Next-generation sequencing technologies have shown that different subpopulations of malignant cells with differing biological behaviors coexist within the same tumor. This concept also extends to metastases and to differences between primary and metastatic tumors. Cancer heterogeneity has deep implications for therapy. In spite of this knowledge, no fundamental steps have been introduced in the clinical field to deal with the problem. It is the objective of this mini-review to analyze possible measures to circumvent the therapeutic problems represented by tumor heterogeneity.
Common misconceptions about cancer are that it is one single disease, that a single genetic mutation is the driver of a tumor and that all parts of a tumor are the same. It may be so at the very beginning, but as said, the single mutation is just the beginning of a cascade of genetic changes that grow out of control. Cancer is not one disease but a group of diseases that share common hallmarks as described by Hanahan and Weinberg [1]. And we may go even further and say that one tumor in a patient is more than one disease in that same patient. So it is a multiplicity of diseases. And in the middle of this, Darwinian evolution of genetic mutations and epigenetic changes provide the explanation for the survival of the most fit clones, that at a certain point become resistant to almost all known treatment protocols.
Cancer may start (and usually does) as a monoclonal disease. But as it advances a multiclonal evolution is built up.
Therefore, two patients harboring the same type of cancer (let’s say a squamous cell carcinoma of the tongue) may show different genetic mutations and different epigenetic changes. Furthermore, different parts of this tumor may show different environments and phenotypes.
But there is another factor that makes this worse: a patient having a cancer may show many different malignant cells with different genetic mutations in the same tumor: this is genetic heterogeneity which makes each cancer unique. And different parts of this tumor may have different environmental conditions. This makes it even more unique.
Hao JJ et al. [2] found that in 13 cases of esophageal squamous cell carcinomas there were an average of 35.8% heterogeneous somatic mutations with strong evidence of intratumoral heterogeneity.
But this concept is not new. Back in 1976 Novell [3] already called our attention to this issue. This idea of cancer as an “evolutionary process driven by stepwise, somatic cell mutations with sequential sub-clonal selection” [4] was a change in the understanding of cancer biology.
As we can see, the knowledge and the problem of tumor heterogeneity is not a new one.
The Gombertzian model of tumor growth that many tumors show at the initial stages [5] and becomes absolutely heterogeneous in the late stages is partially a consequence of tumor heterogeneity. But according to our presumptions, the lack of Gombertzian growth from the beginning is also a consequence of tumor heterogeneity.
Kuukasjärvi T et al. [6] studied the comparative genotypes of breast cancers and their asynchronous metastasis. In the 31 cases studied, 20 of the metastases (69%) showed a high degree of clonal relationship with the corresponding primary tumor, whereas the genetic composition of 9 metastases (31%) differed almost completely from that of the paired primary tumors. In both groups, however, chromosome X inactivation patterns suggested that the metastatic lesions originated from the same clone as the primary tumor. Their conclusion was that an early stem line clone apparently evolved independently in the primary tumor and in its metastasis, eventually leading to multiple, genetically almost completely different, clones in the various tumor locations in a given patient.
To this concept we have to add, that not only metastatic cells may differ from the primary tumor, but also that we may find different genetic types in the same tumor (individual uniqueness of each cancer).
In a case report by De Mattos-Arruda et al. [7] a patient with breast cancer with bone and liver metastasis showed sixteen somatic non-synonymous mutations in the liver metastasis, 9 of which were also detected in the primary tumor sample. Not all mutations identified in the metastasis were reliably identified in the primary tumor.
A first conclusion is that there is extensive genetic diversity among and within tumors.
This is the fundamental reason why conventional chemotherapy and even personalized directed therapy so frequently end up as failures.
Usual chemotherapy is a massive attack on the mitotic system with the capacity of destroying malignant and normal cells. This fact limits its potential due to toxicity to normal cells. This means that it lacks the capacity of destroying 100% of the malignant cells, leading to resistance at a certain stage of tumor progression allowing the development of mutated resistant cells. Greaves and Maley [4] described this process with these words: “Therapeutic intervention may decimate cancer clones, and erode their habitats, but inadvertently provides potent selective pressure for the expansion of resistant variants. The inherently Darwinian character of cancer lies at the heart of therapeutic failure but perhaps also holds the key to more effective control.”
Targeted therapies on the other hand, attack specific characteristics of malignant cells, but the problem that we find here is that not all the malignant cells have the specific target (driver mutation) due to genetic heterogeneity. The cells that do not have the target will survive and continue with their evolution.
It is important to define one term: tumor evolution. It is the process of genetic change and selection of the fittest. This means that those cells harboring pro-survival genetic changes will generate the new clones resistant to the harsh environment and the chemoradiotherapy intents of oncologists. The Darwinian selection of malignant tumors has the capacity to generate these “evolutioned” new clones in matter of weeks or months when a similar evolution in nature may take not only years but centuries.
What is responsible for this accelerated evolution?
Basically genomic instability.
Mutations generating major genomic instability are necessary to explain the accumulation of the multiple mutations seen in cancer and the short time in which they occur [8]. These mutations, which increase the inherent rate of genetic change, are referred to as mutator mutations; cell lineages harboring them are called mutator clones, and cancer cells are said to exhibit a mutator phenotype [9].
We are not going to dwell on how many mutations are needed to develop a malignant phenotype because according to all the published medical literature we need to distinguish between two different pathways leading to the development of a malignant cell:
- Tumorigenesis occurs through a multi-step process (usually 2 to 8 mutations) in which normal cells progress through a series of premalignant phenotypes until an invasive cancer emerges. For example colon adenomas progressing to carcinoma in situ and finally to colorectal carcinoma.
- A one step process in which parts of chromosomes with their genes are transposed to a different chromosome. For example chromosome Philadelphia in chronic myeloid leukemia.
Within the first category we include the seminal discovery of the two hit process by Knudson in the case of retinoblastoma.
So the number of hits or mutations may differ according to the type of cancer we are dealing with, but undoubtedly genetic or chromosomal instability are the vehicles that lead to polyclonal heterogeneity in cancer.
Heterogeneity is thus the main reason why we are unable to eliminate all the malignant cells contained in a tumor or in a patient. The residual malignant cells that through mutations have escaped our treatments are the primum movens of relapses. We have already mentioned the genetic differences usually found between the primary tumor and metastases.
The pathway we are following nowadays in the treatment of advanced cancer has shown to be poorly effective in many cases, when not a total failure.
Ding et al. [10] showed clearly the influence of tumor heterogeneity on the results of treatment and relapse. Working with acute myeloid leukemia patients, they observed two types of clonal evolution patterns. In the first, the original clone gained mutations and progressed towards the relapsed clone. In this case the chemotherapy failed to eradicate all the founding cells, leaving a few alive that evolved incorporating new mutations and producing the relapse. In the second type of clonal evolution a subclone of the original tumor that survived chemotherapy underwent new mutations that were responsible for the relapse. This research shows that relapse is related to new mutations and evolution and that chemotherapy is unable to eradicate all the malignant cells.
We cannot change genetic and behavioral heterogeneity. It is almost always there, when we face advanced cancer and so far, there is no known way to eliminate it. So we must, out of necessity, do something about this.
Our caveats indicate that we have to find a way to circumvent heterogeneity or at least find accessory ways to eliminate surviving cells.
Almost all malignant cells in an advanced stage of evolution have three common characteristics:
- Increased need of energy
- A particularly harsh environment characterized by lack of nutrients, hypoxia and extracellular acidity.
- Tendency to migrate, invade and metastasize.
If, in addition to the classical treatments (surgery, chemoradiotherapy, targeted treatments, immunoregulation, etc.) we employ, we add an attack on the three above elements it is highly possible that the results of our treatments will improve.
There is a limit to what conventional chemotherapy can do, and that limit is defined by the toxicity to normal cells and the development of resistance, so in the best case scenario the number of surviving malignant cells is small. But they exist. Heterogeneity in this case may show clones that are resistant to the drugs employed. In other words, what we are actually doing with chemotherapy in certain cases is killing nonresistant cells and allowing the development of resistant clones. So we are responsible for the selection of the more fit malignant cells. There is also a limit to targeted therapy which is tumor heterogeneity in that there might be malignant clones that do not possess the target.
Independently of heterogeneity, all quickly-growing tumors need more energy than normal tissues. All solid tumors live in, and adapt to, a difficult environment characterized by hypoxia, lack of nutrients and extracellular acidity. And most of the tumors have a tendency to metastasize.
What we are proposing here is that, in addition to the conventional treatments, synergistically targeting all three of these characteristics of malignant tissues would represent an advantage for a better treatment and also would circumvent what we consider one of the main reasons of treatment failure: heterogeneity.
Let’s take a closer look at these concepts.
The three hallmarks described above that are common to the majority of advanced malignant tumors are integrated and interdependent issues, because most tumors are characterized by regions of acidity and hypoxia. Malignant cells typically exhibit altered metabolic patterns marked by increased glucose uptake and elevated glycolysis. Compared to the aerobic metabolism this inefficient energy production, forces tumor cells to upregulate their glycolytic pathway, which increases the production and release of H+-ions. It results in an acidification of the local micro-environment, which promotes apoptosis of normal cells, while cancer cells can survive and proliferate and increase migration. Hence, cancer cells get an evolutionary advantage over normal cells, an advantage that facilitates invasion (Figure 1).
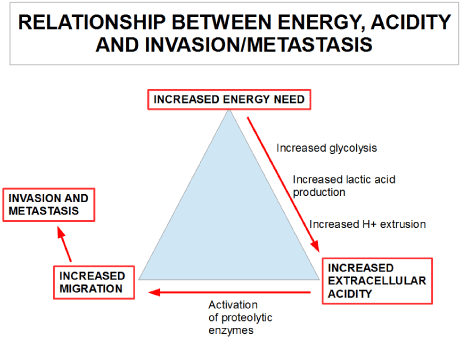
Figure 1. The relationship between increased energy need, increased extracellular acidity and increased migration.
All tumors exhibit a high energy consumption, independently of whether they have one, ten, or one hundred mutations. So energy requirements are independent of the mutations a malignant cell undergoes and as well as of the tumor's genetic instability. On the other hand, energy requirements do depend on the cell's biosynthetic rate. This means that those cells with a high metabolic rate like the majority of malignant cells exhibit, need a greater energy supply. There might be variations in the way the cell gets its energy within a certain tumor. This means many types of preferential metabolic pathways may be used in the same tumor, but they all coincide with the high energy requirement. Some cells utilize glucose and degrade it to lactic acid (aerobic glycolysis), other cells utilizing glucose degrade it to water and carbon dioxide (oxidative phosphorylators), other cells may use lactic acid as the energy source by introducing it in the tricarboxylic acid pathway (oxidative phosphorylators). Finally there are malignant cells that obtain their energy from glutamine (glutaminergic cells).
Malignant cells stop growing when ATP production is reduced to a critical level (1/3 of the normal level). This can be achieved with mitochondrial poisons like metformin, doxycycline, fenformin complemented with bromopiruvate and 2 deoxyglucose.
Could heterogeneity be a problem for these treatments? Basically no, because it does not matter if the cell is following the Warburg effect or the oxidative phosphorylation pathway, the high energy requirement is a universal need that should not be changed by the usual mutations found in cancer. There may be differences in those cells that mainly use the glutaminergic pathway. These cells would escape our energy restriction therapy, but glutaminergic pathways can also be blocked.
Therefore, energy restriction may partially circumvent the heterogeneity problem that permits the survival of resistant clones.
A very simple experiment will show the importance of energy restriction in a malignant cell’s life: those patients who reduce their calorie intake by 30 to 40% show a slowdown in cancer growth.
In 1909 Moreschi found that tumors transplanted into underfed mice did not grow as much as those transplanted into mice fed ad libitum. Soon after that, it was discovered that caloric restriction also negatively affected the growth of spontaneous tumors. In the 1940s Tannenbaum and Baumann showed that this effect was due to the caloric content of the diet independently of the source of calories.
Energy restriction enhances DNA repair and moderates oxidative damage to DNA. Energy restriction also reduces oncogene expression [11].
According to Xue and Michels more recent findings suggest a hormesis hypothesis proposing that caloric restriction creates a low-intensity biological stress on organisms, which may elicit an adaptive response of enhanced maintenance and repair [12].
We have our own simplistic idea: caloric restriction decreases the available energy that the cell can use for growth.
Pearson et al. [13] found one of the possible pathways that led from caloric restriction to anti cancer activity: Nrf2.
2021 Copyright OAT. All rights reserv
Also certain compounds like sulforaphane and resveratrol can mimic the translational modifications found under caloric restriction [13-14].
Foods containing high levels of sulforaphane like broccoli, Brussels sprouts or cabbage may produce a similar effect to caloric restriction regarding Nrf2 . (For further reading about sulforaphane see Thimmulappa et al. [15])
Using metformin or other mitochondrial poisons plus caloric restriction (15% reduction of calorie intake is sufficient for this purpose) and 2-deoxy-glucose would be a non toxic adjuvant therapy that would decrease the survival of cells that are resistant to conventional treatments.
Increased extracellular acidity is a hallmark of cancer cells. It is mainly the result of high lactic acid production and extrusion from malignant cells and an hypoxic environment due to imbalanced vascularization.
The acidification of the local environment is crucial for cancer cells’ invasiveness. While it promotes the death of normal cells, cancer cells can survive and proliferate since they are able to regulate their intracellular pH level through several membrane based transport mechanisms (e.g., Na+/H+ exchanger NHE1, monocarboxylate transporters MCT1/MCT4, voltage gated sodium channels and V-ATPase pumps). The acidification of the microenvironment enhances the activity of proteolytic enzymes like catepsines and metalloproteases and this permits the necessary remodeling of the environment in order for migration and invasion to take place [16].
When microenvironmental acidity is experimentally decreased, migration and metastasis are diminished in a very significant way.
Extracellular acidity is a universal phenomenon which is present in all the highly glycolytic tumors, independently of tumor heterogeneity. Probably the less metabolically active parts of the tumor are less acidic, but we need to target the more metabolically active parts specifically. So tumor heterogeneity, in this sense, is an advantage for the antacid treatment because this allows the more aggressive areas of the tumor to come under attack.
Acidity at the extracellular level is also very heterogeneous, so that different parts of a malignant tumor show different pH values and sometimes these differences are very important [17].
Increasing the pH of the tumor's microenvironment is the first step towards decreasing migration. For this approach the use of repurposed non toxic drugs may make a difference. And we are referring to very simple drugs such as proton pump inhibitors like omeprazole or pantoprazole plus bicarbonate plus acetazolamide.
Then, by targeting proteolytic enzymes like catepsines and metallopreoteases a further reduction of migration may be achieved. There are many non toxic drugs that can decrease metastasis. Zoledronic acid would be an example regarding bone metastasis.
We strongly believe that attacking the three issues that are independent of heterogeneity (energy supply, pH gradient inversion and metastatic cascade), with re-purposed drugs as a complementary treatment to conventional therapies would decrease the resistance born from tumor heterogeneity and reduce relapses and metastases.
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-674. [Crossref]
- Hao JJ, Lin DC, Dinh HQ, Mayakonda A, Jiang YY (2016) Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat Genet 48: 1500-1507. [Crossref]
- Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194: 23-28. [Crossref]
- Greaves M, Maley CC (2012) Clonal evolution in cancer. Nature 481: 306-313. [Crossref]
- Speer JF, Petrosky VE, Retsky MW, Wardwell RH (1984) A stochastic numerical model of breast cancer growth that simulates clinical data. Cancer Res 44: 4124-4130. [Crossref]
- Kuukasjärvi T, Karhu R, Tanner M, Kähkönen M, Schäffer A, et al. (1997) Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res 57: 1597-1604. [Crossref]
- De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, et al. (2014) Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol 25: 1729-1735. [Crossref]
- Loeb LA (1991) Mutator phenotype may be required for multistage carcinogenesis. Cancer Res 51: 3075-3079. [Crossref]
- Beckman RA, Loeb LA (2005) Genetic instability in cancer: theory and experiment. Semin Cancer Biol 15: 423-435. [Crossref]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, et al. (2012) Clonal evolution in relapsed acute myeloid leukemia revealed by whole genome sequencing. Nature 481: 506-510. [Crossref]
- Kritchevsky D (2001) Caloric restriction and cancer. J Nutr Sci Vitaminol (Tokyo) 47: 13-19. [Crossref]
- Xue, Fei, Karin B (2010) Michels. Caloric restriction and cancer. Cancer and Energy Balance, Epidemiology and Overview. Springer New York: 181-199.
- Pearson KJ, Lewis KN, Price NL, Chang JW, Perez E, et al. (2008) Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc Natl Acad Sci U S A 105: 2325-2330. [Crossref]
- Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, et al. (2008) Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab 8: 157-168. [Crossref]
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, et al. (2002) Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 62: 5196-5203. [Crossref]
- Koltai T (2016) Cancer: fundamentals behind pH targeting and the double-edged approach. Onco Targets Ther 9: 6343-6360. [Crossref]
- Sebastian John, Sivakumar KC, Mishra R (2017) Extracellular proton concentrations impacts LN229 glioblastoma tumor cell fate via differential modulation of surface lipids. Front Oncol 7: 20. [Crossref]