposition just beneath the internal limiting membrane. Migration and proliferation of retinal pigment epithelium (RPE) leads to the development of black sunburst spots which likely occur in response to hemorrhage [14]. Reduction in visual acuity is usually from the occlusion of perifoveal capillaries, causing a concave macular depression from RPE degeneration. Occlusions in the choroidal circulation with associated breaks in Bruch’s membrane are known to cause angioid streaks, a phenomenon seen in many retinal diseases including NPSR and PSR.
Nonproliferative or background sickle retinopathy includes the following manifestations [15]:
- Venous tortuosity
- Salmon-patch hemorrhage
- Schisis cavity
- The black sunburst
Venous tortuosity: probably is due to arteriovenous shunting from the retinal periphery. It can occur in many patients with hemoglobin SS and hemoglobin SC disease. Vascular tortuosity is a nonspecific sign that is common to several diseases associated with blood hyperviscosity [16].
Salmon-patch hemorrhages: are superficial intraretinal hemorrhages. They are usually seen in the mid periphery of the retina adjacent to a retinal arteriole. The schisis cavity: is a space caused by the disappearance of the intraretinal hemorrhage. Nonproliferative sickle retinopathy features iridescent spots and glistening refractive bodies in the schisis cavity.
The black sunburst: consists of round chorioretinal scars usually located in the equatorial fundus. These lesions result from pigment accumulated around the vessels. They do not cause any visual symptoms.
Proliferative sickle retinopathy (PSR)
Proliferative sickle retinopathy (PSR) is the most severe ocular change in SCD. The ocular complications of PSR are more characteristically seen in HbC (SC) and SThal disease, rather than in SS disease, and hemoglobin AS and hemoglobin AC disease. As localized vascular occlusion leads to the changes described above in NSPR, chronic changes of local hypoxia and ischemia lead to the upregulation of vascular growth factors, which may result in retinal neovascularization, pre-retinal or vitreous hemorrhage and tractional retinal detachment [17,18].
PSR is progressive. A desirable objective is to treat the neovascular tissue before a vitreous hemorrhage occurs (Table 3). In stage I, the peripheral arteriolar vessels occlude, with anteriorly located avascular vessels evident. Early in the process, the occluded arterioles are dark-red lines, but eventually they turn into silver-wire–appearing vessels.
Table 3. Goldberg classified PSR into 5 different stages.
Goldberg classified PSR into 5 different stages [19] |
Stage I |
Peripheral arterial occlusion. |
Sage II |
Peripheral arteriovenous anastomoses, representing dilated pre-existing capillaries (hairpin loop) |
Stage III |
Neovascular and fibrous proliferation (sea fan) (Fig.2) occurring at the posterior border of nonperfusion. A subsequent white sea fan appearance is due to auto-infarction of the neovasculature. |
Stage IV |
Vitreous hemorrhage |
Stage V |
Tractional retinal detachment |
In stage II, peripheral arteriolar-venular anastomosis occurs as the eye adjusts to peripheral arteriolar occlusion, and blood is diverted from the occluded arterioles into the adjacent venules. Peripheral to these anastomoses, no perfusion is present.
In stage III, new vessel formation occurs at the junction of the vascular and avascular retina. These neovascular tufts resemble sea fans. Initially, the sea fans can be fed by a single arteriole and draining vessel. Later, as the sea fan grows in size, it is difficult to distinguish the major feeding and draining vessels. The sea fans may acquire a glial and fibrotic tissue envelope. This envelope may pull on the vitreous. A full-thickness retinal break, which may lead to total rhegmatogenous retinal detachment, may occur [17,18.
The condition remains asymptomatic until complications such as vitreous hemorrhage (VH) or retinal detachment (RD) occur typically between 20-30 years of age. The significance of PSR as a cause of reduced visual acuity is confounded by the spontaneous regression of the neovascular complexes, which has been described in 20-60% of the cases and often occurs approximately two years after the development of proliferative lesions (Figure 2) [20].
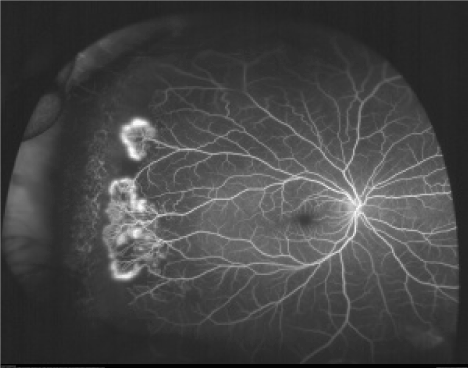
Figure 2. Neovascular and fibrous proliferation (sea fan) (Sea fan formation with neovascularization. Fluorescein angiogram image of an individual with sickle cell retinopathy showing sea fan formation with neovascularization. This image was taken using an Optos P200MA ultra-wide field imaging device [19]).
Posterior retinal and macular vascular occlusions
Retinal artery occlusions are either central or branch. Peripapillary or macular arteriolar occlusions are rare. Retinal vein occlusions also are rare with SCD. Sickle-cell maculopathy occurs as a result of chronic changes in the perifoveal capillary network. It may be present in 10-40% of patients [21,22]. The SS genotype is the most affected. Ischemic changes are chronic and insidious, and the patient may display no symptoms.
Ischemic macular changes are uncommon in angiograms of patients with sickle Hemoglobinopathy. Some patients display low visual acuity and a well-perfused macular capillary network. In these cases, an old ischemic lesion of the photoreceptors and recanalization of the affected capillaries may have occurred [23]. The ischemic macular changes associated with sickle hemoglobinopathy are due to the occlusion of arteriolar circulation around the foveal avascular zone and branches that supply the temporal horizontal raphe [24]. These changes occur primarily in the non-perfused areas and do not cause leaking of fluorescein [23,25].
Patients with sickle-cell disease may also have significantly reduced retinal sensitivity to microperimetry compared to those without focal thinning or to age- and visual acuity-matched controls [26]. Angioid streaks: The incidence of angioid streaks in populations non-selected for sickle-cell is 1-2% [27,28].
Other vascular changes: Occlusion of the central retinal artery may rarely occur in these patients [29,30].
Optic nerve sign: Vascular changes in the optic nerve are transient and consist of dark, small and red dilated capillaries that show occlusion in fluorescein angiography [31,32] without any impact on acuity [33].
Sickle cell retinopathy should be distinguished from the conditions of other diseases that occur with macular ischemia or neovascularization should be considered in the differential diagnosis of sickle-cell retinopathy. [34] A prior history of sickle cell disease helps rule out many of these conditions.
The diseases that occur concomitantly with macular ischemia or neovascularization and should be considered in the differential diagnosis [35], and it could of these conditions to occur simultaneously with NPSR/PSR.
I. Other causes of macular ischemia:
- Diabetic retinopathy
- Retinal vascular occlusion
- Embolic phenomena (e.g., talc retinopathy)
- Infectious diseases (dengue)
II. Other causes of peripheral retinal neovascularization, VH and RD:
- Ischemic vascular disease:
a. Proliferative diabetic retinopathy
b. Branch retinal vein occlusion
c. Ocular ischemic syndrome
d. Retinopathy of prematurity
e. Eales’ disease
f. Familial exudative vitreoretinopathy
g. Chronic myelogenous leukemia
h. Scleral buckle
i. Hyperviscosity syndrome
- Inflammatory disease with possible ischemia:
a. Sarcoidosis
b. Retinal vasculitis
c. Intermediate uveitis
d. Acute retinal necrosis
- Miscellaneous:
a. Incontinentia pigmenti
b. Autosomal dominant vitreoretinochoroidopathy
c. Old RD
d. Hypertensive retinopathy
Presence of traumatic hyphema in African Americans warrants testing the patients for sickle cell hemoglobinopathies due to ocular complications that can result from sickle red blood cells. To control intraocular pressure, use of carbonic anhydrase inhibitors, especially acetazolamide, is contraindicated as it induces systemic acidosis and increases the local concentration of ascorbic acid in the aqueous humor; hence, the resulting low pH enhances sickling of RBCs. Instead of acetazolamide, the use of methazolamide is recommended [36,37]. Scleral buckling surgery also carries higher intra- and post-operative risk with similar complications as pars plana vitrectomy (PPV) in patients with sickle cell disease.
Retinal tear and rhegmatogenous retinal detachments may occur after scatter photocoagulation. The rate of surgical complications in patients with PSR has been reported to be as high as 50% for traction/rhegmatogenous retinal detachment in a recent study [38].
Surgery carries a significant risk of intraoperative and postoperative complications, including severe ocular ischemia, recurrent hemorrhage and elevated eye pressure. In order to minimize the risk of such complications, partial exchange transfusion has been recommended prior to surgery, usually with a target of about 50%-60% normal red cells, but there has never been a controlled study demonstrating the efficacy of this maneuver. Exchange transfusion is also not without its own risks, including various immune-mediated transfusion reactions as well as transmission of infectious diseases such as HIV and hepatitis. When considering an exchange transfusion, a thorough discussion of the risks and benefits should take place. An alternative to exchange transfusion is the use of a hyperbaric chamber to increase blood oxygenation [39], but such equipment is neither convenient nor widely available. In any event, intra-operative and post-operative hyper-oxygenation is indicated in an attempt to reduce the risk of anterior segment ischemia and necrosis.
The only significant clinical findings were two slightly raised lesions in the mid periphery of the left fundus. They had a pale appearance with no associated hemorrhage or pigmentation. Fluorescein angiography (FA) revealed no signs of leakage (specifically, no signs of neovascularization), however there appeared to be patches of nonperfusion within the location of the pale patches identified indicated possible ischemia.
2021 Copyright OAT. All rights reserv
Interestingly, spectral domain optical coherence tomography (OCT) through the area highlighted the pre-retinal nature of the lesion with scattered hyper-reflectivity and shadowing [1].
The complete ophthalmic examination twice a year is recommended for sickle cell patients (SC, SS, S-thal, etc). Baseline fluorescein angiography may be performed to examine blood flow in the retina and choroid [2].
- BP, CBC, ESR, glucose, lipids, protein electrophoresis, TFT, and ECG. Further investigation is directed by clinical indication and may include CRP, serum ACE, anticardiolipin, lupus anticoagulant, autoantibodies (RF, ANA, anti-DNA, ANCA), fasting homocysteine, CXR, and thrombophilia screen (e.g., proteins C and S, antithrombin, factor V Leiden).
- Nonischemic: vein wall staining, microaneurysms, dilated optic disc capillaries.
- Ischemic: as for nonischemic but capillary closure (5–10 disc areas is borderline; >10 is significantly ischemic), hypofluorescence (blockage due to extensive hemorrhage), leakage (CME, NV).
In the acute setting, the diagnosis is not usually in doubt, so the urgent priority is to rule out underlying disease (such as giant cell arteritis [GCA] that extremely rare as a cause of BRAO and does not need investigation in the absence of other supporting evidence) that may threaten the contralateral eye. When presentation is delayed, the clinical picture is less specific and may require ancillary tests.
Most importantly, consider GCA (if age >50 years then get ESR, CRP, CBC, followed by temporal artery biopsy; p. 524). More common causes are atherosclerosis (check BP, blood glucose) and particularly carotid artery disease (may have carotid bruit).
Further investigation is directed by clinical indication and may include PTT, APTT, thrombophilia screen (e.g., proteins C and S, antithrombin, factor V Leiden), antiphospholipid screen, vasculitis autoantibodies (ANA, ANCA), syphilis serology (VDRL, TPHA), blood cultures, ECG, echocardiography, and carotid Doppler scans.
Occlusions Treatment [2]
Central retinal vein occlusion (CRVO): There is no proven treatment. The following are common practice:
- dIOP: if elevated (in either eye).
- Panretinal photocoagulation for neovascularization or high risk.
- Intravitreal triamcinolone acetonide or intravitreal dexamethasone implant for treatment of CME.
- Intravitreal bevacizumab and ranibizumab for treatment of CME and neovascularization.
- Pars plana vitrectomy and endolaser for vitreous hemorrhage secondary to neovascularization.
- Treat underlying medical conditions coordinate care with a PCP.
Branch retinal vein occlusion (BRVO)
- Macular grid laser (after FA): if macular edema, VA _20/40 and no spontaneous improvement by 3–6 months.
- Sectoral PRP: if neovascularization.
- Fill-in PRP: if neovascularization progresses or vitreous hemorrhage.
Central retinal artery occlusion (CRAO)
Treat affected eye (if within 24 hours of presentation).
- dIOP with 500 mg IV acetazolamide, ocular massage AC paracentesis (all common practice, but no proven benefit); ocular massage. Selective ophthalmic artery catheterization with thrombolysis is performed in some centers.
- Protect other eye, e.g., treat underlying GCA with systemic steroids immediately.
Branch retinal arteriolar occlusion (BRAO)
There is no proven treatment for BRAO.
Sickle Cell Retinopathy Treatment
Treatment for eyes which have proliferative retinopathy and at risk for severe visual loss from retinal detachment and bleeding. Given the high rate of spontaneous regression and/or lack of neovascularization progression in the eye, the indications for treatment of retinal neovascularization are not always clear. Usually, the therapeutic intervention is recommended in a bilateral proliferative disease case, spontaneous hemorrhage, large elevated neovascular fronds, the rapid growth of neovascularization, or cases of an eye has lost to proliferative retinopathy. The goal is early treatment aimed at inducing neovascular regression tissue before progression to bleeding and detach of retinal. Techniques such as diathermy, cryotherapy, and laser photocoagulation have all been used to cause involution of neovascular lesions.
- Observation due to neovascularization identified lack on angiography, the treatment plan for this patient was to simply observe.
The patient was told to follow up in several months at time the lesions appear to be maturing with early pigmentation consistent with an early salmon patch hemorrhage. The initial pale appearance has resolved leaving a mottled pigmentation as the hemorrhage resolved. The patient carried the SS sickle trait which carries a better prognosis than SC sickle trait.
- Consider laser photocoagulation in proliferative sickle retinopathy. Its use is controversial, as most sea-fans spontaneously regress. The rationale is to remove the drive to neovascularization by ablating the ischemic retina.
Laser photocoagulation has the fewest side effects. Laser application specific methods which include direct feeder vessels coagulation, local scatter photocoagulation with and without focal ablation of the neovascular frond, and peripheral scatter delivery in 360 degree. Both sector scatter laser photocoagulation and feeder vessel ablation have been shown to be effective in controlled clinical trials, but the associated complications with the high power settings that used in feeder vessel treatment have pushed modern clinicians towards local scatter photocoagulation. Heavy feeder vessel photocoagulation it's just usually used for recalcitrant cases with repetitive bleeding.
- Sea fan treatment: Historically, SF have been treated in a variety of ways (diathermy, cryotherapy and argon/xenon photocoagulation), always aiming to obliterate the neovessels. Diathermy, which is no longer in use, can cause uveitis and anterior segment ischemia. Cryotherapy is effective in the treatment of SF occlusion, although complications (retinal tears and tractional RD) can occur [40].
- Consider vitreoretinal surgery for persistent vitreous hemorrhage (e.g., >6 months) and tractional retinal detachment, although the results are generally disappointing, and specialist perioperative care is required.
Surgical treatment is indicated in complications of PSR (VH and RD). Because patients with sickle-cell disease are susceptible to intra- and postoperative complications, some general precautions must be taken. If retinal detachment and/or non-clearing vitreous hemorrhage is present, surgical intervention is that's including techniques of vitrectomy with or without the scleral buckle placement usually is required. Although modern vitreoretinal microsurgery can improve vision with sickle retinopathy.
Patients of sickle cell retinopathy are underlying a systemic and ocular manifestations of SCD due to vaso-occlusive ischemia that caused by the blocking of blood streams by sickle-shaped erythrocytes. Resultant vaso-occlusive changes lead to several anterior segment complications, such as an Iris changes as atrophy with or without anterior or posterior synechiae, a conjunctival sickling sign that characterized by capillary vessel segmentation, and the development of iris neovascularization (INV) could lead to secondary glaucoma and severe vision loss.
Retinal manifestations of sickle-cell disease can be proliferative or non-proliferative; in the proliferative case, there is a high risk of ocular morbidness. In both non-proliferative and proliferative manifestations are caused by erythrostasis secondary to sickling.
Sickle-cell disease is a complex hematological disorder result in a homozygous condition which known as sickle cell anemia (HbSS). There are various heterozygous forms of Sickle Disease, including sickle-hemoglobin C disease (HbSC) and sickle beta-thalassemia (SThal).
While sickle-cell anemia is associated with life-threatening systemic conditions, ocular complications are relatively moderative. On the other hand, SCD and SThal are associated with more severe ocular disease; the proliferative sickle cell retinopathy incidence in sickle-cell anemia is less than sickle-cell disease and thalassemia.
The sickle-cell trait may experience systemic and ocular complications. Proliferative sickle cell retinopathy is the most vision-threatening complication of the sickle cell disease. Ocular disease due to the sickle cell is more epidemically in SC patients compared to SS patients, the various ocular manifestations of sickle cell disease occur in both forms. Ocular disorder findings placed in the anterior segment as well as the posterior segment. In the inferior bulbar conjunctiva is commonly found Comma-shaped capillary segments, which may be present due to transient dilatation of conjunctival blood vessels by abnormally shaped red blood cells.
Most vascular occlusions that associated with sickle cell disease manifest in the retinal periphery, while branch retinal artery occlusions, central retinal artery occlusions, and choroidal infarction may occur in the posterior pole. The occlusion usually occurs on the level of the precapillary arterioles.
Sickle Hemoglobinopathy is among the most prevalent genetic disorders globally, and it remains a serious public health concern and pathologic risk factors associated considerable morbidity and mortality. Sickle cell Retinopathy patients typically presented after 12 weeks of onset of sudden drop in vision. Sickle retinopathy presents with vitreoretinal complications. It could often be observed and may spontaneously regress. Sickle cell disease leads to Sickle cell retinopathy of peripheral ischemic retinopathy, and it could affect macular area through the same process of peripheral terminal vascular occlusion.
It is necessary to recommend that sickler patients receive early periodic ophthalmological examinations to prevent progression of the disease and early blindness, establish more effective treatment, and assure a good quality of patients’ eyes care.
Multiple ocular complications exist for SCD patients, and continuous assessment is required to detect lesions early enough for effective prophylactic therapy. Laser photocoagulation is important to prevent the development of retinal complications related to neovascularization.
- Jackson H, Bentley CR, Hingorani M, Atkinson P, Aclimandos WA, et al. (1995) Sickle retinopathy in patients with sickle trait. Eye (Lond) 9 : 589-593. [Crossref]
- James C Tsai, Alastair KO Denniston, Philip I Murray, John J Huang, Tamir S Aldad. Oxford American Handbook of Ophthalmology.
- Elagouz M1, Jyothi S, Gupta B, Sivaprasad S (2010) Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol 55: 359-377. [Crossref]
- Araujo JT (1965) Hemoglobinopatias anormais em Sio Paulo. Métodos de estudo. Incidência. J Brasil Med 9: 1264-1283.
- Steinberg MH, Hebbel RP (1983) Clinical diversity of sickle cell anemia: genetic and cellular modulation of disease severity. Am J Hematol 14: 405-416. [Crossref]
- Fox PD1, Dunn DT, Morris JS, Serjeant GR (1990) Risk factors for proliferative sickle retinopathy. Br J Ophthalmol 74: 172-176. [Crossref]
- Mehta JS1, Whittaker KW, Tsaloumas MD (2001) Latent proliferative sickle cell retinopathy in sickle cell trait. Acta Ophthalmol Scand 79: 81-82. [Crossref]
- Roy MS, Rodgers GP, Podgor MJ, Noguchi CT, Nienhuis AW, et al. (1985) Conjunctival sign in sickle cell anaemia: an in-vivo correlate of the extent of red cell heterogeneity. Br J Ophthalmol 69: 629-632. [Crossref]
- Nia J, Lam WC, Kleinman DM, Kirby M, Liu ES, et al. (2003) Retinopathy in sickle cell trait: does it exist? Can J Ophthalmol 38: 46-51. [Crossref]
- Gagliano DA, Goldberg MF (1989) The evolution of salmon-patch hemorrhages in sickle cell retinopathy. Arch Ophthalmol 107: 1814-1815. [Crossref]
- Hasan S, Elbedawi M, Castro O, Gladwin M, Palestine A (2004) Central retinal vein occlusion in sickle cell disease. South Med J 97: 202-204. [Crossref]
- Adam T Gerstenblith, Michael P Rabinowitz. The Wills Eye Manual Office and Emergency Room Diagnosis and Treatment of Eye Disease 6th edition.
- Jampol LM, Condon P, Dizon-Moore R, Serjeant G, Schulman JA (1981) Salmon-patch hemorrhages after central retinal artery occlusion in sickle cell disease. Arch Ophthalmol 99: 237-240. [Crossref]
- Friberg TR, Young CM, Milner PF (1986) Incidence of ocular abnormalities in patients with sickle hemoglobinopathies. Ann Ophthalmol 18: 150-153. [Crossref]
- Hampton Roy. Ophthalmologic Manifestations of Sickle Cell Disease: Overview, Posterior Segment Abnormalities, Anterior Segment Abnormalities, Medscape, available: http://emedicine.medscape.com/article/1918423-overview#showall
- Bonanomi MT, Cunha SL, de Araújo JT (1988) Funduscopic alterations in SS and SC hemoglobinopathies. Study of a Brazilian population. Ophthalmologica 197: 26-33. [Crossref]
- Babalola OE, Wambebe CO (2001) When should children and young adults with sickle cell disease be referred for eye assessment? Afr J Med Med Sci 30: 261-263. [Crossref]
- Gill HS, Lam WC (2008) A screening strategy for the detection of sickle cell retinopathy in pediatric patients. Can J Ophthalmol 43: 188-191. [Crossref]
- Goldberg MF (1971) Natural history of untreated proliferative sickle retinopathy. Arch Ophthalmol 85: 428-437. [Crossref]
- Condon PI, Serjeant GR (1980) Behaviour of untreated proliferative sickle retinopathy. Br J Ophthalmol 64: 404-411. [Crossref]
- Asdourian GK, Nagpal KC, Busse B, Goldbaum M, Patriankos D, et al. (1976) Macular and perimacular vascular remodelling sickling haemoglobinopathies. Br J Ophthalmol 60: 431-453. [Crossref]
- Raichand M, Dizon RV, Nagpal KC, Goldberg MF, Rabb MF, et al. (1978) Macular holes associated with proliferative sickle cell retinopathy. Arch Ophthalmol 96: 1592-1596. [Crossref]
- Stevens TS, Busse B, Lee CB, Woolf MB, Galinos SO, et al. (1974) Sickling hemoglobinopathies; macular and perimacular vascular abnormalities. Arch Ophthalmol 92: 455-463. [Crossref]
- Condon PI, Serjeant GR (1972) Ocular findings in hemoglobin SC disease in Jamaica. Am J Ophthalmol 74: 921-931. [Crossref]
- Stevens TS, Busse B, Lee CB, Woolf MB, Galinos SO, et al. (1974) Sickling hemoglobinopathies; macular and perimacular vascular abnormalities. Arch Ophthalmol 92: 455-463. [Crossref]
- Chow CC, Genead MA, Anastasakis A, Chau FY, Fishman GA, Lim JI. Structural and functional correlation in sickle cell retinopathy using spectral-domain optical coherence tomography and scanning laser ophthalmoscope microperimetry. Am J Ophthalmol. 2011;152(4):704-11.e2.
- Goodman G, Von Sallmann L, Holland MG (1957) Ocular manifestations of sickle-cell disease. AMA Arch Ophthalmol 58: 655-682. [Crossref]
- Jampol LM, Acheson R, Eagle RC Jr, Serjeant G, O'Grady R (1987) Calcification of Bruch's membrane in angioid streaks with homozygous sickle cell disease. Arch Ophthalmol 105: 93-98. [Crossref]
- Liem RI, Calamaras DM, Chhabra MS, Files B, Minniti CP, et al. (2008) Sudden-onset blindness in sickle cell disease due to retinal artery occlusion. Pediatr Blood Cancer 50: 624-627. [Crossref]
- Fine LC, Petrovic' V, Irvine AR, Bhisitkul RB (2000) Spontaneous central retinal artery occlusion in hemoglobin sickle cell disease. Am J Ophthalmol 129: 680-681. [Crossref]
- Condon PI, Serjeant GR (1972) Ocular findings in homozygous sickle cell anemia in Jamaica. Am J Ophthalmol 73: 533-543. [Crossref]
- Goldbaum MH, Jampol LM, Goldberg MF (1978) The disc sign in sickling hemoglobinopathies. Arch Ophthalmol 96: 1597-1600. [Crossref]
- Emerson GG, Lutty GA (2005) Effects of sickle cell disease on the eye: clinical features and treatment. Hematol Oncol Clin North Am 19: 957-973. [Crossref]
- Bonanomi, Maria Teresa Brizzi Chizzotti; Lavezzo, Marcelo Mendes, Sickle Cell Retinopathy: diagnosis and treatment. Arq Bras Oftalmol 2013;76: 320-327
- Ho AC. Hemoglobinopathies. In: Yanoff M, Duker JS, editors. Ophthalmology. 3rd ed.St. Louis: Mosby Elsevier; 2009. p. 626-30.
- Fritch CD (1976) Traumatic hyphema. Ann Ophthalmol 8: 1223-1225. [Crossref]
- Walton W, Von Hagen S, Grigorian R, Zarbin M (2002) Management of traumatic hyphema. Surv Ophthalmol 47: 297-334. [Crossref]
- Chen RW, Flynn HW Jr, Lee WH, Parke DW 3rd, Isom RF, et al. (2014) Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol 157: 870-875. [Crossref]
- Freilich DB, Seelenfreund MH (1972) The use of hyperbaric oxygen in the treatment of retinal detachment in patients with sickle cell disease. Isr J Med Sci 8:1458-1461. [Crossref]
- Harlan JB, Morton F, Goldberg MD. Management and Therapy of Eye Disorders in Sickle Cell Disease