The paper is based on the long discussed question of the origin of circumscribed necroses in glioblastoma multiforme. They were interpreted as an ischemic consequence of a vessel occlusion by thrombotic events or by an endothelial pathology. However, they were referred to the emergence of an avascular area in hyperproliferating zones of tumor, due to the imbalance between the high proliferation rate of tumor cells and the low one of endothelial cells. The hypoxia stimulates angiogenesis and microvascular proliferations can be found around to and at a distance from circumscribed necroses. Later on, with the diffusion of the concept that glioblastomas arise from tumor initiating cells or tumor stem cells, the location of the latter in the tumor was found to occur in perivascular and perinecrotic niches. These sites were recognized as responsible for tumor progression and proliferation. In the past we have already contributed in both fields. Presently we wanted to unify the two concepts showing that the origin of circumscribed necroses and the location of tumor stem cells in perinecrotic niches recognize the same pathogenetic mechanism. Our presentation is not at variance with previous ones, but it aims to be added as a further possible interpretation.
Two main types of necrosis occur in glioblastoma multiforme (GBM): large necroses of thrombotic origin, usually at the tumor center, and circumscribed necroses with pseudo-palisading. These are found in the proliferative areas of the tumor of which they represent the hallmark. Instrumental to necrosis development is hypoxia, variably spread throughout the tumor, to the point that it is a feature of it [1]. Hypoxia is mediated by Hypoxia-Inducible Factor (HIF)-1/2 that is composed of two subunits, an oxygen regulated HIF-α subunit and an oxygen insensitive HIF-β subunit [2]. Under normoxic conditions, HIF-α is rapidly degraded through hydroxylation by the oxygen-dependent prolyl-hydroxylase domain proteins (PHDs), that marks it for ubiquitination and proteasomal degradation [3]. Hypoxia stabilizes HIF-1α by preventing its hydroxylation and degradation, together with HIF-2α. HIF-2α remains elevated under chronic hypoxia, while HIF-1α is only transiently up-regulated [4]. It is of paramount importance that hypoxia is critically involved in the regulation of glioblastoma stem cells (GSCs) [5]. Through HIF-1α, it promotes the expansion of GSCs by the phosphatidylinositol 3-kinase (PI3K)/Akt and extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) pathways, the inhibition of which reduces the fraction of CD133+ GSCs [6]. GSCs are, therefore, activated in perinecrotic regions [5,7] and under hypoxic conditions, when Notch and its target genes Hes1 and Hey2 are successively activated [8]. Blockade of Notch signaling with γ-secretase inhibitors depletes the GSC population, reduces the expression of GSC markers such as CD133, Nestin, Bmi1 and OLIG2 and inhibits the growth of tumor neurospheres and xenografts [9]. Also the key stem cell genes such as Nanog, octamer-binding transcription factor 4 (Oct4) and c-Myc are activated [10]. GSCs can be demonstrated to lie around circumscribed necroses or scattered in the tissue by CD133 positivity or other specific antigens [5,11].
Necrosis is the consequence of hypoxia and to find it in the tissue means that HIF-1/2 must have been active, but this process needs time and it is possible that HIF-1/2 are activated by hypoxia when its consequences are not yet histologically visible in the tissue, but other processes may have been already elicited. An example is given by apoptosis, a frequent phenomenon in GBM. Apoptotic nuclei can be found either scattered in proliferating tumor areas, due to an intrinsic or transcriptional pathway via mitochondria, focused on p53 [12], or they crow in hypoxic areas around necrosis through an extrinsic pathway starting from Tumor Necrosis Factor (TNF), through APO-2 and TNF-related Apoptosis-Inducing Ligand (TRAIL) [13]. It is, however, possible that isolated apoptotic nuclei in a proliferating area are not due to the first type of apoptosis, i.e. the intrinsic one, but to the extrinsic type, consequence of the not yet morphologically evident hypoxia [14], as said before. As a matter of fact, HIF-1α expression can occur not only in or around circumscribed necroses, but also in scattered cells in proliferating areas [15].
Necrosis is the consequence of hypoxia and to find it in the tissue means that HIF-1/2 must have been active, but this process needs time and it is possible that HIF-1/2 are activated by hypoxia when its consequences are not yet histologically visible in the tissue, but other processes may have been already elicited. An example is given by apoptosis, a frequent phenomenon in GBM. Apoptotic nuclei can be found either scattered in proliferating tumor areas, due to an intrinsic or transcriptional pathway via mitochondria, focused on p53 [12], or they crow in hypoxic areas around necrosis through an extrinsic pathway starting from Tumor Necrosis Factor (TNF), through APO-2 and TNF-related Apoptosis-Inducing Ligand (TRAIL) [13]. It is, however, possible that isolated apoptotic nuclei in a proliferating area are not due to the first type of apoptosis, i.e. the intrinsic one, but to the extrinsic type, consequence of the not yet morphologically evident hypoxia [14], as said before. As a matter of fact, HIF-1α expression can occur not only in or around circumscribed necroses, but also in scattered cells in proliferating areas [15].
Circumscribed necroses in GBM are the hallmark of the tumor, but their origin and development have been the object of endless discussion. Recently, they have been carefully described and codified [16,17] as due to an ischemic process around an occluded vessel or with endothelial changes. The consequent hypoxia would stimulate angiogenesis, through HIF-1 and Vascular Endothelial Growth Factor (VEGF). In addition to this hypothesis, another one has been and can be advanced. Necroses may develop in hyperproliferating areas of the tumor, with a high Ki-67/MIB.1 Labeling Index (LI) and a high Nestin expression in comparison with Glial Fibrillary Acidic Protein (GFAP), as a consequence of a focal insufficiency of angiogenesis that becomes inadequate to feed a so large number of tumor cells, due to the imbalance between the high tumor cell proliferation capacity and the low one of endothelial cells [18,19]. This observation does not exclude that inside necroses regressive pathological vessels can occur. In GBMs, beside areas with a high vessel density due to an active neo-angiogenesis, large avascular areas can, therefore, occur where necroses develop. The perinecrotic palisades would be the remnants of the hyperproliferating area that escaped necrosis development.
It is currently known that GSCs are localized in perivascular and perinecrotic niches, expressing CD133, Musashi.1, Nestin, or specific antigens [5,11,20,21], activated by HIF-1. Regardless of the nature of these cells, i.e. whether they are real stem cells or progenitor cells with a stemness hierarchy and of their demonstration after or without sorting, they are also positive for Oct4 and Nanog [22,23] as well as for Sex-Determining Region Y (SRY)-box2 (SOX2) and RE-1-silencing transcription factor (REST) [15,24] (Figure 1). In our experience, they show the same features that characterize the majority of cells of the hyperproliferating areas. The cells of this areas represent the most malignant tumor phenotype after mutation accumulation and tumor microenvironment influence, and they may undergo an embryonic regression re-acquiring properties that are typical of stem cells/progenitors [15,25]. As perinecrotic palisadings could be the remnants of hyperproliferating areas spared by necrosis [15,26], so perinecrotic accumulation of GSC-like cells/progenitors could be the remnants of those that crowded hyperproliferating areas (Figure 2).
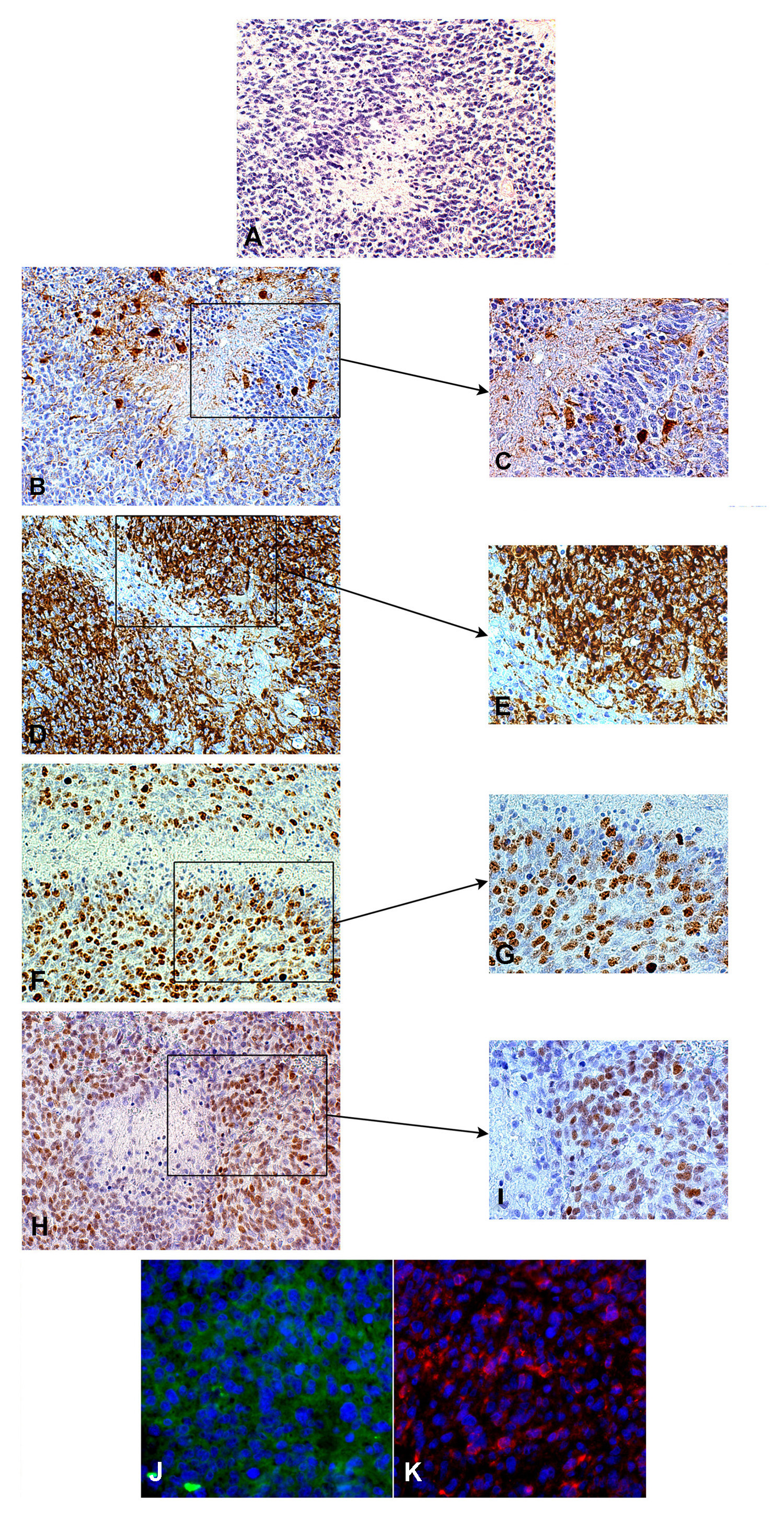
Figure 1. Circumscribed necrosis in a hyperproliferating area, H&E, 200x (A). The hyperproliferating zone bordering necrosis is almost GFAP-negative, GFAP, DAB, 200x (B) and 400x (C). The same area is highly Nestin-positive, Nestin, DAB, 200x (D) and 400x (E). The same area shows a high Ki-67/MIB.1 labeling index, Ki-67/MIB.1, DAB, 200x (F) and 400x (G). Id. for SOX2, SOX2, DAB, 200x (H) and 400x (I). The same area is positive for Musashi.1, cryostat section, immunofluorescence, 400x (J) and highly CD133-positive, cryostat sections, immunofluorescence, 400x (K).
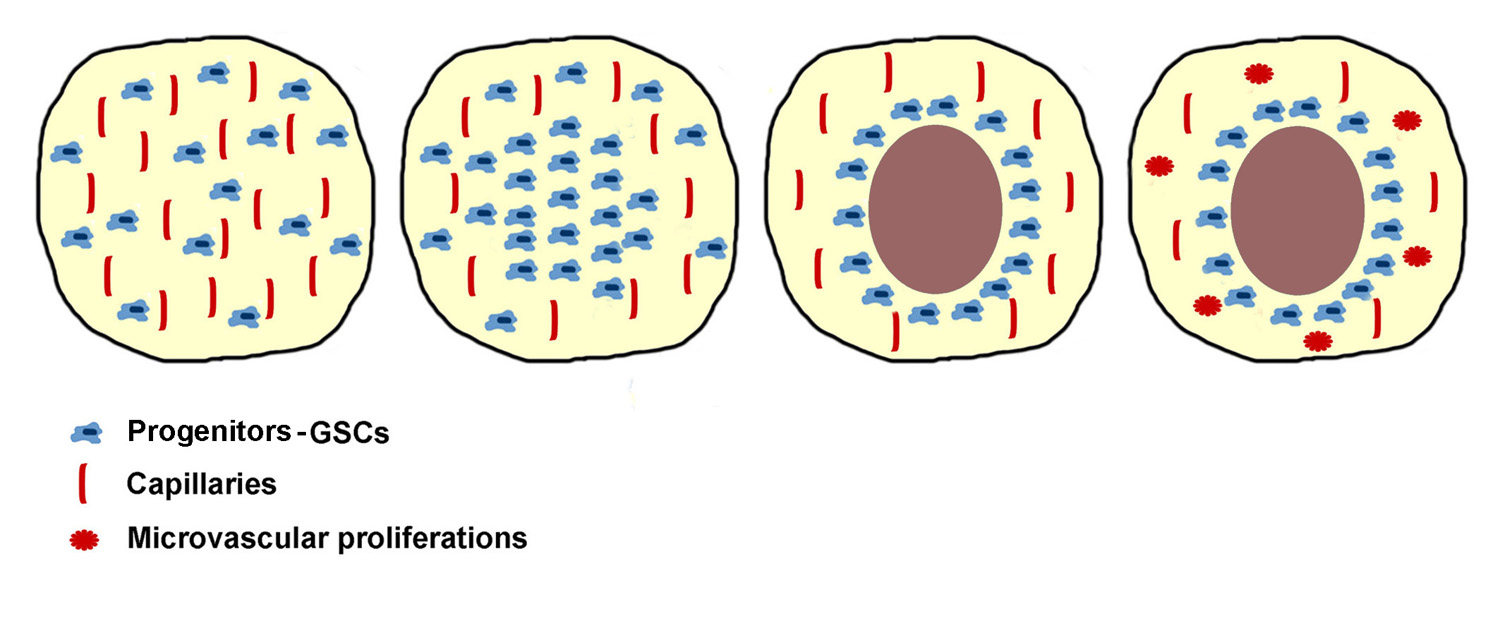
Figure 2. Scheme illustrating the development of a circumscribed necrosis in the avascular zone of a hyperproliferating area with stem cells/progenitors remaining to circumscribe it.
This interpretation is not at variance with those till now proposed for the origin of circumscribed necroses with pseudo-palisadings of GBM, but it adds a possible different understanding of the relationship between GBM and its GSC-like cells. There is a resemblance between our images of perinecrotic SOX2 distribution and that of CCAAT/enhancer
binding protein (C/EBP)-β and signal transducer and activator of transcription 3 (STAT3) found in mesenchymal class of GBM [27], activated by hypoxia and conditioning a bad survival. In culture of neural stem cells (NSCs) they prevent neural differentiation and trigger reprogramming toward an aberrant mesenchymal lineage and they are essential for mesenchymal transformation and glioma aggressiveness [28]. While STAT3 induces astrocyte differentiation and inhibits neuronal differentiation of neural stem/progenitor cells, C/EBP-β promotes neurogenesis and opposes gliogenesis. One wonders how the combined activity of C/EBP-β and STAT3 can be conceived to reprogram NSCs toward an aberrant lineage (mesenchymal)and to oppose the genesis of the normal neuronal and glial lineage. Maybe, their expression in human gliomas is essential to maintain the tumor initiating capacity and the ability to invade the normal brain [29].
The origin of GBM is still under discussion and practically only hypotheses are at our disposal. However, it remains established, since Penfield (1932) and Globus and Kuhlenbeck (1944) [26,30], that gliomas derive from immature glia. The most credited theory is that they derive from the transformed NSCs [31-33], regardless this transformation takes place in the subventricular zone (SVZ) or during migration. Glioma-initiating cells (GICs) and GSCs [34] share with NSCs some properties, i.e. proliferation and self-renewal, and GSCs share with malignant gliomas the genetic alterations.
A possible origin of gliomas is also from mature astrocytes by acquiring stemness properties through a dedifferentiation process, as above mentioned [35,36]. Recently, the hypothesis has been put forward that the origin could be from NG2+ cells that would fit better with tumors arising far from the ventricles such as oligodendroglioma, but also astrocytomas or secondary GBMs [37,38]. Also reactive astrocytes could be candidate for glioma origin [39,40], since they can acquire a stem-like phenotype [41]. GSCs may not represent a cell type, but rather a functional status [42,43], which can be acquired or lost depending on the microenvironment [25]. As stem cells/progenitors in the normal cytogenesis lose stemness properties during differentiation upon a complicated regulation mechanism, dedifferentiating malignant tumor cells can re-acquire them by embryonic regression, through a hierarchy of stem cell or progenitor status (Figure 3).
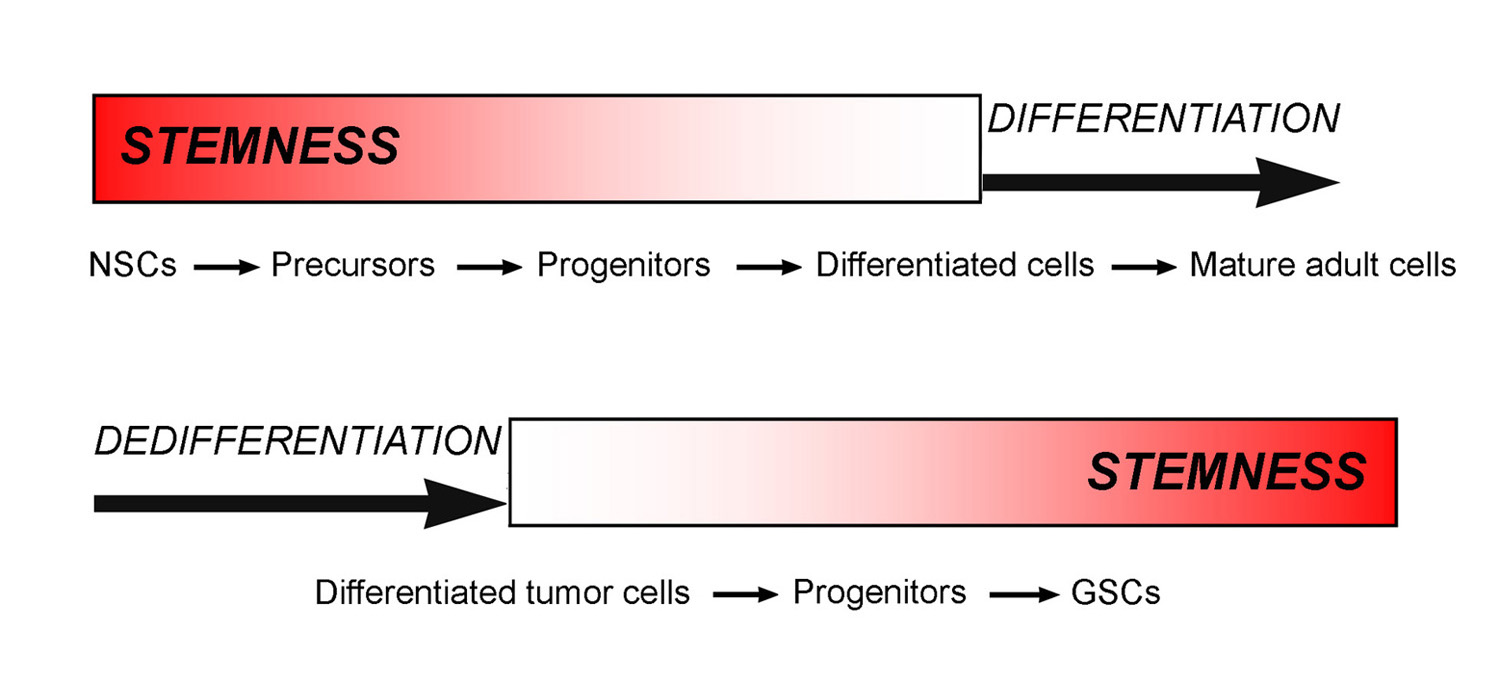
Figure 3. Scheme illustrating the acquisition during normal cytogenesis and the loss in tumor dedifferentiation of stemness properties.
This work was supported by Grant n. 4011 SD/cv 2011-0438 from Compagnia di San Paolo, Turin, Italy.
- Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang WT, et al. (2004) Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res 10: 8177-8184. [Crossref]
- Keith B, Johnson RS, Simon MC (2011) HIFα and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12: 9-22. [Crossref]
- Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92: 5510-5514. [Crossref]
- Holmquist-Mengelbier L, Fredlund E, Löfstedt T, Noguera R, Navarro S, et al. (2006) Recruitment t of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 10: 413-423. [Crossref]
- Seidel S, Garvalov BK, Wirta V, von Stechow L, Schänzer A, et al. (2010) A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 133: 983-995. [Crossref]
- Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, et al. (2009) Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 28: 3949-3959. [Crossref]
- Rankin EB, Giaccia AJ (2008) The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ 15: 678-685. [Crossref]
- Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG (2010) Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol 177: 1491-1502. [Crossref]
- Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, et al. (2010) NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28: 5-16. [Crossref]
- Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN (2009) The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8: 3274-3284. [Crossref]
- Christensen K, Schrøder HD, Kristensen BW (2008) CD133 identifies perivascular niches in grade II-IV astrocytomas. J Neurooncol 90: 157-170. [Crossref]
- Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281: 1309-1312. [Crossref]
- Ashkenazi A, Dixit VM (1999) Apoptosis control by death and decoy receptors. Curr Opin Cell Biol 11: 255-260. [Crossref]
- Mellai M, Schiffer D (2007) Apoptosis in brain tumors: prognostic and therapeutic considerations. Anticancer Res 27: 437-448. [Crossref]
- Schiffer D, Mellai M, Annovazzi L, Caldera V, Piazzi A, et al. (2014) Stem cell niches in glioblastoma: a neuropathological view. Biomed Res Int 2014: 725921. [Crossref]
- Fischer I, Gagner JP, Law M, Newcomb EW, Zagzag D (2005) Angiogenesis in gliomas: biology and molecular pathophysiology. Brain Pathol 15: 297-310. [Crossref]
- Rong Y, Durden DL, Van Meir EG, Brat DJ (2006) 'Pseudopalisading' necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol 65: 529-539. [Crossref]
- Schiffer D, Chiò A, Giordana MT, Mauro A, Migheli A, et al. (1989) The vascular response to tumor infiltration in malignant gliomas. Morphometric and reconstruction study. Acta Neuropathol 77: 369-378. [Crossref]
- Kargiotis O, Rao JS, Kyrits2021 Copyright OAT. All rights reservis in gliomas. J Neurooncol 78: 281-293. [Crossref]
- Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H (2012) The brain tumor microenvironment. Glia 60: 502-514. [Crossref]
- Filatova A, Acker T, Garvalov BK (2013) The cancer stem cell niche(s): the crosstalk between glioma stem cells and their microenvironment. Biochim Biophys Acta 1830: 2496-2508. [Crossref]
- Du Z, Jia D, Liu S, Wang F, Li G, et al. (2009) Oct4 is expressed in human gliomas and promotes colony formation in glioma cells. Glia 57: 724-733. [Crossref]
- Guo Y, Liu S, Wang P, Zhao S, Wang F, et al. (2011) Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology 59: 763-775. [Crossref]
- Conti L, Crisafulli L, Caldera V, Tortoreto M, Brilli E, et al. (2012) REST controls self-renewal and tumorigenic competence of human glioblastoma cells. PLoS One 7: e38486. [Crossref]
- Schiffer D, Mellai M, Annovazzi L, Piazzi A, Monzeglio O, et al. (2012) Glioblastoma cancer stem cells: Basis for a functional hypothesis. Stem Cell Discovery 2: 122-131. [doi: http://dx.doi.org/10.4236/scd.2012.23017].
- Penfield W (1932) Cytology and cellular pathology of the nervous system. New York: PB Hoeber.
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98-110. [Crossref]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, et al. (2010) The transcriptional network for mesenchymal transformation of brain tumours. Nature 463: 318-325. [Crossref]
- Cooper LAD, Gutman D, Chisolm C, Appin C, Kong J, et al. (2012) The Tumor Microenvironment Strongly Impacts Master Transcriptional Regulators and Gene Expression Class of Glioblastoma. Am J Pathol 180: 2108-2119. [Crossref]
- Globus JH, Kuhlenbeck H (1944) The subependymal plate (matrix) and its relationship to brain tumors of the ependymal type. J Neuropathol Exp Neurol 3:1-35.
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, et al. (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63: 5821-5828. [Crossref]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, et al. (2004) Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 64: 7011-7021. [Crossref]
- Visvader JE, Lindeman GJ (2008) Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8: 755-768. [Crossref]
- Singh SK, Clarke ID, Hide T, Dirks PB (2004) Cancer stem cells in nervous system tumors. Oncogene 23: 7267-7273. [Crossref]
- Sanai N, Alvarez-Buylla A, Berger MS (2005) Neural stem cells and the origin of gliomas. N Engl J Med 353: 811-822. [Crossref]
- Assanah M, Lochhead R, Ogden A, Bruce J, Goldman J, et al. (2006) Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci 26: 6781-6790.
- Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L (2009) Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 28: 2266-2275. [Crossref]
- Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, et al. (2010) Non-stem cell origin for oligodendroglioma. Cancer Cell 18: 669-682. [Crossref]
- Dufour C, Cadusseau J, Varlet P, Surena AL, de Faria GP, et al. (2009) Astrocytes reverted to a neural progenitor-like state with transforming growth factor alpha are sensitized to cancerous transformation. Stem Cells 27: 2373-2382. [Crossref]
- Silver DJ, Steindler DA (2009) Common astrocytic programs during brain development, injury and cancer. Trends Neurosci 32: 303-311. [Crossref]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, et al. (2008) Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A 105: 3581-3586. [Crossref]
- Zipori D (2004) The nature of stem cells: state rather than entity. Nat Rev Genet 5: 873-878. [Crossref]
- Vescovi AL, Galli R, Reynolds BA (2006) Brain tumour stem cells. Nat Rev Cancer 6: 425-436. [Crossref]