Epstein–Barr virus (EBV) infection is present in 10% of gastric cancer cases and plays a role in carcinogenesis. HER2 expression has received increasing attention since the efficacy of an anti-HER2 antibody, trastuzumab, in gastric cancer with HER2 overexpression was shown. However, a significantly lower frequency of HER2 overexpression has been observed in EBV+ gastric cancers. The aim of this study was to analyze the association between EBV infection and HER2 expression ingastric cancer cell lines. We used a HER2-positive gastric cancer cell line named JRST. The cells were infected with EBV using the cell-cell contact method and were designated as JRST-EBV. Celllines transfected with a neomycin resistance gene were also established; these were used as controlsand were referred to as JRST-Neo. We analyzed the effects of EBV infection on JRST usingreal-time RT-PCR, western blots, MTT assays, scratch assays, and caspase 3 assays. HER2 mRNAexpression levels were similar in JRST-EBV and JRST-Neo cells. In contrast, HER2 proteinexpression levels were lower in JRST-EBV cells than in JRST-Neo cells. Expression of phospho MAPK and phosphor Akt, both of which are downstream signal molecules of HER2, was alsoreduced in JRST-EBV cells. MTT assays showed that proliferation activity was significantly lower in JRST-EBV cells than in JRST-Neo cells. Scratch assays showed that cell migration was significantlylower in JRST-EBV cells than in JRST-Neo cells. Caspase 3 assays showed that trastuzumab-induced apoptosis was lower in JRST-EBV cells than in JRST-Neo cells. Trastuzumab reduced migration of JRST-Neo cells but not that of JRST-EBV cells. These results suggest that EBV infection plays asuppressive role in HER2 expression in gastric cancer cells, which may explain the mutually exclusive relationship between EBV infection and HER2 expression in human gastric cancer.
EB virus, gastric cancer, HER2,trastuzumab, gene amplification
Gastric cancer (GC) is the third highest cause of global cancer mortality [1]. GC is aheterogeneous disease with multiple environmental etiologies and alternative carcinogenic pathways [2-5]. A recent comprehensive molecular characterization of GC by The Cancer Genome Atlas(TCGA) proposed a molecular classification dividing GC into four subtypes: Epstein-Barr virus (EBV)-positive cancers, microsatellite instability (MSI)-positive cancers, genomically stable cancers, and cancers with chromosomal instability (CIN) [6]. EBV infection is observed in approximately 10% of GC cases [7-14]. EBV-positive (EBV+) GC is characterized by male predominance, predominant location in the proximal stomach, lymphoepithelioma-like histology, relatively good prognosis, and CpG island hypermethylator phenotype (CIMP). In gastric carcinoma cells, EBV promotes cell proliferation through the inductionof insulin-like growth factor-mediated signaling in an autocrine fashion [15]. EBV+ GCs showed frequent PIK3CA and ARID1A mutations, rare TP53 mutations, and recurrent amplification of JAK2, CD274 (also known as PD-L1), PDCD1LG2 (also known as PD-L2), and ERBB2 (HER2) [6]. Recently, an anti-HER2 antibody, trastuzumab (Tmab), was shown to prolong survival of GC patients with HER2 overexpression [16-19]. Although HER2 gene amplification is a major cause of HER2 overexpression, not all GC cases with HER2 amplification showed HER2 overexpression inimmunohistochemical analyses. These results suggest that HER2 expression is regulated not only by gene amplification, but also by other mechanisms. HER2 overexpression (strong intensity, score 3) was observed in only one case (0.8%) among 123 patients with EBV+ GCs [8]. Importantly, the significantly lower frequency of HER2 overexpression observed in EBV+ GCs was independent of Lauren subtype and clinical stage, which further supports the concept of EBV+ GC as a distinct biological entity [6]. EBV-encoded RNA(EBER) 1 reportedly suppressed HER2 expression in an ovarian cancer cell line [20]. However, no reports have shown any association between EBV infection and HER2 overexpression in GC cell lines. The aim of this study was to analyze the association between EBV infection and HER2 expression in GC cell lines by establishing EBV-infected HER2-expressing GC cell lines using the cell-cell contact method [21].
Cell culture and EBV infection using the cell-cell contact method
The HER2-expressing human GC cell line JRST was used. Cells were grown in DMEM (Sigma-Aldrich) containing 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 5% CO2 in air. JRST cells were infected with recombinant EBV using the cell-cell contact method [21]. A Burkitt’s lymphoma cell line, Akata, in which the neomycin resistance gene is inserted into BXLF1 [22], was modified to produce recombinant EBV and was used as a source of the virus. G418-resistant cell clones were isolated by incubation in medium containing 500 ng/ml of G418. After selection with G418, EBV-infected clones were maintained in bulk, but G418 was removed from the medium for the EBV-infected GC cell lines 24 h before performing further experiments. Cell lines transfected with a neomycin resistance gene alone were also established as controls and arereferred to as JRST-Neo. Expression of EBNA1 was examined in acetone: methanol (1:1)-fixed cells by anticomplement immunofluorescence with reference human serum.
Reverse transcription-polymerase chain reactions (RT-PCR)
Expression of EBNA1, LMP2A, BARF0, and EBER1 was analyzed by RT-PCR. TotalRNA was extracted from cell lines. cDNA was synthesized from total RNA, and 400 ng/μl aliquots of cDNA were used to perform PCR. The GAPDH gene was used as an internal control. The sequences of the PCR primers used and the thermal cycling conditions for amplifying each gene have been described previously [21]. PCR products were electrophoresed on 2% agarose gels and visualized by ethidium bromide staining under UV illumination. All PCRs were repeated at leasttwice to validate the results.
Western blot analysis
Protein extracts of cells were prepared by scraping the lysed cells in RIPA lysis buffer and protease inhibitors in TBS. Samples were diluted and the protein concentrations were adjusted for western blot analyses. Proteins were resolved on 7.5% and 10% SDS-PAGE gels for analysis of HER2 and others, respectively. Protein controls were also electrophoresed, and subsequently transferred to a PVDF membrane. Samples were visualized using chemiluminescence with specific antibodies. The antibodies used were HER2 (3B5, Abcam), Path Scan Multiplex Western Cocktail 1 (#7100, Cell Signaling), Akt (#9272, Cell Signaling), ERK1 (K-23, Santa Cruz), and actin (Thermo Fisher Scientific). All antibodies were applied at the concentration suggested by the manufacturer.
Cell proliferation analysis using MTT assays
The cell growth rate was determined by MTT assay using a Cell Counting Kit-8 (Dojindo). Cells were seeded on 96-well plates at 2 × 103 cells per well and cultured with 10% FBS and without G418. After 24, 48, 72, and 96 h of incubation, Cell Counting Kit-8 solution was added to each well and the absorbance was measured at 450 nm.
In vitro migration assay (scratch assay)
Approximately 1 × 106 cells/well were seeded onto 6-well plates and cultured in DMEM with 10% FBS and without G418 overnight at 37°C with 5% CO2. When the cells were semi confluent, a straight scratch was gently made through the central axis of the plate using a 200-μl pipette tip. The plates were rinsed with PBS and culture media were replaced. The initial widths of the scratches were immediately measured. After 24, 48, 72, and 96 h of incubation, the distance thatthe remaining cells migrated across the scratch line was measured, as described previously [22]. All scratch assays were performed in duplicate.
Sensitivity analysis for Tmab therapy
A caspase 3 assay and a scratch assay were used for Tmab (20 µg/ml) sensitivity analyses.
Statistical analysis
The software program JMP was used for all statistical analyses. All P-values were two-sided and statistical significance was set at P< 0.05.
Establishment of EBV-infected GC cells
We established an EBV-infected GC cell line using the cell-cell contact method. JRSTcells were infected with EBV carrying a neomycin-resistance gene, and G418-resistant cell clones were isolated by incubation in medium containing 500 ng/ml of G418. The latency 1 type of EBV infection was confirmed in JRST-EBV cells by RT-PCR for EBNA1, LMP2, BARF0, and EBER1 and immunofluorescence staining for EBER1 (Figure 1).
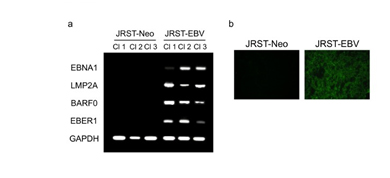
Figure 1. Establishment of EBV-infected GC cells. (a) RT-PCR for EBNA1, LMP2A, BARF0,and EBER1 in JRST-EBV and JRST-Neo cells. (b) Immunofluorescence staining for EBNA1 inJRST-Neo and JRST-EBV cells. Cl, clones.
Alteration of HER2 expression after EBV infection
Changes in HER2 mRNA expression after EBV infection were analyzed by real-time RT-PCR. HER2 mRNA expression did not differ between JRST-EBV and JRST-Neo cells (relative quantity of HER2: 1.3 vs. 1.1, P = 0.35). Subsequently, western blot analysis was performed to clarify changes in HER2 protein levels after EBV infection. Expression of HER2 protein was lower in JRST-EBV cells than in JRST-Neo cells (Figure 2). To confirm this change, we performed immunofluorescence staining for HER2. Staining for HER2 was also lower in JRST-EBV cells than in JRST-Neo cells (Figure 2).
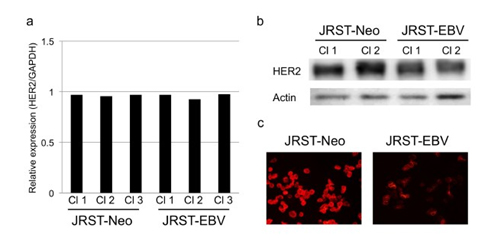
Figure 2. Alteration of HER2 expression after EBV infection. (a) RT-PCR for HER2 inJRST-Neo and JRST-EBV cells. (b) Western blot analysis of HER2 in JRST-Neo and JRST-EBVcells. (c) Immunofluorescence staining for HER2 in JRST-Neo and JRST-EBV cells. Cl, clones.
The effects of EBV infection on HER2 downstream signals
Expression of HER2 downstream signals such as MAPK and Akt was analyzed by western blot analysis. Although MAPK and Akt expression remained unchanged, expression of phospho-MAPK and phospho-Akt was lower in JRST-EBV cells than in JRST-Neo cells (Figure 3).
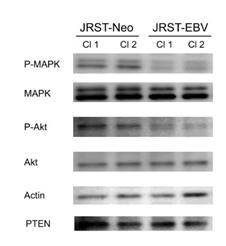
Figure 3. The effects of EBV infection on HER2 downstream signals. Expression ofphospho-MAPK, MAPK, phospho-Akt, Akt, and PTEN was analyzed in JRST-Neo and JRST-EBVcells using western blot analysis. Cl, clones.
However, expression of PTEN wa2021 Copyright OAT. All rights reserv
The effects of EBV infection on cell proliferation and migration
Next, we analyzed the effects of EBV infection on the proliferation and migration of GC cells. In the MTT assay, cell proliferation was significantly lower in JRST-EBV cells than in JRST-Neo cells (P< 0.01) (Figure 4). To evaluate the effects of EBV infection on the migration of GC cells, a scratch assay was performed. JRST-EBV cells showed significantly lower migration activity than JRST-Neo cells (216 nm vs. 316 nm, P=0.03) (Figure 4).
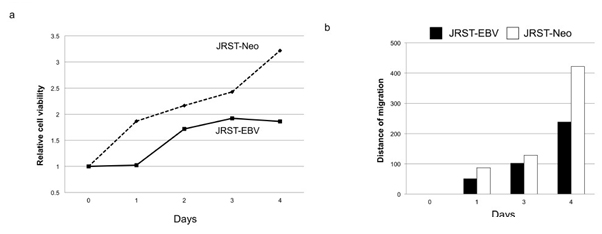
Figure 4. Cell proliferation and migration after EBV infection. Cell proliferation and migrationwere analyzed in JRST-Neo and JRST-EBV cells using an MTT assay (a) and a scratch assay (b).
Reduced sensitivity for Tmab therapy in EBV-infected GC cells
Tmab sensitivity was tested using a caspase 3 assay and a scratch assay. The caspase 3 assay showed that Tmab-induced apoptosis was lower in JRST-EBV cells than in JRST-Neo cells (Figure 5). Tmab reduced migration of JRST-Neo cells but not that of JRST-EBV cells (Figure 5).
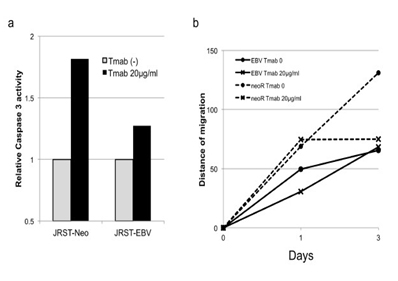
Figure 5. Reduced sensitivity for Tmab therapy in EBV-infected GC cells.Tmab sensitivity wastested using a caspase 3 assay (a) and a scratch assay (b) in JRST-Neo and JRST-EBV cells.
EBV+ GCs reportedly show a latency I (or II) type of EBV infection and express EBNA1, LMP2A, BARTs, and EBER1/2 [9]. In this study, we successfully infected the HER2-expressing GC cell line JRST with EBV using the cell-cell contact method [21]. Moreover, the latency 1 type ofEBV infection was confirmed by RT-PCR and immunofluorescence staining. These results suggest that JRST cells infected with EBV are an appropriate model for the analysis of EBV infection and HER2 expression in GC.
Interestingly, EBV infection downregulated HER2 expression at the protein level, but not the mRNA level. In the ToGA study, 22% of HER2 amplification cases showed no overexpression ofHER2 protein in immunohistochemical analysis [16]. Our results further support the notion that suppressors of HER2 expression may be present in GC. EBV infection may be involved in the discrepancy observed between HER2 amplification and HER2 overexpression in GC.
The effects of EBV infection on HER2 downstream signals were analyzed; phospho-MAPK and phospho-Akt expression were found to be downregulated, suggesting that this downregulation affects tumor phenotype. EBV infection reportedly induces loss of PTEN expression through CpG island methylation of its promoter, leading to activation of the PI3K-AKT signaling pathway, in GC [23]. However, EBV infection did not affect PTEN expression in JRST cells.
We then analyzed the functional significance of EBV infection. EBV downregulated the proliferation and migration activity of JRST GC cells. These results suggest that the HER2 signaling plays an important role in HER2-expressing JRST GC cell lines, and therefore its downregulation by EBV infection resulted in the reduction of proliferation and migration activity. Finally, the effects of EBV infection on sensitivity to Tmab were analyzed. EBV infection downregulated the antitumor effects of Tmab, suggesting that HER2 suppression by EBV resulted in
Tmab resistance. There are three major mechanisms of resistance to Tmab: alterations of HER2 structure or surroundings, dysregulation of downstream signaling effectors such as PIK3CA mutation or PTEN inactivation, and interactions of HER2 with other membrane receptors [19]. In this study, EBV infection did not affect PTEN expression in JRST. Further analysis is necessary to address the issue in GC.
Chuang et al. reported that EBNA1 might act as a transforming suppressor of the HER2gene in an ovarian cancer cell line [20]. Two viral oncoproteins, SV40 large T antigen and adenovirus-5 E1a gene product, also reportedly repress HER2 expression [24,25]. Since EBNA1 is structurally and functionally different from these viral oncoproteins, EBNA1 presumably represses HER2 expression through a distinct mechanism [20]. Chuang et al. suggested that the repression of HER2 by EBNA1 may be mediated by an indirect protein-protein interaction mechanism because no EBNA1-specific binding sequences are found on the HER2 promoter element [20]. Further analysisis necessary to address these issues in GC.
Our results suggest that EBV infection plays a suppressive role in HER2 expression in GC cells, which may explain the mutually exclusive relationship observed between EBV infection and HER2 expression in human GC [8,12]. In clinical settings, Tmab may not be promising for the vast majority of patients with EBV+ GC. On the other hand, expression of JAK2, PD-L1, and PD-L2 was increased in EBV+ GC cases with amplification of the relevant genes, suggesting that JAK2 inhibitors, PD-L1/L2 and/or PD-1 antagonists, as well as PI3K inhibitors, show promise for treatment of this subgroup of GCs [6]. Using EBV-infected JRST and other GC cell lines, analyses of the effects of EBV infection on these molecular alterations are also promising.
The authors declare no competing interests.