Abstract
Labs are routinely ordered for patients admitted to the emergency department (ED) for uncomplicated Vaso Occlusive Events (VOE), however, there are no “standard” screening practices. The objective of this study was to evaluate the utility of one measure, reticulocyte count, in cases of uncomplicated VOE in Sickle Cell Disease (SCD). To assess the value of screening reticulocyte count we completed a retrospective database and chart review of patients with SCD presenting in the ED with pain suggestive of VOE. The setting for the study was an urban tertiary hospital with an active sickle cell program including a dedicated Sickle Cell Center (SCC). SCD patients presenting in the ED are treated using a standardized clinical pathway (SCP). Over the eight-year study period, 346 patients with SCD presented in the ED with VOE pain and 1509 patient encounters were included in the study. No participants presented with reticulocytopenia as identified by the initial reticulocyte count. This study demonstrates that a reticulocyte count is not necessary in patients presenting with uncomplicated VOE without the presence of other complicating factors. Our results support a focused testing strategy utilizing reticulocyte count evaluation in complicated SCD presentations only. The results obtained in this study are in concordance with similar studies. The strength of this study lies in the long duration of evaluation and the use of a clinical pathway to screen for high-risk criteria in a prospective fashion. Even more restrictive testing strategies might be employed to further streamline the management of low risk patients, improve ED workflow processes, and reduce costs. One such strategy may be earlier evaluation of the serum hemoglobin via point-of-care testing at triage; however, further studies are needed to examine the reliability and practicality in applying such an approach.
Key words
sickle cell disease, vaso-occlusive events, reticulocyte count, screening practices
Introduction
Labs are routinely ordered for patients admitted to the emergency room for uncomplicated vaso-occlusive events (VOE), however, there are no “standard” screening practices. Review articles and expert opinions regarding routine testing vary widely in their recommendations [1-3]. In addition, recent literature has called into question the need for routine screening laboratory analysis in clinically uncomplicated VOE [1,4,5]. The objective of this study was to evaluate the utility of one measure, reticulocyte count, in cases of uncomplicated VOE in Sickle Cell Disease.
Background
Sickle Cell Disease (SCD) is the most commonly inherited genetic disease in the United States. Approximately 1 in 400 or an estimated 100,000 African Americans in the US have SCD [2,6]. Individuals with SCD are routinely encountered in the clinical practice of Emergency Medicine (EM) [2,3]. The most common reason patients with SCD seek treatment in the Emergency Department (ED) is for VOE, one of the hallmarks of SCD, which is characterized by severe pain that may affect any body part or system [2,3,5,7]. Triggers for VOE vary widely and can include infection, psychological stressors, and exercise. However, in more than 50% of cases, a discernible cause is not identified [2,4]. Other complications of SCD, some life threatening, may co-occur with VOE, such as infection. An uncomplicated VOE is defined as sickle cell pain without associated clinical indicators such as fever, acute chest syndrome, etc. The task of the Emergency Physician is to diagnose life-threatening complications while also treating the VOE [2,6,8]. Because many of the more serious complications present similarly to VOE or have nonspecific signs and symptoms, a high index of suspicion must be maintained as there is an increased risk of morbidity and mortality associated with delays in diagnosis and treatment [2,8].
While the pathophysiology of SCD is complex, it is generally agreed that it is a chronic inflammatory condition which results in varying degrees of anemia, hemolysis, and pain which are intimately tied to the distinctive changes in erythrocyte morphology and intravascular behavior [2,3,6,9]. This change causes a compensatory erythropoiesis to replace the red blood cells (RBCs), and subsequently introduces reticulocytes (immature RBCs) into the system. Individual reticulocytes are transient, lasting only a few days before maturation, but reticulocytes serve as a marker for bone marrow function [5,6]. When bone marrow fails to respond with increased RBC production the reticulocyte count drops, this condition is known as transient red cell aplasia, aplastic crisis, or reticulocytopenia [3]. When reticulocytopenia occurs, SCD patients are at risk for life-threatening anemia secondary to precipitous drops in hemoglobin from a baseline anemic state. In its most severe form, reticulocytopenia can lead to cardiovascular collapse and death [3,5,6]. Reticulocytopenia is one of several SCD-specific complications that must be considered during evaluation for VOE and is typically suspected only after laboratory values consistent with the diagnosis are seen [2,3,6,8]. Routine laboratory screening includes complete blood count (CBC) and reticulocyte count to evaluate for occult serious illness [4,8].
Methods
To assess the value of screening reticulocyte count we completed a retrospective database and chart review of SCD patients presenting in the ED with pain suggestive of VOE. The setting for the study was an urban tertiary hospital with an annual average ED census of 90,000 patients per year. Located within the hospital system is an active sickle cell program including a dedicated Sickle Cell Center (SCC) with a statewide outreach program.
SCD patients presenting in the ED are treated using a standardized clinical pathway (SCP) that utilizes a screening process to identify patients presenting with uncomplicated VOE. The inclusion and exclusion criteria for participation in the pathway are presented in Table 1. If the inclusion criteria are met and no exclusion criteria are present, the initial 24 hours of care is performed in the ED observation unit. If any exclusion criteria are present, the patient is admitted to the Hematology/Oncology inpatient service after initial evaluation by ED physicians. The observation unit treatment protocol uses patient controlled analgesia (PCA) delivered and oral narcotics. Predetermined endpoints of the SCP observation unit treatment include pain not adequately controlled within 24 hours despite protocol treatment and diagnosis of complicated presentation (development of exclusion criteria). Patients meeting these endpoints are admitted to the Hematology-Oncology service for inpatient care. All patients discharged from the SCP have a follow-up evaluation either by phone or in the SCC within 48 hours.
Inclusion Criteria |
Exclusion Criteria |
SCD (Hb-SS, -SC, and -Sβ-Thalassemia) |
Fever (>38.5 oC) |
Pain consistent with VOE |
Hypotension (BP <90/60) |
|
Hypoxia (oxygen saturation <90%) |
|
Pregnancy |
|
Altered mental status or other neurologic symptoms |
|
Pneumonia (defined as any new infiltrate identified on chest radiograph obtained due to clinical suspicion of the disease) |
Table 1. Inclusion and exclusion criteria for inclusion in the ED based Observation Unit Protocol.
As part of the observation unit’s ongoing quality assessment, a database was developed to track hospital admissions, resource utilization, and other service monitors such as time to care and discharge follow-up. This database was used to identify patients with SCD treated at our institution during the study period.
The study sample included all SCD patients seen in the ED over the eight-year study period. Patients who were not followed at the SCC were excluded from the study since follow-up data would not be reliably available for analysis. The primary outcome assessed was diagnosis of reticulocytopenia as defined by an initial reticulocyte count of <1%, or a diagnosis of reticulocytopenia either at discharge or at the subsequent follow up visit to the SCC. Initial reticulocyte count obtained on arrival to the ED was compared to the final diagnosis of reticulocytopenia. A single trained reviewer using a standardized data collection process reviewed charts. A second reviewer examined a 20% random sample of the data collected for accuracy. The study was approved by the Institutional Review Board.
Results
Over the eight-year study period 346 SCD patients presented in the ED with VOE pain. Limiting the analysis to those followed by the SCC during the study period yielded 192 patients. The 192 patients accounted for 1885 ED visits, an average of 9.8 ED visits over the study period per patient or approximately 1.2 visits per patient per year. Of the 1885 visits, 376 of these visits had an Observation Unit Protocol exclusion criterion. These 376 ED visits resulted in direct admissions. There were 1509 encounters treated in the ED using the Observation Unit Protocol (Figure 1). 677 of these encounters were admitted to the hospital and 832 were discharged to home.
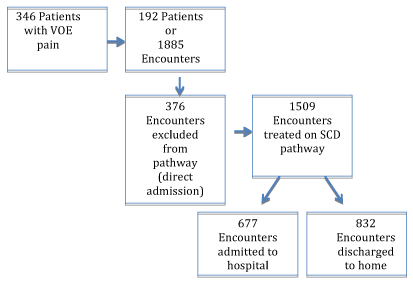
Figure 1. Patients' outcomes based on Observation Unit Protocol inclusion and exclusion criteria.
Of the 376 visits with an exclusion criteria, 5 (1.3%) were identified in the ED or during admission with reticulocytopenia (Table 2).
Patient |
WBC |
Hb |
Reticulocyte count |
Heart Rate |
High-Risk Criteria |
1 |
2.2 |
1.6 |
0.2 |
106 |
AMS |
2 |
12.7 |
8.6 |
14.3 |
95 |
Pneumonia |
3 |
6.7 |
10.4 |
4.6 |
114 |
Fever |
4 |
6.8 |
8.4 |
0.5 |
125 |
Fever |
5 |
8.7 |
10.8 |
6.4 |
114 |
Fever |
Table 2. Patients with high-risk criteria and a diagnosis of aplastic anemia.
Of the 1509 visits in the Observation Unit that did not have exclusion criteria, none presented with reticulocytopenia as identified by the initial reticulocyte count. Two patients developed reticulocytopenia after the initial Observation Unit treatment. Both of these patients had an elevated reticulocyte count during their ED evaluation and therefore did not meet the definition of reticulocytopenia. One of these patients presented with a hemoglobin of 8.3 mg/dl (baseline 14 mg/dl) and a reticulocyte count of 8.3%; the other presented with hemoglobin of 3.9 mg/dl (baseline 8.3 mg/dl) and a reticulocyte count of 4.7% (Table 3).
Patient |
WBC |
Hb |
Reticulocyte count |
Heart Rate |
High-Risk Criteria |
1 |
11.8 |
8.3 |
8.3 |
99 |
None |
2 |
37 |
3.9 |
4.7 |
102 |
None |
Table 3. Patients without high-risk criteria who developed aplastic anemia after admission to the hospital.
The results obtained in this study are in concordance with similar studies evaluating the usefulness of routine laboratory testing in the SCD population [1,4]. The strength of this study lies in the long duration of evaluation and the use of a clinical pathway to screen for high-risk criteria in a prospective fashion.
2021 Copyright OAT. All rights reserv
Discussion
The utility of the reticulocyte count is to differentiate between reticulocytopenia and splenic sequestration as the cause of acute worsening of anemia in SCD patients [6]. Once identified, the diagnosis of reticulocytopenia then serves as a proxy to signify a change in management between the two entities: reticulocytopenia requires inpatient evaluation with serial reticulocyte counts, possible transfusion, and consideration of exotic therapies while splenic sequestration does not mandate inpatient evaluation if patients respond to ED treatment [3]. In reality, however, these management strategies are practitioner-dependent and show great variability between providers and institutions. Assuming, however, that identification of reticulocytopenia does alter patient management, the question remains whether it is useful to obtain a reticulocyte count in low-risk patients as determined by clinical parameters. The true incidence of reticulocytopenia has not been well established, but recent literature suggests that it is quite rare and possibly declining secondary to newer maintenance treatment modalities in SCD, further limiting the utility of routine screening [3-5].
Bernard et al. assert that routine evaluation of the complete blood count and reticulocyte count rarely affects clinical decision-making in uncomplicated VOEs and that it is ineffective to screen for occult illness based on a few studies with very limited data [2,3,5]. Our results support this assertion. This study demonstrates that a reticulocyte count is not necessary in patients presenting with uncomplicated VOE without the presence of one or more exclusion criteria. When a patient is admitted to the hospital or when high-risk clinical criteria are present, evaluating for reticulocytopenia using a reticulocyte count is indicated. After reviewing the two additional patients that were diagnosed with reticulocytopenia after admission, this study suggests addition of a new exclusion criteria: an observed drop of hemoglobin of at least 2g/dL or the presence of severe anemia (defined as Hb <6 g/dL). The addition of this exclusion criterion is intuitive, as it would mirror the standard management upon identifying such a significant initial laboratory finding by prompting further workup. We found extremely low overall incidence of reticulocytopenia paired with evidence supporting the inability to identify reticulocytopenia in patients without other clinical features suggesting higher risk and thereby necessitating the need for expanded laboratory evaluation.
Limitations
This study has several limitations. First, the study included only adult patients and did not include pediatric patients. This was, however, intentional due to the structure of the SCP at the institution and recognition that similar evaluations have previously been conducted in a pediatric population [1]. Second, the sample represents a specific urban demographic and the generalizability of these results to other populations may be limited. Third, the incidence of reticulocytopenia identified was quite low and may have been falsely lowered secondary to exclusion of patients not following up at the SCC. Last, the analysis also sought to identify patients in whom management changes would likely be instituted based on abnormal laboratory findings only, but it is unclear whether identification of reticulocytopenia did indeed alter management in any significant way since this endpoint was not specifically assessed.
Conclusion
Increasing healthcare costs, expansion of health care coverage, and high cost of evaluation of low risk SCD patients with routine laboratory studies contribute to a significant burden on our health care system [10]. Our results support a focused testing strategy utilizing reticulocyte count evaluation in complicated SCD presentations only. The majority of these complicated presentations are identified using exclusion criteria that are easily applied during initial triage evaluation. The single remaining exclusion criterion may be applied after further risk stratification by an ED physician after completion of an appropriate history and physical. The newly suggested exclusion criterion of severe anemia or significant drop in hemoglobin may also be applied as a branch point which serves to identify the need for further testing. Even more restrictive testing strategies might be employed to further streamline the management of low risk patients, improve ED workflow processes, and reduce costs. One such strategy may be earlier evaluation of the serum hemoglobin via point-of-care testing at triage; however, further studies are needed to examine the reliability and practicality in applying such an approach.
References
- Chapman JI, El-Shammaa EN, Bonsu BK (2004) The utility of screening laboratory studies in pediatric patients with sickle cell pain episodes. Am J Emerg Med 22: 258-263. [Crossref]
- Glassberg J (2011) Evidence-based management of sickle cell disease in the emergency department. Emerg Med Pract 13: 1-20. [Crossref]
- Redding-Lallinger R, Knoll C (2006) Sickle cell disease--pathophysiology and treatment. Curr Probl Pediatr Adolesc Health Care 36: 346-376. [Crossref]
- Bernard AW, Venkat A, Lyons MS (2006) Best evidence topic report. Full blood count and reticulocyte count in painful sickle crisis. Emerg Med J 23: 302-303. [Crossref]
- Koshy M, Leikin J, Dorn L, Lebby T, Talischy N, et al. (1994) Evaluation and Management of Sickle Cell Disease in the Emergency Department (An 18-year Experience): 1974--1992. Am J Ther 1: 309-320. [Crossref]
- Brandow AM, Liem R (2011) Sickle Cell Disease in the Emergency Department: Atypical Complications and Management. Clin Pediatr Emerg Med 12: 202-212. [Crossref]
- Shapiro BS, Benjamin LJ, Payne R, Heidrich G (1997) Sickle cell-related pain: perceptions of medical practitioners. J Pain Symptom Manage 14: 168-174. [Crossref]
- Hampton R, Balasa V, Bracey SEA (2005) Emergencies in Patients With Inherited Hemoglobin Disorders—An Emergency Department Perspective. Clin Ped Emerg Med 6: 138-148.
- Conran N, Franco-Penteado CF, Costa FF (2009) Newer aspects of the pathophysiology of sickle cell disease vaso-occlusion. Hemoglobin 33: 1-16. [Crossref]
- Lanzkron S, Carroll CP, Haywood C Jr (2010) The burden of emergency department use for sickle-cell disease: an analysis of the national emergency department sample database. Am J Hematol 85: 797-799. [Crossref]