Abstract
Titanium dioxide (TiO2) is one of the most applied nanomaterial and widely used in different food and non-food products as additive or coating material. In vitro and in vivo studies have shown that TiO2 nanoparticles (NPs) induce cytotoxicity and inflammation in Caco-2 cells, but the detailed mechanisms how TiO2 NPs interact with the cell and mediate their inflammatory potential remains unclear. Our recent studies on Caco-2 cells have shown that exposure to TiO2 NPs induce an inflammatory response via an epidermal growth factor receptor (EGFR)-mediated pathway. In the current study we demonstrate that 10 nm TiO2 NPs induce the activation of extracellular signal-regulated kinases (ERK) and the expression of ERK target genes Chemokine (C-C motif) ligand 2 (CCL2) and Chemokine (C-X-C motif) ligand 3 (CXCL3). This activation is dependent on EGFR, as treatment of EGFR kinase inhibitor BIBX 1382 attenuates phosphorylation of ERK1/2 and the mRNA expression of CCL2 and CXCL3. Moreover, TiO2 NPs induce the activation of the transcription factor ELK1. We thus suggest TiO2 NPs induce an EGFR/ERK/ELK1-mediated pathway.
Key words
epidermal growth factor receptor, extracellular signal-regulated kinases, intestinal epithelial cells, titanium dioxide nanoparticles
Introduction
Titanium dioxide (TiO2) is one of the most applied nanomaterial and widely used in the food and non-food industry as additive or coating material [1]. TiO2 nanoparticles (NPs) are characterize by its high refractive index, photo catalytic activity and stability and thus used as white pigment in cosmetics, personal care products, paints, coatings, or in food and pharmaceuticals as additive E171. Additionally, TiO2 NPs are used in water and air purification or medical applications because of its photo catalytic and antibacterial attributes [2,3]. The growing production and wide range of applications of TiO2 NPs will cause an increasing release into the environment and is assumed to raise the exposure especially to the gastrointestinal tract [4]. Based on current contents in food and personal care products Weir and colleagues could estimate an exposure to TiO2 NPs of approximately 1 mg/kg*day in adults and up to 2−3 mg/kg*day in children [2].
Because of their ability to induce inflammation, cytotoxicity and genotoxicity in different cell types in vitro and in vivo [5] the effect of TiO2 NPs on gut homeostasis is a major concern [6] . Studies using the human colon carcinoma cell line Caco-2 have shown that TiO2 NPs induced oxidative damage [7] influenced metabolic activity and cytotoxicity [8,9] produced reactive oxygen species (ROS) [10,11] and finally induced the expression of Interleukin 8 (IL8) [12] in part, through the activations of nuclear factor (NF)-κB and p38 mitogen activated protein kinase (MAPK) pathways [13] . Little is known about the detailed mechanisms how TiO2 NPs interact with the cell and mediate their inflammatory potential. Several studies discuss a possible activation of toll-like receptors (TLR) [14] or suggest an endocytic uptake [15-18] since NPs have been detected intracellular [19-21]. Moreover, our recent findings have shown that cellular response to TiO2 NPs in Caco-2 cells are dependent on the activation of epidermal growth factor receptor (EGFR), mediated by endocytosis-related structures [12]. The underlying precise signaling cascade between EGFR activation and expression of different inflammatory markers in intestinal epithelial cells remains unknown. Studies on various cell lines declare effects to TiO2 NPs on signaling pathways such as p38 MAPK and extracellular signal-regulated kinases 1/2 (Erk 1/2) in neutrophils [22]. c-Jun N-terminal kinases (JNK)/p38-caspase-8-Bid pathway in lymphocytes [23] . NF-κB pathway in kidney cells of mice [24] apoptotic pathways in bronchial epithelial cells via activation of caspase-8 and -3, -7 [25] and mitochondrial-mediated pathways in mouse splenocytes [26]. In macrophage-like cells TiO2 NPs response is depended on active cathepsin B [27] and in breast, lung and liver cells by Fas upregulation and Bax activation [28] . Induction of these pathways and expression of downstream genes such as IL8 or IL-1β, are initiated by the generation of ROS or via binding of TiO2 NPs to TLR4 at the cell membrane and further activation of NF-κB [14,29]. Besides these findings, studies using different types of NPs have shown an activation of the ERK signaling pathway [30,31] with a previous activation of EGFR in epithelial cells [32-36].
So far, there has been no evidence for TiO2 NP-induced inflammatory response through EGFR activation and further induction of ERK signaling pathway and downstream mediators in Caco-2 cells. In the current study, Caco-2 cells were exposed to 10 nm TiO2 NPs which induce a transient activation of ERK1/2 and the ERK-related transcription factor ELK1. This activation was accompanied with an increased expression of Chemokine (C-C motif) ligand 2 (CCL2) and Chemokine (C-X-C motif) ligand 3 (CXCL3) genes. We could further demonstrate that TiO2 NP-induced ERK1/2 signaling is mediated through EGFR activation and increased after IκB-α inhibition.
Material and methods
Preparation of TiO2 NPs
The preparation and characterization of the used TiO2 NPs were described in our previous study [13]. Briefly, titanium (IV) dioxide (anatase) particles were obtained from Alfa Aesar (Alfa Aesar GmbH & Co KG, Germany). Particles were used with a nominal size of 10 nm (Stock Number 44690, Lot B19T020; specific surface area 120 m2 g-1) and a measured size of 14.4 nm [13]. The particles form agglomerates in a size of 673 × 455 nm [13]. Stock solution was prepared with deionized water to a final concentration of 2 mg ml-1. The dispersion was sonicated at 23 KHZ and 150 W (MSE Ltd, United Kingdom) for two minutes and finally autoclaved. Immediately before treatment of the cells, the dispersion was diluted in cell culture medium as indicated below.
Cell culture
Human colon adenocarcinoma cell line Caco-2 (Toni Lindl, Munich, Germany) between passage 34 and 52 were used in this study. Cells were cultured in Caco-2 medium (45% Dulbecco’s Modified Eagle Medium (DMEM), low glucose, 45% Ham’s F12, 9% FCS, 0.9% non-essential amino acids (all PAA Laboratories GmbH, Austria) and Insulin (10 µg ml-1) (Biochrome AG, Germany)) at 37°C and 5% CO2.
Western blot analyses
Caco-2 cells were seeded in 6-well plates (8.87 cm-2 per well) (Sarstedt, Germany) at a density of 1-2×10E5 cells cm-2. At confluency, prepared medium with 10 nm TiO2 NPs were added to the cells at a final concentration of 40 μg cm-2 cell growth surface or remain untreated. After indicated time cells were washed twice with cold PBS and lysed 20 min on ice in RIPA buffer (Sigma-Aldrich, U.S.) added with inhibitors (1 mM PMSF, 1 mM Na3VO4, 1x complete protease inhibitor cocktail (Santa Cruz Biotechnology, U.S.), 1x phosphatase inhibitor cocktail 3 (Sigma-Aldrich, U.S.)). Lysates were clarified by centrifugation (12.000 g for 6 min) and protein concentration was measured using bicinchoninic acid assay (BCA) reagent. 40 μg protein from each preparation were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (Carl Roth GmbH + Co.KG, Germany). Membranes were blocked in 5% (w/v) BSA/TBST (20 mM Tris–HCl (pH 7.6), 150 mM NaCl, and 0.1% (v/v) Tween 20) for 1 h at room temperature. Primary antibody incubation p-ERK1/2 1:200 (sc-7383, Santa Cruz Biotechnology, U.S.) in 5% BSA/TBST took place at 4°C overnight, followed by secondary antibody incubation 1:500 in TBST (anti-mouse HRP (PAB10782, Abnova, Taiwan)) for 2 h at room temperature. The antibody was detected with enhanced chemiluminescence (ECL) reagent (MBL International Corporation, U.S.) and visualized with Fusion solo S (Vilber Lourmat Deutschland GmbH, Germany). Densitometry of the bands was analyzed using FusionCapt Advance Solo 4s software and the ratio of p-ERK to total protein, detected by ponceau staining, determined. Expressions of the treated samples were set in relation to the untreated control sample. Experiments were performed at least three times.
Electrophoretic mobility shift assay (Emsa)
Binding of ELK1 was investigated by non-radioactive EMSA using LightShift EMSA Optimization and Control Kit (Thermo Fisher Scientific Inc., U.S.) according to manufacture’s protocol. In detail, Caco-2 cells were seeded in 6-well plates (Sarstedt AG & Co, Germany) at a density of 1-2×10E5 cells cm-2. At confluency, cells were stimulated with 10 nm TiO2 NPs at a final concentration of 40 μg cm-2 cell growth surface or remain unstimulated. After indicated time cells were washed twice with ice cold PBS and lysed 20 min on ice in lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 1 mM PMSF, 1x complete protease inhibitor cocktail (Santa Cruz Biotechnology, U.S.)) adapted from [37]. Cells were scraped, placed in a microlitre tube and centrifuged 20 min at 11.000 g at 4°C to separate cytosolic and nuclear fractions. Supernatant was decanted and pellet was suspended in extraction buffer to lyse nuclei (20 mM HEPES, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25 % (v/v) glycerol, 1 mM DTT, 1 mM PMSF, 1x complete protease inhibitor cocktail, [37]. 2 µg of nucleus extraction were used for binding reaction with biotin-labeled DNA for ELK1: 5'-TTTGCAAAATGCAGGAATTGTTTTCACAGT-3' [38]. Samples were separated by 6 % native polyacrylamide gel and transferred to a nylon membrane (GE Healthcare, U.K.). Biotin were detected with peroxidase-coupled streptavidin and ECL (Thermo Fisher Scientific Inc., U.S.) and visualized and quantified with Fusion solo S (Vilber Lourmat Deutschland GmbH, Germany). Densitometry of the binding complex was analyzed using FusionCapt Advance Solo 4s software and expressions of the treated samples were set in relation to the untreated control sample. Experiments were performed at least three times.
Immunocytochemistry
Caco-2 cells were seeded in removable 12-well chambers (ibidi GmbH, Germany) at a density of 1-2×10E5 cells cm-2. At confluency prepared medium with 10 nm TiO2 NPs were added to the cells at a final concentration of 40 μg cm-2 cell growth surface or remain untreated. After indicated incubation time cells were washed with PBS and fixed with cold 4 % paraformaldehyde in PBS for 30 min. After washing, cells were permeabilized with 0.1% Trion X-100 in PBS for 10 min and blocked with 10% donkey serum in PBS for 1 h. Primary antibody p-ERK1/2 (sc-7383, Santa Cruz Biotechnology, U.S.) was diluted 1:100 in 10% donkey serum in PBS and cells were incubated overnight at 4°C. Following PBS wash, cells were incubated 1 h at RT with secondary antibody Alexa Fluor®546 (A10036, Molecular Probes®, U.S.) 1:200 in 10% donkey serum in PBS. After washing with PBS cells were mounted with Dapi-Fluoromount-G™ clear mounting media (Southern Biotech™, U.S.). Pictures were acquired with a confocal laser scanning microscope TCS SP8 (Leica Microsystems GmbH, Germany).
m RNA expression analysis
For mRNA expression analysis cells were seeded in 24-well or 6-well culture plates (Sarstedt AG & Co., Germany) at a density of 1-2×10E5 cells cm-2. After reaching confluency, 10 nm TiO2 particles were added at a final concentration of 40 μg cm-2 cell growth surface or remain untreated. At indicated time points cells were washed twice with PBS and proceed to RNA extraction. RNA was extracted using the GeneJET RNA Purification Kit (Thermo Fisher Scientific GmbH, Germany) and first strand cDNA synthesis was prepared using RevertAid™ H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific GmbH, Germany) as described by the manufacturer. Real-time PCR was performed on a 7500 Real-Time PCR Systems (Life Technologies Inc., U.S.) using HOT FIREPol® EvaGreen® qPCR Mix Plus ROX (Solis BioDyne, Estonia). Sequences of primers (used at 0.2 µM) were as follows: B2M (Beta-2 microglobulin) forward: GCAAGGACTGGTCTTTCTATCT, reverse: TAACTATCTTGGGCTGTGACAAA; CCL2 (Chemokine (C-C motif) ligand 2) forward: CCCAAAGAAGCTGTGATCTTCA; reverse: TCTGGGGAAAGCTAGGGGAA, CXCL3 (chemokine (C-X-C motif) ligand 3) forward: CCCAAACCGAAGTCATAGCCA, reverse: ACCCTGCAGGAAGTGTCAA; GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) forward: AGAGCACAAGAGGAAGAGAGAG, reverse: GGTTGAGCACAGGGTACTTTATT. Parameters for qPCR were: 95°C for 15 min, 40 cycles of 10 s 95°C, 30 s 60°C and 30 s 72°C. After cycling, melting curve analysis was performed. Expressions of the different genes was normalized to the expressions of GAPDH and B2M and compared to control using delta delta Ct method [39]. Experiments were performed four times.
Cells treatment
Inhibitor of IκB-a phosphorylation Bay11-7082 (InvivoGene, USA), inhibitor of p38α and p38β MAPK SB202190 (InvivoGene, USA) and inhibitor of EGFR kinases BIBX 1382 (Santa Cruz Biotechnology, Inc., U.S.) were dissolved in DMSO to obtain a stock solution at 50 mM. Epidermal growth factor (EGF) at a concentration of 100 ng ml-1 was used as positive control in the EGFR/ERK signaling pathway as described [40]. Confluent monolayer of Caco-2 cells were incubated for 1 h with 50 µM BIBX 1382, 5 µM SB202190 or 30 min with 50 µM Bay11-7082 and subsequently stimulated with NPs, EGF or left unstimulated. After treatment for indicated time, cells were washed twice with cold PBS and processed as described above. Results of the treated cells are presented as fold change to untreated cells.
Data analysis
Results are presented as means ± standard error (SEM). Treated samples are presented as fold change to untreated controls. Statistical analysis was carried out using GraphPad Prism 4 software (GraphPad Software, Inc., U.S.) and comparisons of the means were performed by ANOVA followed by Tukey's multiple comparison test. Differences were considered to be statistically significant at p<0.05.
Results and discussion
Nanosized TiO2 particles and their impacts on in vivo and in vitro systems are currently researched intensively in biomedical studies. There are different effects described on cellular level in various cell types. TiO2 NPs shown an impact on oxidative stress, inflammation or apoptosis [5], but the precise cellular mechanisms are still unknown. Several studies discuss a possible activation of toll-like receptors (TLR) [14,29] or suggest an endocytic uptake [15-18] since NPs have been detected intracellular [19- 21]. Moreover, our recent findings have shown that cellular response to TiO2 NPs in Caco-2 cells are dependent on the activation of epidermal growth factor receptor (EGFR), mediated by endocytic-related structures [13]. Activation of EGFR initiates the induction of downstream signaling pathways, including ERK, JNK and p38 MAPK pathways [41,42]. The present study attempted to provide an insight of the signaling pathway responsible for the NP-induced inflammatory response.
TiO2 NPs activate ERK1/2 kinases
ERK is part of the Ras/Raf/MEK/ERK signal transduction cascade, which is activated following stimulation by various extracellular factors besides EGF like insulin, other growth factors, osmotic stress, cytokines or trans membrane G-protein coupled receptors [43]. To investigate the role of ERK in the NP-induced inflammatory response, p-ERK expression was determined by western blot analyses and immunocytochemie. As shown in Figure 1 expression of p-ERK1/2 in Caco-2 cells was induced after treatment with 10 nm TiO2 NPs compared to untreated control at any time point (Figure 1A to F). Western blot analyses revealed exposure to 10 nm TiO2 induce a transient activation of p-ERK1/2 (Figure 2A). Levels of p-ERK1/2 consequently increased in a time dependent manner until 60 min after NPs exposure and is significantly induced at 60 min by 10.2±3.1 compared to untreated control cells (Figure 2B). Activation of ERK1/2 faded 90 min after NPs exposure. It was shown EGF treatment increase the levels of p-ERK1/2 peaking at 2, 5 and 10 min [44]. Therefore the observed induction of ERK1/2 by NPs is slightly slower than with EGF. If the NP-induced ERK1/2 activation resulted from an activation of EGFR or another mechanism is not known. To go more into detail in this issue we further used inhibitors which intervene the ERK and NF-κB pathways.
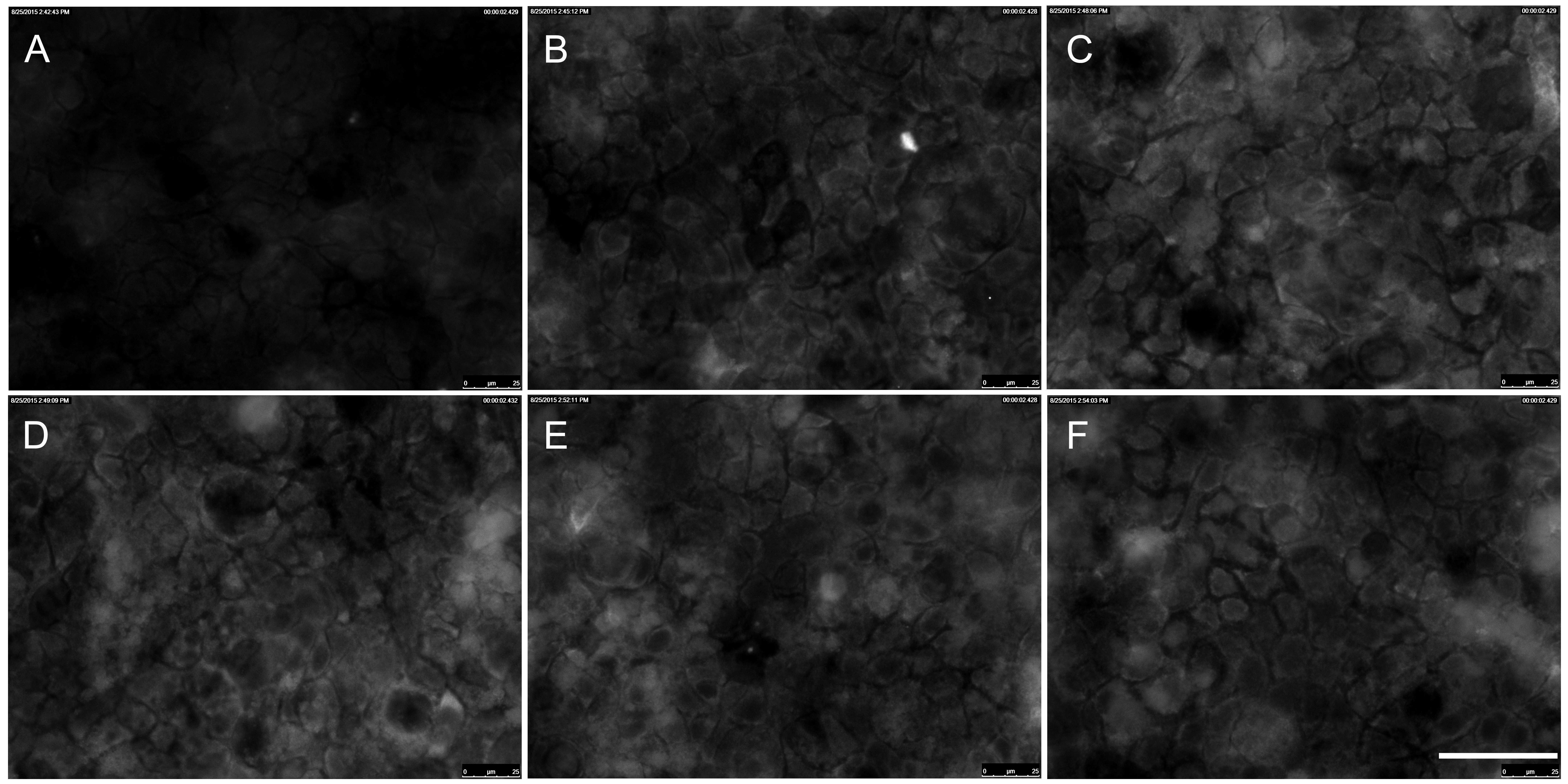
Figure 1. Immunocytochemical staining of p-ERK1/2 in Caco-2 cells exposed to TiO2 particles. Caco-2 cells were stimulated with 10 nm TiO2 (concentration of 40 µg cm-2 of the cell growth surface) for A – 0 min = untreated control, B – 15 min, C – 30min, D – 45 min, E – 60 min, F – 90 min. Scale bar corresponds to 50 µm.
Activation of EKR1/2 is dependent on EGFR
To analyses signal transduction of the NP-induced inflammatory response, we used three different inhibitors after stimulation with 10 nm TiO2 and EGF. In our recent studies we have shown that NP-induced inflammatory response was dependent on EGFR, NF-κB and p38 MAPK activation [13].
It is known activation of EGFR leads to the phosphorylation of ERK1/2 [41]. As shown by western blot analysis p-EKR1/2 was induced 30 min after NPs and EGF stimulation by factor 2.5 ± 0.4 and 4.2 ± 0.9 compared to untreated control cells (Figure 3A). Densitometric analysis of western blots revealed that treatment with BIBX1382 (EGFR inhibitor) lead to a decreased p-ERK1/2 by 68 % in NP-treated cells and by 83 % in EGF-treated cells compared to the stimulated specimens without inhibitor treatment (Figure 3B). These results shown that NP-induced activation of ERK1/2 is dependent on activation of EGFR. Study with ultrafine carbon black particles support our observations, which shown a transactivation of EGFR and further stimulation of ERK in human bronchial epithelial cells [45].
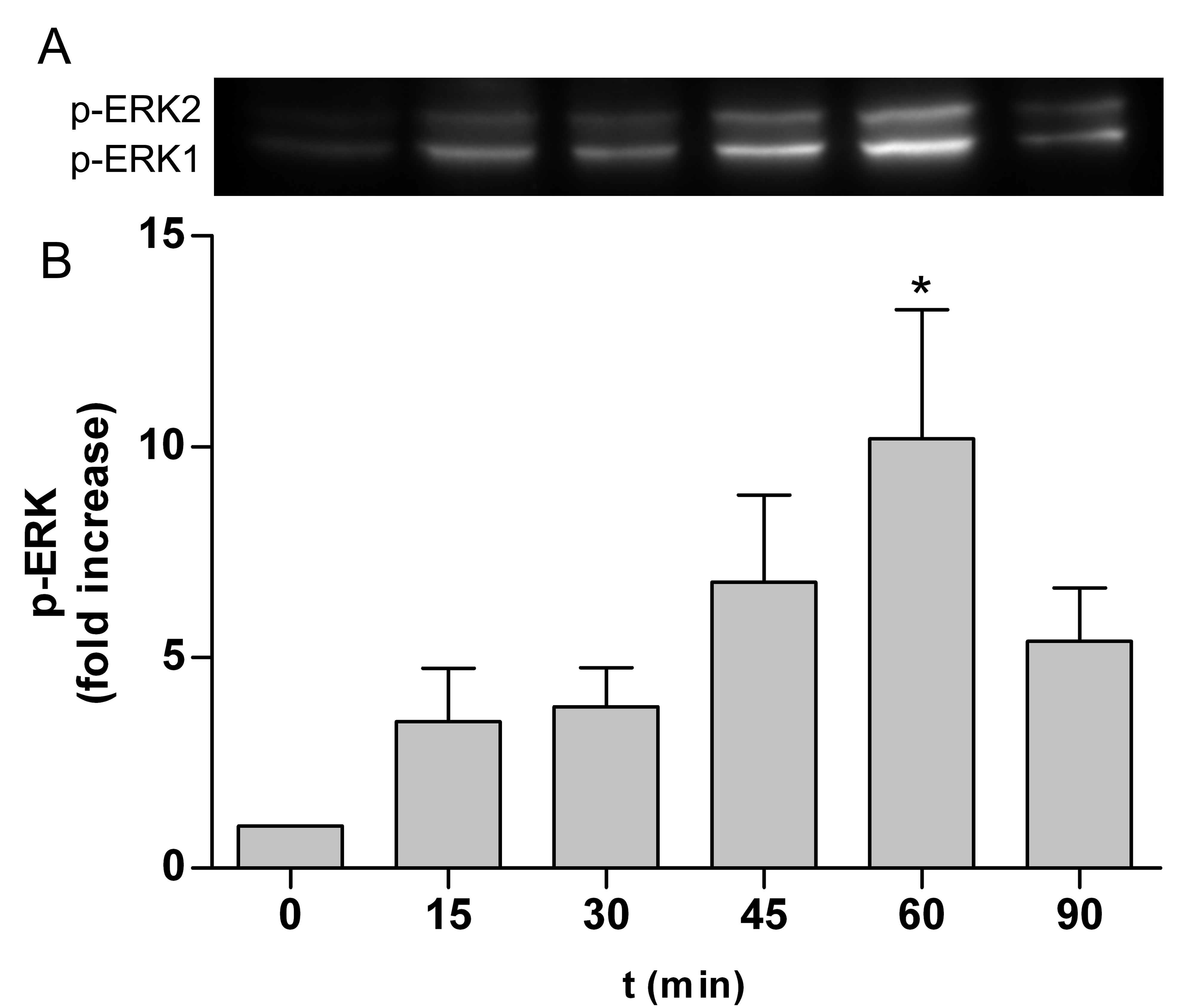
Figure 2. TiO2 NPs induced activation of p-ERK1/2. Caco-2 cells were stimulated with 10 nm TiO2 (concentration of 40 µg cm-2 of the cell growth surface) for 15, 30, 45, 60 and 90 min. A – Phosphorylated ERK1/2 was evaluated by western blot analyses. B – Samples are presented as fold change to unstimulated sample = 0 min. Expression of p-ERK1/2 was normalized to total protein expression measured by densitometry. Shown are representative western blots of four independent experiments. Data presented the mean ± SEM. *p<0.05 significantly different to untreated control, One-way ANOVA followed by Tukey's multiple comparisons test.
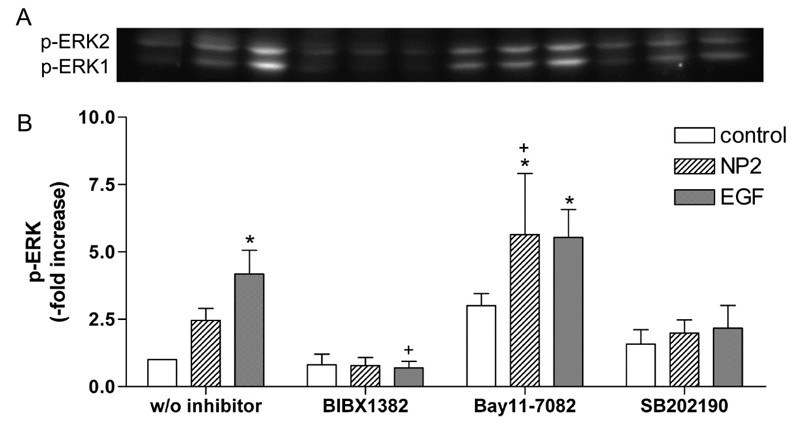
Figure 3. Effects of different inhibitors on NP- and EGF-induced ERK1/2 phosphorylation. Caco-2 cells were pre-treated with 50 µM BIBX 1382, 5 µM SB202190 or 50 µM Bay11-7082 and exposed to 10 nm TiO2 (co<ncentration of 40 µg cm-2 of the cell growth surface), 100 ng ml-1 EGF for 30 min or remained untreated (w/o inhibitor). A – Phosphorylated ERK1/2 was evaluated by western blot. B – Densitometric analyses of the treated samples are presented as fold change to the untreated control sample. Expression of p-ERK1/2 was normalized to total protein expression. Shown are representative western blots of three independent experiments. Data presented the mean ± SEM. Two-way ANOVA followed by Tukey's multiple comparison test (p<0.05). Asterisks (*) represent significant differences to the untreated control w/o inhibitor, plus (+) represent significant differences to NP2- or EGF-treated cells w/o inhibitor, respectively.
As published earlier, the NP-induced inflammatory response can be inhibited by BAY11-7082 [13]. Using the same inhibitor at identical concentration, which inhibited the phosphorylation of IκB-α, we shown that p-ERK1/2 is elevated in comparison to untreated cells. Inactivation of IκB-α phosphorylation by BAY11-7082 resulted in a significant increase of p-ERK1/2 in NP2- and EGF-treated specimens by factor 5.7 ± 2.3 and 5.5 ± 1.0, respectively (Figure 3A and 3B). Thus manipulation of the NF-κB pathway confound ERK signaling through a possible feedback mechanism. Different studies already demonstrated interactions between NF-κB and ERK signaling pathway [46- 48]. Having shown p38 MAPK is also involved in NP-induced inflammatory response [13], we analyze whether p38 MAPK is required for NP-induced ERK activation. Treatment with p38α and p38β MAPK inhibitor SB202190 did not show an influence on NP-induced activation of ERK1/2 (Figure 3A and B). Activation of p38 MAPK is therefore not required for the NP-induced activation of ERK1/2, but is involved in the NP-induced expression of IL8 mRNA as shown in our previous study [13]. Thus, results demonstrate exposure to 10 nm TiO2 NPs induce the activation of both p38 MAPK and NF-κB/ERK pathways. If this activation is responsible for the induction of ERK-related gene expression was further investigated.
TiO2 NPs induced binding of ELK1
The best-studied nuclear target of phosphorylated ERK is the transcription factor ELK1 [28] Stimulation with 10 nm TiO2 increased the binding of ELK1 to ELK1-specific biotin-labeled oligos up to 45 min after TiO2 exposure (Figure 4A) and declined 60 min after NPs treatment. Densitometric analyses revealed a 1.8 ± 0.5-fold increase of the binding complex compared to untreated control. This time course and amount is typical for activated ELK1 [49].
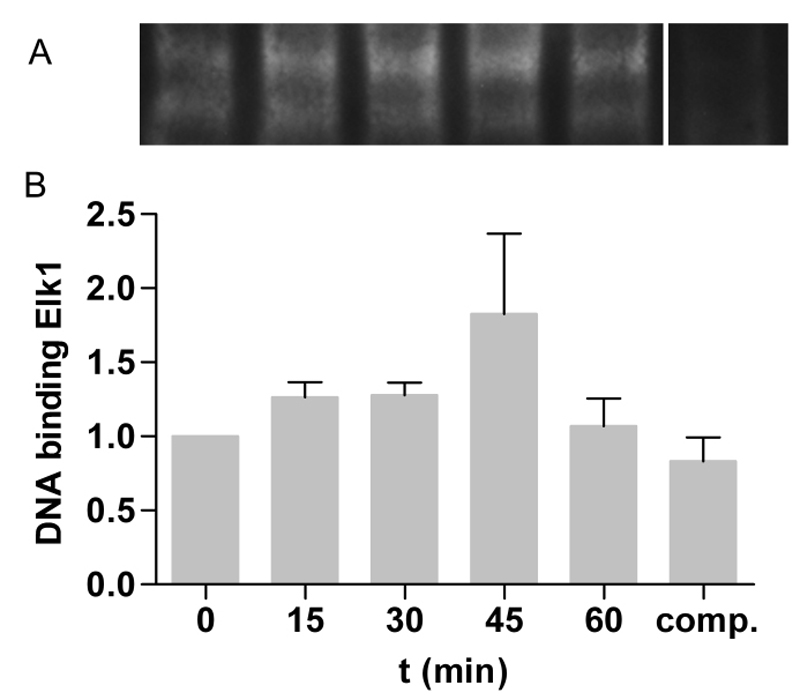
Figure 4. TiO2 NPs induced binding capacity of ELK1. A – Caco-2 cells were stimulated with 10 nm TiO2 (concentration of 40 µg cm-2 of the cell growth surface) for 15, 30, 45 and 60 min. ELK1 binding was measured by EMSA and shown as binding complex of ELK1 and ELK1-specific biotin-labeled oligos. For competition reaction (comp.) 1000-fold molar excess of unlabled oligos were used. B – Binding capacity was quantified by densitometry and treated samples are presented as fold change to untreated control. Shown are representative EMSA of three independent experiments. Data presented the mean ± SEM.
NP-induced activation of EGFR/ERK/ELK1 signaling pathway is related to the expression of CCL2 and CXCL3
It has been shown that activation of ERK1/2 induce the expression of CCL2 and CXCL3 mRNA [50]. Expression analysis revealed an increase by factor 11.0 ± 2.6 and 12.6 ± 5.3 3 h after NPs exposure of CCL2 and CXCL3 mRNA, respectively (Figure 5A and B). Pre-incubation with EGFR kinase inhibitor BIBX 1382 decreased the NP-induced expression by 77 % and by 70 % of CCL2 and CXCL3 mRNA. 6 h after NPs treatment mRNA expression of CCL2 and CXCL3 was induced by 4.8 ± 0.4-fold and by 3.3 ± 0.5-fold, respectively. Treatment with BIBX 1382 resulted in a 54% and 58% reduction of NP-induced expression of these genes. The results demonstrate that NP-induced expression of ERK target genes is dependent on the activation of EGFR. It was shown that the expression of CXCL3 and CCL2 mRNA was increased after different inflammatory stimuli and was part of an inflammatory response, mediated by EGFR [51,52]. EGFR-dependent ERK1/2 activation and downstream signaling shown regulatory effects on cellular transformation, intestinal cell migration and wound healing [50,53-55]. Moreover, Unfried et al. demonstrated that carbon NPs induce proliferation via ERK1/2 signaling in lung epithelial cells [56]. This activation was dependent on EGFR. If the TiO2 NP-induced activation of these pathways in Caco-2 cells promote cell migration and transformation remains unknown. Further experiments are needed to clarify this.
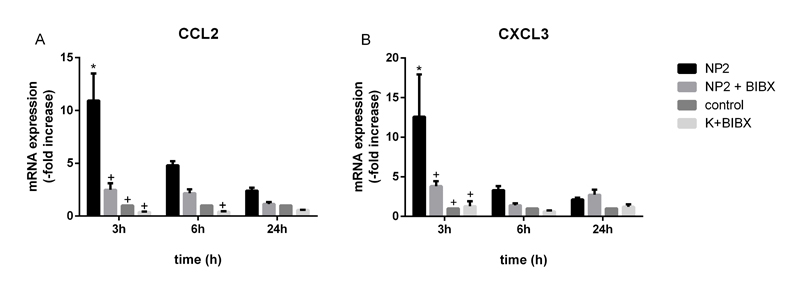
Figure 5. mRNA expression analyses of CXCL3 and CCL2 in Caco-2 cells. Caco-2 cells were pre-treated with 50 µM BIBX 1382 and exposed to 10 nm TiO2 (concentration of 40 µg cm-2 of the cell growth surface), or remained untreated for indicated time. A – Expression of CCL2 mRNA and B – expression of CXCL3 mRNA was determined and values of the treated samples are presented as fold change to the untreated control sample at the same time point. Data presented the mean ± SEM of ≤ 3 independent experiments. Two-way ANOVA followed by Tukey's multiple comparisons test (p<0.05). Asterisks (*) represent significant differences to untreated control, plus (+) represent significant differences to NP2-treated cells of the same time point.
Conclusion
We demonstrated that exposure to 10 nm TiO2 NPs induced the activation of ERK1/2 and the expression of ERK1/2 target genes Chemokine (C-C motif) ligand 2 (CCL2) and Chemokine (C-X-C motif) ligand 3 (CXCL3). This cellular response to TiO2 NPs is dependent on the activation EGFR. Furthermore, TiO2 NPs induce the activation of the transcription factor ELK1 and thus suggest an EGFR/ERK/ELK1 signaling pathway. IκB-α was shown to interact in the NP-induced ERK1/2 signaling cascade as treatment with inhibitor BAY11-7082 increase the NP-induced phosphorylation of ERK1/2.
Declaration of interest
The authors declare that they have no competing interests.
Acknowledgements
We kindly thank Silvia Kaschner and Kevin Pohl for excellent technical support. This work was supported by the German Federal Ministry of Food and Agriculture.
References
- Schrand AM, Rahman MF, Hussain SM, et al. (2010) Metal-based nanoparticles and their toxicity assessment. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2: 544–568.
- Weir A,Westerhoff P, Fabricius L, Hristovski K, von Goetz N (2012) Titanium dioxide nanoparticles in food and personal care products. Environ Sci Technol 46: 2242-2250. [Crossref].
- Pelaez M, Nolan NT, Pillai SC, et al (2012) A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl Catal B Environ 125: 331–349.
- Gottschalk F,Sonderer T, Scholz RW, Nowack B (2009) Modeled environmental concentrations of engineered nanomaterials (TiO(2), ZnO, Ag, CNT, Fullerenes) for different regions. Environ Sci Technol 43: 9216-9222. [Crossref].
- Shi H, Magaye R, Castranova V, Zhao J (2013) Titanium dioxide nanoparticles: a review of current toxicological data. Part Fibre Toxicol 10: 15. [Crossref].
- Lomer MC,Thompson RP, Powell JJ (2002) Fine and ultrafine particles of the diet: influence on the mucosal immune response and association with Crohn's disease. Proc Nutr Soc 61: 123-130. [Crossref].
- Barone F, De Berardis B, Bizzarri L, et al. (2011) Physico-chemical characteristics and cyto-genotoxic potential of ZnO and TiO 2 nanoparticles on human colon carcinoma cells. J Phys Conf Ser 304:012047.
- Gerloff K, Albrecht C, Boots AW, et al. (2009) Cytotoxicity and oxidative DNA damage by nanoparticles in human intestinal Caco-2 cells. Nanotoxicology 3: 355–364.
- Gerloff K, Fenoglio I, Carella E, Kolling J, Albrecht C, et al. (2012) Distinctive toxicity of TiO2 rutile/anatase mixed phase nanoparticles on Caco-2 cells. Chem Res Toxicol 25: 646-655. [Crossref].
- Fisichella M, Berenguer F, Steinmetz G, et al. (2012) Intestinal toxicity evaluation of TiO2 degraded surface-treated nanoparticles: a combined physico-chemical and toxicogenomics approach in caco-2 cells. Part Fibre Toxicol 9: 18.
- Ivask A,Titma T, Visnapuu M, Vija H, Kakinen A, et al. (2015) Toxicity of 11 Metal Oxide Nanoparticles to Three Mammalian Cell Types In Vitro. Curr Top Med Chem 15: 1914-1929. [Crossref].
- Chalew TEA, Schwab KJ (2013) Toxicity of commercially available engineered nanoparticles to Caco-2 and SW480 human intestinal epithelial cells. Cell Biol Toxicol 29: 101–116.
- Krüger K, Cossais F, Neve H, Klempt M (2014) Titanium dioxide nanoparticles activate IL8-related inflammatory pathways in human colonic epithelial Caco-2 cells. J Nanoparticle Res 16: 2402.
- Mano SS,Kanehira K, Taniguchi A (2013) Comparison of cellular uptake and inflammatory response via toll-like receptor 4 to lipopolysaccharide and titanium dioxide nanoparticles. Int J Mol Sci 14: 13154-13170. [Crossref].
- Geiser M, Rothen-Rutishauser B, Kapp N, et al (2005) Ultrafine Particles Cross Cellular Membranes by Nonphagocytic Mechanisms in Lungs and in Cultured Cells. Environ Health Perspect 113: 1555–1560.
- He B, Lin P, Jia Z, Du W, Qu W, et al. (2013) The transport mechanisms of polymer nanoparticles in Caco-2 epithelial cells. Biomaterials 34: 6082-6098. [Crossref].
2021 Copyright OAT. All rights reserv
- Bannunah AM,Vllasaliu D, Lord J, Stolnik S (2014) Mechanisms of nanoparticle internalization and transport across an intestinal epithelial cell model: effect of size and surface charge. Mol Pharm 11: 4363-4373. [Crossref].
- Gitrowski C,Al-Jubory AR2, Handy RD3 (2014) Uptake of different crystal structures of TiOâ‚‚ nanoparticles by Caco-2 intestinal cells. Toxicol Lett 226: 264-276. [Crossref].
- Koeneman BA, Zhang Y, Westerhoff P, Chen Y, Crittenden JC, et al. (2010) Toxicity and cellular responses of intestinal cells exposed to titanium dioxide. Cell Biol Toxicol 26: 225-238. [Crossref].
- Brun E, Barreau F, Veronesi G, Fayard B, Sorieul S, et al. (2014) Titanium dioxide nanoparticle impact and translocation through ex vivo, in vivo and in vitro gut epithelia. Part Fibre Toxicol 11: 13. [Crossref].
- Janer G,Mas del Molino E,Fernández-Rosas E,Fernández A,Vázquez-Campos S2 (2014) Cell uptake and oral absorption of titanium dioxide nanoparticles. Toxicol Lett 228: 103-110. [Crossref].
- Gonçalves DM,Chiasson S, Girard D (2010) Activation of human neutrophils by titanium dioxide (TiO2) nanoparticles. Toxicol In Vitro 24: 1002-1008. [Crossref].
- Kang SJ, Kim BM, Lee YJ, et al. (2009) Titanium dioxide nanoparticles induce apoptosis through the JNK/p38-caspase-8-Bid pathway in phytohemagglutinin-stimulated human lymphocytes. Biochem Biophys Res Commun 386: 682–687.
- Gui S, Zhang Z, Zheng L, et al (2011) Molecular mechanism of kidney injury of mice caused by exposure to titanium dioxide nanoparticles. J Hazard Mater 195: 365–370.
- Hussain S,Thomassen LC, Ferecatu I, Borot MC, Andreau K, et al. (2010) Carbon black and titanium dioxide nanoparticles elicit distinct apoptotic pathways in bronchial epithelial cells. Part Fibre Toxicol 7: 10. [Crossref].
- Li N, Duan Y, Hong M, Zheng L, Fei M, et al. (2010) Spleen injury and apoptotic pathway in mice caused by titanium dioxide nanoparticules. Toxicol Lett 195: 161-168. [Crossref].
- Morishige T, Yoshioka Y, Tanabe A, et al (2010) Titanium dioxide induces different levels of IL-1ß production dependent on its particle characteristics through caspase-1 activation mediated by reactive oxygen species and cathepsin B. Biochem Biophys Res Commun 392: 160–165.
- Yoo KC,Yoon CH, Kwon D, Hyun KH, Woo SJ, et al. (2012) Titanium dioxide induces apoptotic cell death through reactive oxygen species-mediated Fas upregulation and Bax activation. Int J Nanomedicine 7: 1203-1214. [Crossref].
- Chen P, Kanehira K, Taniguchi A (2013) Role of toll-like receptors 3, 4 and 7 in cellular uptake and response to titanium dioxide nanoparticles. Sci Technol Adv Mater 14: 015008.
- Park EJ,Yi J, Chung KH, Ryu DY, Choi J, et al. (2008) Oxidative stress and apoptosis induced by titanium dioxide nanoparticles in cultured BEAS-2B cells. Toxicol Lett 180: 222-229. [Crossref].
- Totlandsdal AI, Refsnes M, Låg M (2010) Mechanisms involved in ultrafine carbon black-induced release of IL-6 from primary rat epithelial lung cells. Toxicol In Vitro 24: 10-20. [Crossref].
- Sydlik U, Bierhals K, Soufi M, et al.(2006) Ultrafine carbon particles induce apoptosis and proliferation in rat lung epithelial cells via specific signaling pathways both using EGF-R. Am J Physiol Lung Cell Mol Physiol 291: L725–L733.
- Peuschel H, Sydlik U, Haendeler J, et al. (2010) C-Src-mediated activation of Erk1/2 is a reaction of epithelial cells to carbon nanoparticle treatment and may be a target for a molecular preventive strategy. Biol Chem 391: 1327–1332.
- Weissenberg A,Sydlik U, Peuschel H, Schroeder P, Schneider M, et al. (2010) Reactive oxygen species as mediators of membrane-dependent signaling induced by ultrafine particles. Free Radic Biol Med 49: 597-605. [Crossref].
- Comfort KK,Maurer EI, Braydich-Stolle LK, Hussain SM (2011) Interference of silver, gold, and iron oxide nanoparticles on epidermal growth factor signal transduction in epithelial cells. ACS Nano 5: 10000-10008. [Crossref].
- Skuland T, Øvrevik J, Låg M, et al.(2014) Silica nanoparticles induce cytokine responses in lung epithelial cells through activation of a p38/TACE/TGF-a/EGFR-pathway and NF-?? signalling. Toxicol Appl Pharmacol 279: 76–86.
- Zhang L, Li N, Caicedo R, Neu J (2005) Alive and dead Lactobacillus rhamnosus GG decrease tumor necrosis factor-alpha-induced interleukin-8 production in Caco-2 cells. J Nutr 135: 1752-1756. [Crossref].
- Hennenberg M, Strittmatter F, Beckmann C, et al. (2012) Silodosin Inhibits Noradrenaline-Activated Transcription Factors Elk1 and SRF in Human Prostate Smooth Muscle. PLoS One 7: 1–8.
- Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30: e36. [Crossref].
- Kaulfuss S, Burfeind P, Gaedcke J, Scharf J-G (2009) Dual silencing of insulin-like growth factor-I receptor and epidermal growth factor receptor in colorectal cancer cells is associated with decreased proliferation and enhanced apoptosis. Mol Cancer Ther 8: 821–833.
- Jorissen RN,Walker F, Pouliot N, Garrett TP, Ward CW, et al. (2003) Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res 284: 31-53. [Crossref].
- Lemmon MA,Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141: 1117-1134. [Crossref].
- Roskoski R Jr1 (2012) ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 66: 105-143. [Crossref].
- Samak G, Aggarwal S, Rao RK (2011) ERK is involved in EGF-mediated protection of tight junctions, but not adherens junctions, in acetaldehyde-treated Caco-2 cell monolayers. Am J Physiol Gastrointest Liver Physiol 301: G50–G59.
- Tamaoki J, Isono K, Takeyama K, et al. (2004) Ultrafine carbon black particles stimulate proliferation of human airway epithelium via EGF receptor-mediated signaling pathway. 1127–1133.
- Jijon HB, Panenka WJ, Madsen KL, Parsons HG (2002) MAP kinases contribute to IL-8 secretion by intestinal epithelial cells via a posttranscriptional mechanism. Am J Physiol Cell Physiol 283: C31–41.
- Kuwahara I,Lillehoj EP, Lu W, Singh IS, Isohama Y, et al. (2006) Neutrophil elastase induces IL-8 gene transcription and protein release through p38/NF-{kappa}B activation via EGFR transactivation in a lung epithelial cell line. Am J Physiol Lung Cell Mol Physiol 291: L407-416. [Crossref].
- Lin CH,Yu MC, Chiang CC, Bien MY, Chien MH, et al. (2013) Thrombin-induced NF-κB activation and IL-8/CXCL8 release is mediated by c-Src-dependent Shc, Raf-,and ERK pathways in lung epithelial cells. Cell Signal 25: 1166-1175. [Crossref].
- Jeng Y-J, Watson CS (2009) Proliferative and anti-proliferative effects of dietary levels of phytoestrogens in rat pituitary GH3/B6/F10 cells - the involvement of rapidly activated kinases and caspases. BMC Cancer 9: 334.
- Schweppe RE,Cheung TH, Ahn NG (2006) Global gene expression analysis of ERK5 and ERK1/2 signag reveals a role for HIF-1 in ERK5-mediated responses. J Biol Chem 281: 20993-21003. [Crossref].
- Pastore S,Mascia F, Mariotti F, Dattilo C, Mariani V, et al. (2005) ERK1/2 regulates epidermal chemokine expression and skin inflammation. J Immunol 174: 5047-5056. [Crossref].
- Rada B, Gardina P, Myers TG, Leto TL (2011) Reactive oxygen species mediate inflammatory cytokine release and EGFR-dependent mucin secretion in airway epithelial cells exposed to Pseudomonas pyocyanin. Mucosal Immunol 4: 158–171.
- Yang SK, Eckmann L, Panja A, Kagnoff MF (1997) Differential and regulated expression of C-X-C, C-C, and C-chemokines by human colon epithelial cells. Gastroenterology 113: 1214-1223. [Crossref].
- Frey MR, Golovin A, Polk DB (2004) Epidermal growth factor-stimulated intestinal epithelial cell migration requires Src family kinase-dependent p38 MAPK signaling. J Biol Chem 279: 44513–44521.
- Dise RS, Frey MR, Whitehead RH, Polk DB (2008) Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol 294: G276–85.
- Unfried K,Sydlik U, Bierhals K, Weissenberg A, Abel J (2008) Carbon nanoparticle-induced lung epithelial cell proliferation is mediated by receptor-dependent Akt activation. Am J Physiol Lung Cell Mol Physiol 294: L358-367. [Crossref].