Abstract
Neuronal activation and regional cerebral blood flow are tightly linked by physiological mechanisms that are still not fully understood. In particular, the “initial dip”, the “early response” that precedes the hyperemic phase of hemodynamic response, remains a rather elusive subject for both experimental work and theoretical studies. This paper presents a hypothesis for cellular mechanisms of neurovascular coupling that are presumed to function at the level of brain capillary vessels. The hypothesis postulates a novel view on the link between neurovascular coupling and anatomical and functional properties of astrocytes, and proposes a unified basis for mechanisms involved in functional hyperemia and cerebral autoregulatory response (to perfusion pressure drop or intracranial pressure change). Supporting evidence and potential implications are discussed.
Key words
neurovascular coupling, functional hyperemia, cerebral hemodynamics, initial dip, astrocyte, cerebral autoregulation
Introduction
“One is forced to conclude that a more likely explanation will emerge from future research that examines the manner in which a change in blood flow may regulate an effect of the activity change [1]”.
“The findings indicate that regulation of cerebral blood flow by cerebral arteries is not entirely complete and that additional physiological mechanisms may participate in the regulation of capillary perfusion and oxygen supply in the brain [2]”.
“The initial dip may seem to be such an elusive phenomenon that so much experimental work and theoretical speculation focused on it cannot possibly be worth the effort. Yet the initial dip is a thread that connects to many fundamental unanswered questions in the critical area of neurovascular coupling. A better understanding of why the initial dip occurs, and what it signifies when it does occur, will lay a more solid foundation for fMRI studies of brain function [3]”.
In the mammalian brain, focal changes in neuronal activity are coupled to regional changes in cerebral blood flow (CBF) by physiological mechanisms collectively called neurovascular coupling [1,4-6]. Functional hyperemia, or the increased regional CBF that occurs on neuronal activation, serves as the basis of functional brain imaging techniques [1,6], for example, positron emission tomography (PET) and functional magnetic resonance imaging (fMRI), that detect signal changes related to various aspects of brain hemodynamic response (HDR): changes in CBF, cerebral blood volume (CBV), and blood oxygenation level. It is generally agreed that functional hyperemia is a response to increased metabolic demands of the activated neuronal tissue, but questions regarding the coupling/uncoupling of regional CBF and oxidative metabolism and tight coupling between CBF and glucose metabolism are still being debated [1,7]. Despite the great success in functional imaging and the continuous progress in research, many details about cellular mechanisms of neurovascular coupling are still largely unknown or poorly understood [3,5,6,8].
The epigraph quotes citing [1-3] are intended to emphasize certain intriguing aspects of the neurovascular coupling phenomenon. I) The exact purpose that is served by the regional CBF increase is currently not clear, and other possible functions, not related to metabolic demands, have been suggested [1]. II) The high spatial specificity attained at the level of cortical columns and layers in high-resolution optical and fMRI studies [9,10], as well as findings of intravital microscopy of functional responses of individual capillaries [2], suggest that neurovascular coupling operates at the level of small arterioles and, perhaps, even capillary vessels [2,11]. III) The elusive “initial dip”, the somewhat controversial phenomenon also known as the “fast response” [3] or “early response”, that was first reported in optical intrinsic signal imaging (OISI) in animal studies [12] and was subsequently found in some, but not all fMRI studies, is known to be better localized to the area of neural activation compared to the later hyperemic response phase [13]. The initial dip may be viewed as the key probe for answering fundamental questions of cerebral hemodynamics [3].
This paper presents a hypothesis on cellular mechanisms of neurovascular coupling that are postulated to operate at the level of brain capillaries. The hypothesis was formulated in an attempt to address important questions arising from consideration of the three topics outlined above. The proposed theoretical model describes the hypothetical sequence of events in the neurovascular unit and the expected effects that propagate from neurons to astrocytes, and to endothelial and red blood cells in the capillary.
Hypothesis
“Perivascular tone” and its modulation by astrocytic end-feet
The central premise of the hypothesis is that brain parenchymal cells (neurons and astrocytes) can dynamically modulate capillary wall compliance and, consequently, capillary lumen cross section area. The hypothesis postulates that the effect of this modulation is not negligible under normal physiological conditions, but on the contrary has a special role in neurovascular coupling. The modulatory effect on passive compliance of a vessel is further referred to as “perivascular tone” to distinguish it from active myogenic vascular tone maintained by smooth muscle cells (SMC) in arterial vessels. Astrocytic perivascular end-feet (AEF) (Figure 1), which ensheath almost the entire surface of capillary vessels [14-18], are hypothesized to play the key role in transduction of cellular events and biochemical signals into changes of mechanical properties of the capillary vessel wall. Perivascular tone is envisioned to be realized as follows. Cellular volume of astrocytes, and particularly the volume of AEF compartment (which is known to change in vivo in response to various events affecting the osmotic balance [19]), influences the composition of the environment immediately surrounding the capillary vessel, namely, its relative occupancy by extracellular space/interstitial fluid (ECS/ISF) and/or intracellular space (ICS) of AEF. The distensibility of a capillary vessel is influenced not only by elastic properties of endothelium, but also, to a large degree, by the elastic environment of the capillary bed [20]. Fung et al. provide detailed biomechanical analysis comparing “tube in fluid” versus “tunnel in gel” model of a capillary (Figure 2), and show that the rate of change of the capillary diameter with intraluminal pressure (ILP) significantly differs between the two cases: a capillary behaves as an elastic or as a rigid tube, respectively. These simplified examples are at the opposite ends of the spectrum, whereas cerebral capillaries are known to be moderately distensible [21,22]. In the proposed hypothesis, compliance of a capillary wall is expected to vary as a function of the ECS/ICS composition of the immediate environment, which is determined by an enlarged or shriveled AEF.
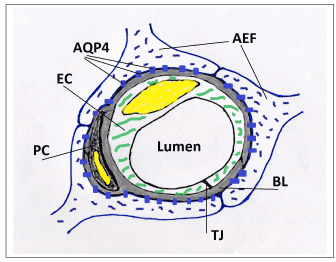
Figure 1. A schematic drawing of a cross-section of a cerebral capillary showing astrocytic end-feet (AEF) with aquaporin-4 channels (AQP4), fused gliovascular basal lamina (BL), endothelial cell (EC), pericyte (PC), and tight junction (TJ).
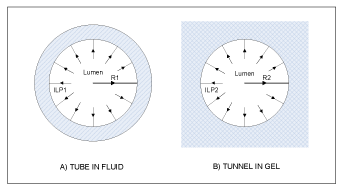
Figure 2. Schematic cross sectional drawings of “tube in fluid” and “tunnel in gel” cases illustrating the two opposite ends of the spectrum of biomechanical models of an elastic capillary and its environment (based on [20]).
Perivascular tone is envisioned to have both a quasi-static, slowly varying component, which is determined by astroglial steady-state cellular volume (and therefore by factors affecting the osmotic balance of astrocytes), and a dynamical, transient component. In response to rapid transient osmotic changes, the volume of AEF compartment is hypothesized to be able to change more rapidly compared to the astrocyte’s cell body volume, and therefore to be able to spatially “focus” the effect of these changes to the immediate environment of capillaries. It is of note for further discussion that the distention of a cerebral capillary is driven by the transmural pressure gradient that is equal to ILP minus intracranial pressure (ICP), where ILP is determined by the cerebral perfusion pressure (CPP) and its attenuation by the resistance of the upstream vascular tree. To summarize, it is expected that, for a fixed ILP and ICP, the capillary will have a smaller diameter and stiffer walls when astrocytic cellular volume, particularly in end-feet, is larger (“swollen” astrocytes – “high” perivascular tone), as opposed to having a larger diameter and more elastic walls when the volume is smaller (“shrunk” astrocytes – “low” perivascular tone).
Coupling of perivascular tone to cerebral blood flow
By modulating mechanical properties of the vessel wall, perivascular tone is hypothesized to influence the capillary’s resistance to flow. The distribution of red blood cells (RBC) in the capillary and the pattern of RBC flow are expected to change in response to changes of perivascular tone. These changes will affect the rates of production of endothelium-dependent vasoactive mediators: vasodilators like nitric oxide (NO), endothelium-derived hyperpolarization factor (EDHF), and certain prostaglandins/prostanoids, as well as vasoconstrictors such as endothelin-1 [23]. This is likely to be realized by graded involvement of the following two previously known mechanisms, in combination: i) deoxygenation- and deformation-related release of ATP from RBC [24-26], which stimulates NO synthesis by endothelial NO synthase (eNOS), and the release of S-Nitrosothiol (SNO, endothelium-independent vasodilators) from RBC [27]. ii) RBC and plasma interactions with glycocalix and endothelial shear receptors [28,29]. It is a well-known fact that basal tonic production of NO, for instance, has an important role in the regulation of cerebral blood flow under baseline resting conditions [23,30]. Therefore it is straightforward to infer that perivascular tone, by influencing the production of endothelium-dependent vasoactive mediators, will affect vasodilation/vasoconstriction of the feeding precapillary arterioles, which in turn will affect vascular tone of the upstream vessels by means of ubiquitous vascular mechanisms of vasodilatory propagation (conducted vasodilation and shear stress-/flow-mediated vasodilation [23,31]). As a result, perivascular tone is hypothesized to have an influence on baseline cerebral blood flow. In addition, the proposed hypothesis postulates that production of vasodilators (NO, EDHF, prostanoids) by endothelium of capillaries in activated neurovascular units will increase in response to functional “tightening” of perivascular tone, as described below.
Coupling of neuronal activation to perivascular tone: Functional activation, an increase in neuronal synaptic or spiking activity caused by sensory stimulation or a cognitive task, leads to a rapid release of potassium ions K+ from neurons into ECS, where excessive potassium is transported into neighboring astrocytes performing potassium homeostasis function (potassium buffering) [32-35]. In the process, chloride ions Cl− are also usually accumulated in astrocytic cytosol by Na–K–Cl cotransporters [34]. In addition to ionic uptake, the largest part of glutamate neurotransmitter molecules released at the excitatory synapses is also actively transported into astrocytes (Figure 3) (where glutamate is oxidatively degraded to aspartate/lactate, amidated to glutamine or converted into other metabolites) [36,37]. Osmotically obligated water follows into astrocytic ICS [34], leading to activity-dependent cellular volume changes in response to functional activation [38-43]. The main influx of water driven by the osmotic gradient, is hypothesized to occur via AQP4 channels at AEF membrane, where astrocyte’s membrane permeability to water is the largest [44]. As postulated above, volume changes in response to rapid transient osmotic imbalances are expected to be more pronounced at the perivascular end-feet. As a result, neuronal activation (glutamatergic synaptic activity and/or potassium-releasing intensive spiking activity) is hypothesized to be directly and “tightly” coupled to mechanical properties of the capillary vessels feeding the activated neurovascular unit. Namely, functional activation is expected to cause stiffening of a vessel wall (an increased perivascular tone), and the onset of activation is expected to cause an initial decrease in the diameter of vessels’ lumen (until the effect is counterbalanced by an increased perfusion that brings an increase in ILP). After cessation of activation, perivascular tone is expected to return to baseline level due to osmoregulatory and homeostatic properties of astrocytes [45- 47]. Astrocytic cellular events involved in potassium and glutamate uptake are highly energy dependent, and glutamate oxidative metabolism is also oxygen dependent [34,36], therefore hemodynamic response is linked to increased metabolic demands, but in the proposed mechanism of neurovascular coupling, glucose and oxygen consumption are not considered to be the primary driving factors. The hypothesized events of the triphasic hemodynamic response (HDR) [13] are described in further details below.
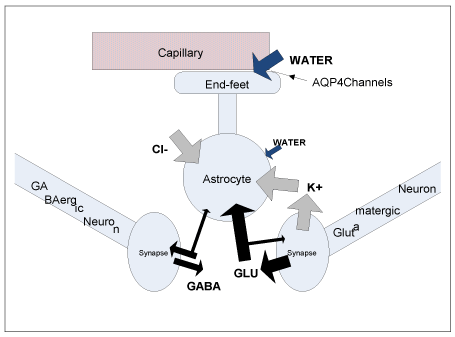
Figure 3. A schematic drawing illustrating neurotransmitter, ion and water transport in astrocytes and neurons during neuronal activation (fluxes of neurotransmitters are based on Figure 1 from [36]; different widths of the arrows are intended to illustrate the relative magnitude of the GABA and Glutamate fluxes on release and uptake from neuron/astrocyte).
HDR phase 1, the initial dip: changes in RBC flow
During the initial phase of HDR that follows right after the onset of activation, rapid changes in astrocytic cellular volume are hypothesized to cause both stiffening of the capillaries and a reduction in their distention. RBC distribution and flow in the capillary are expected to change in response to increased resistance to flow, as compared to baseline. RBC capillary transit time is likely to increase due to higher energy losses in transient deformations of an RBC moving through a tortuous capillary path with heterogeneous geometry of the vessel wall [2] (see also discussion in [48]). An increase of capillary hematocrit, i.e., of the linear density of RBCs flowing through the capillary in a single file, is also expected to occur [2,49], which translates into a local increase of total hemoglobin (Hb) concentration ([HbT]). A longer RBC transit time is usually associated with a more complete deoxygenation of Hb in a blood cell [50]; therefore deoxy-Hb concentration ([HbR]) is expected to rise. Velocity of RBC and plasma is not likely to increase at this phase, because the flow can be redistributed across other capillaries in a multi-channel network that also has thoroughfare channels [2]. The RBC flux through the capillary is expected to fall, which, combined with increased rates of aerobic metabolism, will lead to a decrease of oxygen concentration (PO2) in the tissue.
HDR phase 2, the overshoot: upstream arterial dilations
Compared to baseline, increased quantities of vasodilators are expected to diffuse from the capillaries of functionally activated neurovascular units to precapillary arterioles, where this change will be sensed by SMC and will result in the relaxation of vascular tone. Relaxation occurs with a delay after the onset of activation because of the diffusive process. Relaxation of small arterioles decreases vascular resistance and leads to an increase of shear rate in the upstream generation of arterioles, and therefore dilation propagates upstream in a well-known cascade of endothelium- and shear-dependent vasodilation [23,31]. In addition, conductive vasodilation can also play a role in upstream propagation of dilation from small arterioles [51]. Because of lowered vascular resistance in supplying arteries and arterioles, the drop in hydrostatic pressure is reduced, and ILP in the capillaries increases, counteracting the effect of functional modulation of perivascular tone and distending the capillary vessels. This in turn leads to a decline in the production of vasodilatory mediators. If stimulation is sustained, a steady state is expected to be reached in HDR, i.e., a plateau in CBF response. The apparent “excessiveness” of the CBF response, metaphorically described as “watering the garden for the sake of one thirsty flower” [3,52], thus receives a plausible explanation in the proposed theoretical model: since the effect of increased regional CBF is brought about by the necessity to counteract an increased resistance to flow in activated capillaries (the “thirsty flower”) by increasing the ILP there, and because the vessel network is branched and multi-tiered, there is no other, more spatially specific way to increase ILP at the target, but to increase it in the whole tier of capillaries (“the garden”).
HDR phase 3, the undershoot: returning to baseline perivascular tone
Cessation of neuronal activation is hypothesized to be associated with the return of perivascular tone to baseline due to the capacity of astrocytes to homeostasis. This suggests that activated capillaries will further distend under present ILP in response to restored relaxed compliance of the vessel wall, and their resistance to flow will drop. As a result, a decrease below baseline in production of vasodilatory mediators by capillary endothelium is expected to happen, which after a diffusion-mediated delay will cause tension of vascular tone in precapillary arterioles and then a cascade of vasoconstrictive events further upstream (by the same physiological pathways as in the vascular relaxation cascade described above). The regional CBF, the resistance to flow of activated capillaries, and the rate of vasodilatory mediators production in these capillaries are all expected to return to their respective baseline in approx. the same time frame. Several cycles of damped oscillation around baseline can be expected while flow, resistance of arterial tree, and capillary bed resistance settle to their baseline rest state in a not-precisely synchronized manner. Cerebral blood volume (CBV) is hypothesized to return to baseline with an additional delay, as compared to CBF, due to a contribution from venous compartment, as described by the balloon model [53]; this is hypothesized to cause the undershoot in the HDR signal.
The control system view
Figure 4 presents the proposed theoretical model of neurovascular coupling as a feedback control system. The capillaries of activated neurovascular units are considered as the system “plant” which is perturbed by the functional modulation of perivascular tone, while the upstream arterial vasculature is regarded as the controller that responds to the error signal (vasoactive mediators) “measured” by interactions of the flowing red cells with capillary endothelium (sensors) in the “plant.” It must be remembered that the diagram is oversimplified, whereas the hypothesized “real control system” is heterogeneous and distributed, therefore its settled states and responses are determined by the intricate interplay of various system parts and compartments. For instance, at the peak or plateau of the CBF response one should not expect the resistance to flow in the “activated” capillaries to reach exactly the same value as before activation, because the contribution of responses from neighboring capillaries, which are also affected by the flow increase, must be also taken into account. In such a system, it is natural to expect a hysteresis in both states and responses.
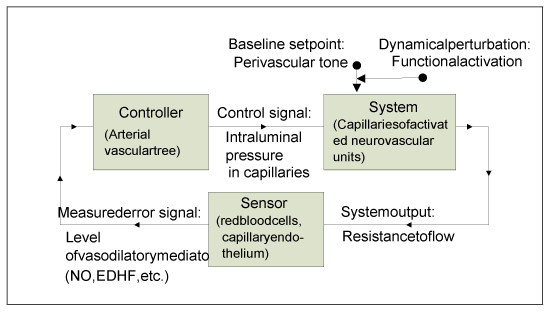
Figure 4. The hypothesized theoretical model of neurovascular coupling viewed as a feedback control system.
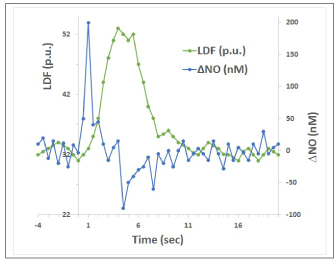
Figure 5. Illustration of temporal dynamics of NO and CBF changes in brain tissue (based on Figure 1 in [76]; LDF is laser-Doppler flowmetry measure of CBF).
A possible link to cerebral autoregulatory mechanisms
As stated above, the hypothesis considers the events of vasodilatory cascade to be governed by basic vascular physiological pathways that are common to all vascular beds. In addition, it is hypothesized here that functional hyperemia shares common roots with the mechanisms of cerebral autoregulatory response to a perfusion pressure drop. While cerebral autoregulation pathways are still a subject of debate [54-56], there is growing evidence that the endothelium and the release of vasoactive substances play the key role in the autoregulatory “metabolic control pathway” [56]. It is hypothesized here that when CPP, and by consequence ILP, drop, or when ICP increases, the resistance to flow in capillaries increases because they become less distended by a decreased transmural pressure gradient (ILP-ICP), and, from this point, the cerebral autoregulatory response and the functional hyperemic response develop further along the same basic scenario, notwithstanding the difference in systemic/regional CBF involved.
Summary
At the risk of oversimplification, the hypothesis can be summarized through the following sequence of events: 1) Neuronal activation intensifies the uptake of potassium and glutamate into astrocytes. 2) Water is driven into astrocytes by osmosis, and astrocytic cellular volume increases, particularly at perivascular end-feet, where the aquaporin “water-highway” is strategically placed. 3) perivascular end-feet tightly ensheath capillaries, therefore when the end-feet “clutch” “engages” around the capillary endothelial tube, the vessel wall becomes stiffer and less yielding to distention by blood pressure in the lumen. 4) A stiffer and narrower capillary resists the flow of red cells and increases the capillary hematocrit. 5) Mechanically-stressed endothelial and red cells enlist help from vasodilatory mechanisms that evolved to prevent hypoperfusion of microcirculation and are common to all vascular beds. 6) Vasodilatory mediators diffuse to precapillary arterioles and dilate them. 7) By common vasoactive pathways, vessel dilation propagates upstream to penetrating arterioles and pial arteries. 8) A drop in arterial vascular resistance increases the regional cerebral blood flow and also increases the luminal pressure in the capillaries of the dilatory signal origin. 9) An increased pressure distends the “clutched” capillaries and decreases the resistance to flow there, thus closing the feedback loop and returning the production of vasodilators (“error signal”) to near baseline rates. It is the pressure in “activated” capillaries, not the flow, which serves as feedback “control signal.” The overabundance of the flow response is caused by the vessel network structure, where an upstream dilation cannot be spatially specific enough to address perfusion deficit in activated neurovascular units only. According to the hypothesis, factors affecting the baseline cellular volume of astrocytes will also affect: i) the baseline CBF, by changing the baseline perivascular tone of the capillaries; ii) the magnitude of CBF response to functional activation, by changing the “expansion capacity” of AEF (i.e., the range of AEF volume change from baseline to the upper limit that is achievable in normal conditions before astrocytic mechanisms of regulatory volume decrease [45-47,57-60] are activated).
Supporting evidence
Distinct structural and functional properties of astrocytes
Studies of perivascular AEF structure revealed an almost complete coverage (>99% of the surface area) of the capillary vessel wall by the endfeet [14,61]. Because of their juxtaposition to blood vessels, AEF have been long suggested as a key cite for neurovascular signaling at arteriolar vessels [16,17] and, recently, also at pericytes in capillaries [11,62,63]. Polarized expression of major water channels, aquaporins-4 (AQP4) [44], which are known to be abundantly and predominantly expressed at the AEF membrane facing the basal lamina of the capillary [16] (Figure 1), and experiments on AQP4 knockout (KO) mice identify perivascular AEF as the site of primary bidirectional transport of water across the brain-blood interface [44,64,65]. The function of AEF AQP4 is generally thought to be associated, in normal physiological conditions, with efflux of metabolically produced water into the blood stream, under osmotically induced (in concert with functionally associated ion channels expressed on AEF membrane) gradients [66]. The proposed hypothesis is consistent with this view, when baseline resting state or activation steady state are considered. During the initial phase, a reversal of the (bidirectional) water transport from efflux to influx, under transient reversal of osmotic gradients during initial accumulation of ions and neurotransmitters into astrocytes, appears to be physiologically plausible. An influx of water into AEF is thought to lead to a more pronounced increase in AEF compartment volume, as compared to cell body volume, because of resistance to water flow through a relatively thin process leading from AEF to soma (end-feet: mean diameter 4–8 µm [17], end-feet processes: up to 1 µm wide [67]). Considering an AEF that was initially 0.3 µm thick [14], and assuming that it inflated to 0.5 µm due to water influx (an uptake of water volume in the range of 4 x 8 x 0.2 µm3 = 6.4 fL, or 0.3 pmol water), then for a capillary with 0.4 µm endothelial tube thickness (estimated from [44,64]), the ratio of the percent change of vessel radius per unit pressure (dR/R)/dP (a measure of wall compliance) before/after influx can be estimated as the inverse ratio of combined AEF-endothelium wall thickness before and after, which is equal to 1.3 in this example (0.9 µm/0.7 µm) (see [20]; it is assumed here for simplicity that AEF and endothelial cells have the same elastic property, and that ECS is filled with ISF). That is, a relatively small increase in AEF volume (∆ 0.2 µm in AEF thickness) is estimated to be, in principle, capable of changing the vessel wall compliance by 30% (measured as percent change of vessel radius per unit pressure).
Astrocyte cellular volume changes
The functional role of astrocytes in homeostasis of ions and water is well-known [34,65,64,68]. Increase in astrocytic cellular volume (swelling) is often mentioned in the context of brain pathophysiology [64,65,69]. In cellular/cytotoxic edema, astrocytes swell in response to osmotic balance disruptions by pathological processes that usually do not incur a breakdown of the blood-brain barrier [69]. In “ionic edema” [69], astrocytes gain water from the vascular compartment, because of increased activation of ion transport processes. In observations of pathological astroglial swelling, it is often mentioned that the volume of perivascular AEF is particularly increased [19,64,65], sometimes even causing a complete collapse of capillary vessels and an interruption of perfusion [70]. The distinct role of AEF AQP4 channels in osmotically induced swelling is confirmed by AQP4 KO [65] and AQP4 adapter protein disruption [64] experiments on mice. Activity-dependent glial swelling is a well-known phenomenon in studies with brain slices [38,40-43] (see [39] for a detailed model of astrocytic mechanisms explaining activity-induced volume changes). Activity-induced volume changes have been also shown to critically depend on AEF AQP4 pool [38]. By cautious extrapolation of the findings describing astrocytic volume changes in pathology-induced osmotic disturbances and in activation experiments on brain slices, it is reasonable to expect certain smaller scale volume changes during in vivo functional activation, and, taking into account the proven critical role of perivascular AQP4 channels, it is reasonable to expect that these cellular volume changes will originate in the AEF compartment.
The initial dip and time courses of oxy- and deoxy-Hb concentration
The origins of the “initial dip” [12,52] in OISI and fMRI signal and the hypothesis of Malonek and Grinvald [52], that the initial dip is caused by increased oxygen extraction (with conversion of HbO2 to HbR) prior to HDR CBF increase, have been subjects of active debate [3,4]. As pointed out by Buxton in [2], optical imaging does not show a drop in oxy-Hb that would be expected if an increase in deoxy-Hb content was caused by increased oxygen extraction. A recent multi-wavelength optical imaging study [71] revealed that the initial dip is dominated by an increase in [HbT] and did not find an increase in [HbR]. Another recent study [72] using high spatiotemporal two-dimensional spectroscopic optical imaging found that, during the initial phase of HDR, both [HbT] and [HbO2] increased above baseline, whereas [HbR] dropped. These recent findings are not entirely consistent with a hypothesis that the initial dip is caused by increased conversion of HbO2 to HbR, but they are consistent with the initial phase of HDR in the model proposed in this paper, where an initial increase in [HbT] and [HbO2] can be explained by early changes in arterial blood volume during upstream propagation of vasodilation that precedes the dilation of the capillaries. Likewise, an increased deoxy-Hb content reported by other experiments [4,52], as well as the reported decrease of oxygen tension (PO2) in tissue, are well explained by the proposed model as effects of increased capillary hematocrit and [HbR] and of decreased RBC flux. These effects are expected to be subtle, which may suggest the reason why the initial dip is such an “elusive” phenomenon [3].
Functional reactivity of cerebral capillaries
Intravital microscopic observations of blood flow in individual cerebral capillaries suggest “the presence of a physiological regulatory mechanism of cerebral capillary flow that may involve communication among various microvascular and parenchymal cells and utilize locally acting endothelial and parenchymal mediators such as endothelium-derived relaxing factor or nitric oxide” [2], which is also emphasized in the epigraph quote by A. Hudetz [2]. The findings show that while all cerebral capillaries are perfused at all times (i.e., there is no functional recruitment per se, as in skeletal muscle microcirculation), there is significant heterogeneity of flow within each capillary network [2], and that functional activation decreases this heterogeneity (in the steady state response). It is reported that in a small fraction of capillaries perfusion by red cells is sometimes observed to stop, seemingly at random, for a period of no more than several seconds [2]. Prolonged stalls and slow 0.1 Hz fluctuations of RBC flow are also reported in a two-photon microscopy (2PM) study [73], where an activity-dependent decrease in capillary resistance to flow was observed (again, in the steady state response). A more recent 2PM study [74] reports stimulation-induced capillary responses in olfactory bulb glomeruli, the responses that consisted of both increases and decreases in RBC flow. Another recent 2PM study [75] describes functional reactivity of capillaries to steady-state somatosensory stimulation, where capillary responses were characterized by overall capillary dilation and a decreased heterogeneity in vessel volumes, but with occasional decreases in capillary volume and RBC flux and speed. Taken together, the above findings on resting and activity-induced microcirculatory patterns are well consistent with the mechanism of “parenchymal/perivascular” modulation of the capillary vessel wall proposed in the hypothesis, and with the distention of capillaries by increased CBF.
The time course of nitric oxide production
Measurements of the temporal dynamics of brain tissue NO during somatosensory stimulation are reported in [76]. Figure 5 here reproduces a sketch of the pattern of NO and CBF changes based on Fig.1 published by Buerk et al.. Measured NO increased immediately after stimulation onset and reached a peak within 0.4 s, and then returned close to prestimulus baseline after 2 s. The CBF started to rise after a 1 s delay, and reached a peak shortly before the end of the 4 s stimulation period. A poststimulus undershoot was consistently observed in NO after the end of stimulation, when CBF was returning to baseline. It is easy to notice that the time course of measured tissue NO is entirely consistent with the expected “error signal” (endothelial production of vasodilatory mediators that diffuse in the tissue), as described above in the theoretical model of the feedback mechanism, whereas CBF time course follows the response of the “controller.” The negative dip in NO after the end of the stimulus is entirely expected in the proposed framework, as described above with regard to HDR phase 3.
Evidence for a possible link to cerebral autoregulation
It is of note that the discussion here is concerned only with the cerebral autoregulatory response to a perfusion pressure drop or to an increased ICP, and does not consider the response to perfusion pressure increase, which is generally thought to be dependent on myogenic pathway [6,56]. If there are common physiological pathways shared by functional hyperemia and cerebral autoregulation, as described in the hypothesis, then cerebral autoregulation must also be dependent on endothelial production of vasodilatory mediators. This is confirmed by multiple studies [77-80]. In addition, a study of control system dynamics of neurovascular coupling and cerebral autoregulation [81] demonstrated that both control systems can be described with identical parameters.
Discussion
The central premise of the proposed hypothesis is the existence and the role of the AEF perivascular tone under normal baseline physiological conditions and during functional activation. This premise is testable and falsifiable. Experimental verification will require delicate in vivo experiments, perhaps involving intravital microscopy [2] and 2PM. Contrary to 2PM studies [73-75] that are mostly concerned with the hyperemic phase of response and with sustained stimulation, the suggested experiments should focus on time resolved initial fast response, which could be technologically difficult. Quantitative modeling could provide some clues with regard to the magnitude of the expected effect, but would otherwise have limited confirmatory value because of the complexity of the involved microcirculation. If the effect of astroglial end-feet volume change on red blood cell flux through capillaries proves to be negligible under normal physiological conditions, then the proposed hypothesis will be refuted. Indirect evidence in support of or against the hypothesis may come from experiments on hemodynamics of aquaporin deficient mice (AQP4 KO [65] or the α-syn(-/-) type with genetically disrupted AQP4 adapter protein [64]). The proposed theoretical model predicts that the timing and the magnitude of functional hyperemic response will be strongly affected in AQP4 KO and α-syn(-/-) mice; the response would be expected to develop slower and have a smaller magnitude, as compared to wild type animals.
Perivascular tone is not expected to have any significant contribution in vessels that have substantial presence of perivascular space (PVS), which is filled with ISF, between the basement membrane of the vascular wall and the glial basement membrane at AEF, e.g., in precapillary arterioles or postcapillary venules [82]. In the capillaries, the PVS is closed, because the two basement membranes fuse and form a “gliovascular membrane” [82], therefore a stronger coupling of perivascular tone to the vessel wall is expected.
In this theoretical model, the existence of Negative BOLD (blood oxygen level dependent) effect [83] in fMRI is not surprising. Increased GABAergic inhibitory synaptic activity is expected to lead to a decrease in glutamate release by excitatory glutamatergic synapses, and therefore to a decrease in glutamate uptake by astrocytes, as compared to baseline resting state, which is expected to lead to “relaxation” of perivascular tone and ultimately to a reduction of regional CBF by vascular pathways described in the model.
A potentially promising area for further consideration in the framework of the proposed model is exploring why in pre-ictal periods vascular events can precede neuronal events [4], and how abnormalities in astrocytic perivascular tone may be connected to epilepsy disease state.
If the effect of the astroglial “perivascular tone” does not prove to be negligible, the proposed theoretical model will invite researchers to explore a new dimension in neurovascular coupling mechanisms, which is likely to be shaped by the complexity of the biomechanics and dynamics of the microcirculation.
Conclusions
The proposed theoretical model has a potential advantage of explaining the following features of cerebral functional hyperemia: 1) the origin of the “hyperemic” overabundance in the CBF response; 2) the tight coupling of regional CBF to glucose metabolism and its apparent uncoupling from oxygen metabolism; 3) the time course of oxy-, deoxy- and total hemoglobin concentration during the initial dip; 4) the currently unknown feedback mechanism that determines the magnitude of the settled CBF response; 5) the time course of nitric oxide measured in activated brain tissue; 6) variability and slow fluctuations of baseline hemodynamics in the brain. The hypothesis sets forth a novel view on the link between neurovascular coupling and anatomical and functional properties of astrocytes, and proposes a unified basis for mechanisms involved in functional hyperemia and cerebral autoregulatory response to perfusion pressure drop. The formulation of this hypothesis was driven by an attempt to understand the inner workings of pathways and events that may be responsible for neurovascular coupling phenomenon, possibly looking ”outside the box” at the risk of going beyond the framework of current expert opinion in the field. It is the author’s sincere hope that this paper will draw the attention of researchers to some potentially overlooked aspects of intriguing neurovascular coupling problem, and will stimulate research in new directions.
References
- Raichle ME, Mintun MA (2006) Brain work and brain imaging. Annu Rev Neurosci 29: 449-476. [Crossref]
- Hudetz AG (1997) Blood flow in the cerebral capillary network: a review emphasizing observations with intravital microscopy. Microcirculation 4: 233-252. [Crossref].
- Buxton RB (2001) The elusive initial dip. Neuroimage 13: 953-958. [Crossref]
- Vanzetta I, Grinvald A (2008) Coupling between neuronal activity and microcirculation: implications for functional brain imaging. HFSP J 2: 79-98. [Crossref]
- Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci 5: 347-360. [Crossref]
- Drake CT, Iadecola C (2007) The role of neuronal signaling in controlling cerebral blood flow. Brain Lang 102: 141-152. [Crossref]
- Paulson OB, Hasselbalch SG, Rostrup E, Knudsen GM, Pelligrino D (2010) Cerebral blood flow response to functional activation. J Cereb Blood Flow Metab 30: 2-14. [Crossref]
- Riera JJ, Sumiyoshi A (2010) Brain oscillations: ideal scenery to understand the neurovascular coupling. Curr Opin Neurol 23: 374-381. [Crossref]
- Moon CH, Fukuda M, Park SH, Kim SG (2007) Neural interpretation of blood oxygenation level-dependent fMRI maps at submillimeter columnar resolution. J Neurosci 27: 6892-6902.[Crossref]
- Tian P, Teng IC, May LD, Kurz R, Lu K, et al. (2010) Cortical depth-specific microvascular dilation underlies laminar differences in blood oxygenation level-dependent functional MRI signal. Proc Natl Acad Sci U S A 107: 15246-15251. [Crossref]
- Hamilton NB, Attwell D, Hall CN (2010) Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenergetics 2. [Crossref]
- Frostig RD, Lieke EE, Ts’o DY, Grinvald A (1990) Cortical functional architecture and local coupling between neuronal activity and the microcirculation revealed by in vivo high-resolution optical imaging of intrinsic signals. Proceedings of the National Academy of Sciences 87: 6082–6086.
- Chen-Bee CH, Agoncillo T, Xiong Y, Frostig RD (2007) The triphasic intrinsic signal: implications for functional imaging. J Neurosci 27: 4572-4586. [Crossref]
- Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP (2010) The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58: 1094-1103. [Crossref]
- El-Khoury N, Braun A, Hu F, Pandey M, Nedergaard M, et al. (2006) Astrocyte end-feet in germinal matrix, cerebral cortex, and white matter in developing infants. Pediatr Res 59: 673-679.[Crossref]
- Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier: an overview: Structure, regulation, and clinical implications. Neurobiol Dis 16: 1–13. [Crossref]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M (2003) Signaling at the gliovascular interface. J Neurosci 23: 9254-9262. [Crossref]
- Nakazawa E, Ishikawa H (1998) Ultrastructural observations of astrocyte end-feet in the rat central nervous system. J Neurocytol 27: 431-440. [Crossref]
- Risher WC, Andrew RD, Kirov SA (2009) Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia 57: 207-221. [Crossref]
- Fung YC, Zweifach BW, Intaglietta M (1966) Elastic environment of the capillary bed. Circ Res 19: 441-461. [Crossref]
- Atkinson JL, Anderson RE, Sundt TM Jr (1990) The effect of carbon dioxide on the diameter of brain capillaries. Brain Res 517: 333-340. [Crossref]
- Duelli R, Kuschinsky W (1993) Changes in brain capillary diameter during hypocapnia and hypercapnia. J Cereb Blood Flow Metab 13: 1025-1028. [Crossref]
- Andresen J, Shafi NI, Bryan RM Jr (2006) Endothelial influences on cerebrovascular tone. J Appl Physiol (1985) 100: 318-327. [Crossref]
- Arciero JC, Carlson BE, Secomb TW (2008) Theoretical model of metabolic blood flow regulation: roles of ATP release by red blood cells and conducted responses. Am J Physiol Heart Circ Physiol 295: H1562-1571. [Crossref]
- Wan J, Ristenpart WD, Stone HA (2008) Dynamics of shear-induced ATP release from red blood cells. Proc Natl Acad Sci U S A 105: 16432-16437. [Crossref]
- Jensen FB (2009) The dual roles of red blood cells in tissue oxygen delivery: oxygen carriers and regulators of local blood flow. J Exp Biol 212: 3387-3393. [Crossref]
- Diesen DL, Hess DT, Stamler JS (2008) Hypoxic vasodilation by red blood cells: evidence for an s-nitrosothiol-based signal. Circ Res 103: 545-553. [Crossref]
- Weinbaum S, Zhang X, Han Y, Vink H, Cowin SC (2003) Mechanotransduction and flow across the endothelial glycocalyx. Proc Natl Acad Sci U S A 100: 7988-7995. [Crossref]
- Pahakis MY, Kosky J2021 Copyright OAT. All rights reserve of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem Biophys Res Commun 355: 228-233.[Crossref]
- Toda N, Ayajiki K, Okamura T (2009) Cerebral blood flow regulation by nitric oxide: recent advances. Pharmacol Rev 61: 62-97. [Crossref]
- Pohl U, De Wit C, Gloe T (2000) Large arterioles in the control of blood flow: role of endothelium-dependent dilation. Acta Physiol Scand 168: 505-510. [Crossref]
- Chen KC, Nicholson C (2000) Spatial buffering of potassium ions in brain extracellular space. Biophys J 78: 2776-2797. [Crossref]
- Walz W (2000) Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 36: 291-300. [Crossref]
- Walz W (2003) Potassium homeostasis in the brain at the organ and cell level. In: Hertz, L., editor. Non-Neuronal Cells of the Nervous System: Function and Dysfunction; vol. 31 of Advances in Molecular and Cell Biology. Elsevier p. 595–609.
- Kofuji P, Newman E (2004) Potassium buffering in the central nervous system. Neuroscience 129: 1043–1054. [Crossref]
- Schousboe A, Waagepetersen HS (2003) Role of astrocytes in homeostasis of glutamate and GABA during physiological and pathophysiological conditions. In: Hertz, L., editor. Non-Neuronal Cells of the Nervous System: Function and Dysfunction; vol. 31 of Advances in Molecular and Cell Biology. Elsevier; 2003, p. 461–474.
- Schousboe A (2003) Role of astrocytes in the maintenance and modulation of glutamatergic and GABAergic neurotransmission. Neurochem Res 28: 347-352. [Crossref]
- Kitaura H, Tsujita M, Huber VJ, Kakita A, Shibuki K, et al. (2009) Activity-dependent glial swelling is impaired in aquaporin-4 knockout mice. Neurosci Res 64: 208-212. [Crossref]
- Østby I, Øyehaug L, Einevoll GT, Nagelhus EA, Plahte E, et al. (2009) Astrocytic mechanisms explaining neural-activity-induced shrinkage of extraneuronal space. PLoS Comput Biol 5: e1000272. [Crossref]
- MacVicar BA, Hochman D (1991) Imaging of synaptically evoked intrinsic optical signals in hippocampal slices. J Neurosci 11: 1458-1469. [Crossref]
- Andrew RD, MacVicar BA (1994) Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience 62: 371-383. [Crossref]
- MacVicar BA, Feighan D, Brown A, Ransom B (2002) Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. Glia 37: 114-123. [Crossref]
- Holthoff K, Witte O (1996) Intrinsic optical signals in rat neocortical slices measured with near- infrared dark-field microscopy reveal changes in extracellular space. J Neurosci 16: 2740–2749. [Crossref]
- Amiry-Moghaddam M, Ottersen OP (2003) The molecular basis of water transport in the brain. Nat Rev Neurosci 4: 991-1001. [Crossref]
- Olson JE, Sankar R, Holtzman D, James A, Fleischhacker D (1986) Fleischhacker, D.. Energy-dependent volume regulation in primary cultured cerebral astrocytes. J Cell Physiol 128: 209–215. [Crossref]
- Olson JE, Fleischhacker D, Murray WB, Holtzman D (1990) Control of astrocyte volume by intracellular and extracellular Ca2+. Glia 3: 405-412. [Crossref]
- Pasantes-Morales , Murray RA, Lilja L, Morán J (1994) Regulatory volume decrease in cultured astrocytes. I. Potassium- and chlorideactivated permeability. Am J Physiol 266: C165–C171. [Crossref]
- Secomb TW, Hsu R, Pries AR (2002) Blood flow and red blood cell deformation in nonuniform capillaries: effects of the endothelial surface layer. Microcirculation 9: 189-196. [Crossref]
- Dzwinel W, Boryczko K, Yuen DA (2003) A discrete-particle model of blood dynamics in capillary vessels. J Colloid Interface Sci 258: 163-173. [Crossref]
- Hudetz AG (1997) Cerebral microcirculation. In: Welch, K., Caplan, L., Reis, D., Siesjo, B., Weir, B., editors. Primer On Cerebrovascular Diseases. Academic Press p 45–51.
- Gustafsson F, Holstein-Rathlou N (1999) Conducted vasomotor responses in arterioles: characteristics, mechanisms and physiological significance. Acta Physiol Scand 167: 11-21. [Crossref]
- Malonek D, Grinvald A (1996) Interactions between electrical activity and cortical microcirculation revealed by imaging spectroscopy: Implications for functional brain mapping. Science 272: 551–554. [Crossref]
- Buxton RB, Wong EC, Frank LR (1998) Dynamics of blood flow and oxygenation changes during brain activation: The balloon model. Magn Reson Med 39: 855–864. [Crossref]
- Paulson OB, Strandgaard S, Edvinsson L (1990) Cerebral autoregulation. Cerebrovasc Brain Metab Rev 2: 161-192. [Crossref]
- Townsend P, Knowles MG (1999) The cerebral circulation. Curr Anaesth Crit Care10: 77–82.
- Franco Folino A (2007) Cerebral autoregulation and syncope. Prog Cardiovasc Dis 50: 49-80. [Crossref]
- Pasantes-Morales H, Moran J, Schousboe A (1990) Volume-sensitive release of taurine from cultured astrocytes: properties and mechanism. Glia 3: 427-432. [Crossref]
- Morales-Mulia S, Vaca L, Hernandez-Cruz A, Pasantes-Morales H (1998) Pasantes-Morales, H.. Osmotic swelling-induced changes in cytosolic calcium do not affect regulatory volume decrease in rat cultured suspended cerebellar astrocytes. J Neurochem 71: 2330–2338. [Crossref]
- Pasantes-Morales H, Morales Mulia S (2000) Influence of calcium on regulatory volume decrease: role of potassium channels. Nephron 86: 414-427. [Crossref]
- Pasantes-Morales H, Cardin V, Tuz K (2000) Signaling events during swelling and regulatory volume decrease. Neurochem Res 25: 1301-1314. [Crossref]
- Hawkins BT, Davis TP (2005) The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57: 173-185. [Crossref]
- Peppiatt CM, Howarth C, Mobbs P, Attwell D (2006) Bidirectional control of CNS capillary diameter by pericytes. Nature 443: 700-704. [Crossref]
- Kamouchi M, Ago T, Kitazono T (2011) Brain pericytes: emerging concepts and functional roles in brain homeostasis. Cell Mol Neurobiol 31: 175-193. [Crossref]
- Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, et al. (2003) An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A 100: 2106-2111. [Crossref]
- Verkman AS, Binder DK, Bloch O, Auguste K, Papadopoulos MC (2006) Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim Biophys Acta 1758: 1085-1093. [Crossref]
- Nicchia GP, Nico B, Camassa LM, Mola MG, Loh N, et al. (2004) The role of aquaporin-4 in the blood-brain barrier development and integrity: studies in animal and cell culture models. Neuroscience 129: 935-945. [Crossref]
- Sasaki H, Sato F, Mannen H (1989) Morphological analysis of single astrocytes of the adult cat central nervous system visualized by HRP microinjection. Brain Res 501: 339-354. [Crossref]
- Wang DD, Bordey A (2008) The astrocyte odyssey. Prog Neurobiol 86: 342-367. [Crossref]
- Kimelberg HK (1995) Current concepts of brain edema. Review of laboratory investigations. J Neurosurg 83: 1051-1059. [Crossref]
- R., B.. Injury and cell function. In: Reilly, P., R., B., editors. Head Injury. London: Chapman and Hall; 1997, p. 124.
- Sirotin YB, Hillman EM, Bordier C, Das A (2009) Spatiotemporal precision and hemodynamic mechanism of optical point spreads in alert primates. Proc Natl Acad Sci U S A 106: 18390-18395.[Crossref]
- Berwick J, Johnston D, Jones M, Martindale J, Martin C, et al. (2008) Fine detail of neurovascular coupling revealed by spatiotemporal analysis of the hemodynamic response to single whisker stimulation in rat barrel cortex. J Neurophysiol 99: 787-798. [Crossref]
- Kleinfeld D, Mitra PP, Helmchen F, Denk W (1998) Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proc Natl Acad Sci U S A 95: 15741-15746. [Crossref].
- Chaigneau E, Oheim M, Audinat E, Charpak S (2003) Two-photon imaging of capillary blood flow in olfactory bulb glomeruli. Proc Natl Acad Sci U S A 100: 13081-13086. [Crossref]
- Stefanovic B, Hutchinson E, Yakovleva V, Schram V, Russell JT, et al. (2008) Functional reactivity of cerebral capillaries. J Cereb Blood Flow Metab 28: 961-972. [Crossref]
- Buerk DG, Ances BM, Greenberg JH, Detre JA (2003) Temporal dynamics of brain tissue nitric oxide during functional forepaw stimulation in rats. Neuroimage 18: 1-9. [Crossref]
- Kobari M, Fukuuchi Y, Tomita M, Tanahashi N, Takeda H (1994) Role of nitric oxide in regulation of cerebral microvascular tone and autoregulation of cerebral blood flow in cats. Brain Res 667: 255-262. [Crossref]
- Preckel MP, Leftheriotis G, Ferber C, Degoute CS, Banssillon V, et al. (1996) Effect of nitric oxide blockade on the lower limit of the cortical cerebral autoregulation in pentobarbital-anaesthetized rats. Int J Microcirc Clin Exp 16: 277-283. [Crossref]
- Jones SC, Radinsky CR, Furlan AJ, Chyatte D, Perez-Trepichio AD (1999) Cortical NOS inhibition raises the lower limit of cerebral blood flow-arterial pressure autoregulation. Am J Physiol 276: H1253-1262. [Crossref]
- Jones SC, Easley KA, Radinsky CR, Chyatte D, Furlan AJ, et al. (2003) Nitric oxide synthase inhibition depresses the height of the cerebral blood flow-pressure autoregulation curve during moderate hypotension. J Cereb Blood Flow Metab 23: 1085-1095. [Crossref]
- Rosengarten B, Huwendiek O, Kaps M (2001) Neurovascular coupling in terms of a control system: validation of a second-order linear system model. Ultrasound Med Biol 27: 631-635.[Crossref]
- Owens T, Bechmann I, Engelhardt B (2008) Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol 67: 1113-1121. [Crossref]
- Boorman L, Kennerley AJ, Johnston D, Jones M, Zheng Y, et al. (2010) Negative blood oxygen level dependence in the rat: a model for investigating the role of suppression in neurovascular coupling. J Neurosci 30: 4285-4294. [Crossref]